Advancing beyond current generation dye-sensitized solar cells
Thomas W.
Hamann
ab,
Rebecca A.
Jensen
a,
Alex B. F.
Martinson
a,
Hal
Van Ryswyk
ac and
Joseph T.
Hupp
*a
aNorthwestern University, 2145 Sheridan Rd, Evanston, Illinois 60208, USA. E-mail: j-hupp@northwestern.edu
bMichigan State University, Department of Chemistry, East Lansing, MI 48824-1322, USA
cHarvey Mudd College, 301 Platt Boulevard, Claremont, CA 91711, USA
First published on 8th July 2008
Abstract
The most efficient dye-sensitized solar cells (DSSCs) have had essentially the same configuration (nanoparticle TiO2 sensitized with [Ru(4,4′-dicarboxy-2,2′-bipyridine)2(NCS)2] in contact with I3−/I−) for the last 17 years. In this article we outline the strategies for improving each of the three major photo-relevant components of a DSSC, review literature reports consistent with these strategies and suggest future directions. Finally we explore the potential of future generation DSSCs for advancing energy-conversion performance.
 Thomas Hamann | Thomas Hamann is an Assistant Professor of Chemistry at Michigan State University. From 2006 to mid-2008 he was a postdoctoral fellow in the Department of Chemistry at Northwestern University. He obtained a BA in chemistry from the University of Texas, an MS in chemistry from the University of Massachusetts, and a PhD in chemistry at California Institute of Technology. His research interests and expertise center on solar energy conversion with semiconductor-liquid junctions. |
Rebecca Jensen is a Northwestern University graduate student pursuing a PhD in inorganic chemistry. Her work comprises the design and synthesis of porphyrinic dyes for photoelectrode sensitization. She obtained a BS degree in chemistry from Western Washington University in 2005. |
Alex Martinson is a 2008 graduate of the PhD program in Chemistry at Northwestern. His graduate work focused on the fabrication and characterization of new architectures for dye-sensitized solar cells. He now holds a Director’s Postdoctoral Fellowship in the Materials Science Division of Argonne National Laboratory where he investigates the effects of alternative electrode geometries on a variety of photovoltaics. |
Hal Van Ryswyk is Professor of Chemistry at Harvey Mudd College in Claremont, California. He earned a BA in chemistry at Carleton College and a PhD at the University of Wisconsin-Madison. He recently completed a sabbatical year at Northwestern. His research interests and expertise include photoelectrochemistry and porphyrin chemistry. |
Joseph Hupp holds a Morrison Professorship at Northwestern University where he serves as chair of the University’s Department of Chemistry. Prior to joining NU in 1986, he earned a BS degree from Houghton College and a PhD from Michigan State. He did postdoctoral work at the University of North Carolina. His current research is focused on photoelectrochemical energy conversion and on the design and synthesis of functional molecular materials. |
1. Introduction
A. Background
The worldwide demand for energy is expected to double by the year 2050 and triple by the end of the century.1 An abundant supply of energy is necessary for global political, economic and environmental stability. There is growing concern, however, that the production of oil will soon not be able to keep up with the growing demand leading to dire economic consequences.2–5 In addition, the combustion of fossil fuels has been implicated in anthropogenic global warming, which is predicted to produce widespread environmental damage.6 The development of carbon-free sources of energy that are scalable to meet increasing societal demands is therefore one of the major scientific challenges of this century.7–9Light from the sun is arguably the ideal source of energy. The solar flux striking the earth contains 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times the average global power usage, and is the largest single source of clean energy which is readily available.1 While technologies have been developed to harness solar energy efficiently, they are not yet an economically viable alternative to fossil fuels.7 The abundant supply and environmental friendliness of solar energy make the efficient and cost-effective conversion of solar radiation into electricity a compelling scientific goal.
000 times the average global power usage, and is the largest single source of clean energy which is readily available.1 While technologies have been developed to harness solar energy efficiently, they are not yet an economically viable alternative to fossil fuels.7 The abundant supply and environmental friendliness of solar energy make the efficient and cost-effective conversion of solar radiation into electricity a compelling scientific goal.
Dye-sensitized solar cells (DSSCs) have attracted much attention as they offer the possibility of extremely inexpensive and efficient solar energy conversion. In 1991 O'Regan and Grätzel published a remarkable report: a 7% efficient DSSC based on nanocrystalline TiO2.10 Subsequent work by the Grätzel group quickly (1993) pushed the efficiency to 10%.11 The maximum efficiency, however, plateaued over the following 15 years with a current record of ∼11%.12–19 While the three major components of the photo-related portion of the DSSC—dye, redox shuttle and photoanode—have been investigated independently over the last 15 years, the most efficient device remains essentially unchanged from its conception.
In the most studied and most efficient devices to date, light is absorbed by a ruthenium-based molecular dye, e.g. [Ru(4,4′-dicarboxy-2,2′-bipyridine)2(NCS)2] (N3), that is bound to a photoanode viacarboxylate moieties.11,13,20 The photoanode is composed of a ∼12 μm thick film of transparent 10–20 nm diameter TiO2nanoparticles covered with a ∼4 μm thick film of much larger (∼400 nm diameter) particles that scatter photons back into the transparent film.15,16 Following light absorption, the excited dye rapidly injects an electron into the TiO2. The injected electron diffuses through the sintered particle network to be collected at the front-side transparent conducting oxide (TCO) electrode, while the dye is regenerated viareduction by a redox shuttle, I3−/I−, dissolved in a solution. Diffusion of the oxidized form of the shuttle to the counter electrode completes the circuit. There are also processes which thwart the successful operation of a DSSC: the excited dye can decay (either radiatively or nonradiatively) before it injects an electron, the injected electron can recombine with the oxidized dye before the dye is regenerated, or the redox shuttle can intercept an electron from the photoanode before it is collected, Fig. 1b.
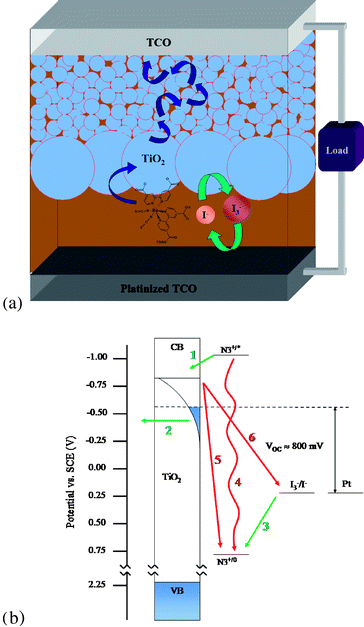 | ||
| Fig. 1 (a) Schematic of a typical DSSC (see Fig. 3 for detailed dye structure). (b) Approximate energy diagram of a conventional DSSC. Desirable processes (1. electron injection, 2. charge collection, 3. regeneration) are shown with green arrows and deleterious processes (4. luminescence or nonradiative decay, 5. recombination, 6. interception) are shown with red arrows. | ||
To clarify a potentially confusing point, “interception” as used here encompasses electrolyte capture of electrons that have been injected by the dye into the photoanode (photocurrent) as well as electrons (majority carriers) injected from the back contact (the potential-dependent dark current). The two processes are conceptually distinct and conditions will certainly be attained where only one process contributes (e.g. only dark current in the absence of illumination) or where neither contributes (e.g. illumination at short-circuit, with 100% current-collection efficiency). At open circuit, the photocurrent (approximately potential independent) is exactly offset by the dark current, resulting in zero overall current. It is convenient to think of this as compensation of the photo-generated current by an interception pathway that involves (only) capture of the electrons injected by the back contact, and indeed this is mathematically correct (i.e. “superposition” holds). In practice, we have no way of distinguishing photo-injected electrons from “dark” electrons once they have entered the photoanode—nor does the electrolyte. From a design perspective, the microscopic indistinguishability of dark electrons from photo-injected electrons implies that strategies that alter the rate constant for the interception of photo-injected electrons will identically affect the rate constant for the interception of back-contact-injected electrons.
B. Route to improvement
The overall solar conversion efficiency, η, is a product of the short-circuit current density, Jsc, the open-circuit photovoltage, Voc, and the fill factor, FF, according to: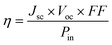 | (1) |
The most straightforward way to increase Jsc is to absorb a greater fraction of the incident light. The optical gap of the Ru dye in the most efficient DSSC to date is ∼1.8 eV, allowing it to absorb essentially all the light out to ∼700 nm.21 Increasing the photocurrent density requires decreasing the optical gap to extend the dye's absorption into the near-infrared. There are two ways to narrow the dye's optical gap: lower the energy of the LUMO or raise the energy of the HOMO. From an electrochemical perspective, these are approximately equivalent to shifting the dye+/* potential to less negative values or making the dye+/0 potential less positive.
We assume the dye's excited state potential cannot be shifted significantly in the positive direction without impairing unity charge injection efficiency—there must be good energy matching between the electron donor (excited dye) and acceptor (semiconductor conduction band) in order for injection to occur at a sufficiently rapid rate.22 This is a subtle point for N3-based cells. The observation of sub-50 fs injection dynamics and ca. 25 ns excited-state decay times, would seem to suggest that there is ample room for lowering the LUMO before injection yields are appreciably diminished. As pointed out by Haque and co-workers,22 the polydispersity of the injection dynamics (including a ca. 10% component with nanosecond dynamics) prevents the observed fast-injection behavior from being exploited. A significant advance would be the discovery of conditions or systems displaying uniformly rapid injection dynamics, as the fast dynamics indeed could then be exploited to enhance photovolatge output and/or other cell performance parameters. The alternative strategy would be to shift the ground-state potential negative; this idea is explored in the dye section below.
The other way to increase the efficiency is to increase the Voc. The Voc is the difference between the Nernstian potential of the solution and the semiconductor's quasi-Fermi level (sometimes referred to as the electron chemical potential).23 At open circuit, the rates of electron injection and recombination/interception are equal, and their values determine the steady-state electron concentration in the semiconductor and therefore its quasi-Fermi level. The quasi-Fermi level, and thus Voc, can be increased by increasing the rate of electron injection (Jsc) or decreasing recombination/interception (the dark current density; strictly speaking, the dark current density depends only on interception). The quasi-Fermi level is extremely unlikely to be higher than the conduction band edge, leaving approximately 200 mV room for improvement in Voc by increasing the quasi-Fermi level. In accord with the diode equation, the photovoltage is expected to increase by ∼60 mV with each order of magnitude increase in injection rate or reduction in dark current density. Thus, large changes in injection or interception are necessary in order to make modest improvements in the photovoltage. It is important to note that the rate of electron injection, not the microscopic dynamics, is what is potential-determining. Since the best cells already inject at essentially the maximum rate, i.e. a rate that matches the light-harvesting rate, increasing the injection rate constant will not change the photovoltage. Improved light harvesting (discussed below), however, can produce larger photovoltages.
The second way to increase Voc is to make the solution potential more positive. In optimal devices, the dye+/0 (N3+/0) potential differs from the I3−/I− potential by approximately 550 mV. Thus, there is theoretically much room for improvement in Voc with alternative redox couples possessing more positive potentials. This strategy is explored in the redox couple section below.
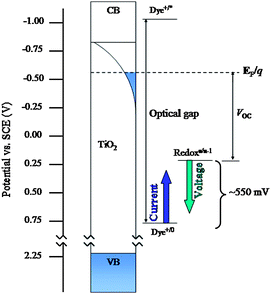 | ||
| Fig. 2 Approximate energy diagram showing two strategies for improving the efficiency of DSSCs discussed: shifting the dye's ground state potential to a less positive value (while holding the excited-state potential fixed) and/or shifting the redox shuttle's electrochemical potential to a more positive value. The first strategy allows for more light collection and greater photocurrent density. The second allows for greater photovoltage. Note that energies on the electrochemical potential scale vary in the direction opposite to the vacuum scale. | ||
A DSSC comprises four major components—the dye, redox shuttle, semiconducting photoanode, and dark electrode—making it essentially modular. In the standard configuration, these components have been well-optimized to maximize the product of Jsc, Voc and FF. It is therefore not surprising that any variation of the configuration has led to worse overall photovoltaic performance. In order to make significant improvements in device efficiency, however, it is necessary to alter at least one, and very likely two or three, of the four major components. (The performance of the fourth component, the dark electrode, is unlikely to be affected significantly by changes in the other components.) Below, we consider specific ways to improve independently the dye, redox shuttle and photoanode, discuss literature reports consistent with the strategies outlined, and highlight the pitfalls encountered thus far. Finally, we discuss what we believe are achievable efficiencies in future-generation DSSCs and how they may come about.
2. Dyes
A. Background
In DSSCs, the adsorbed dyes act as light-harvesting intermediates, absorbing visible and near-infrared solar radiation and efficiently injecting the resultant excited-state electrons into the conduction band of the proximal semiconductor. To produce a photocurrent density, the energy of the dye excited-state necessarily must be higher than the conduction band edge. High quantum efficiency for injection is achieved when the dye LUMO is both energetically matched and reasonably strongly coupled to the underlying semiconductor. The dye should absorb strongly from the blue end of the visible spectrum to the near infrared. Virtually all of the solar irradiance reaching the earth's surface falls between the wavelengths of 300 and 2500 nm, with roughly half of the available power (and roughly a third of the available photons) in the range of 400 to 750 nm (see Fig. 4a).1As noted above, the power conversion efficiency increases with increasing Jsc, all else being equal. The anticipated Jsc for a given dye and cell design can be determined by integrating the incident photon-to-current efficiency, IPCE, with the AM 1.5 terrestrial solar spectrum. The IPCE is given by
| IPCE(λ) = LHE(λ) × Φinj × ηc = LHE(λ)×APCE |
To date, the most efficient (>10%) DSSCs have incorporated the ruthenium polypyridyl complex N3 (Fig. 3a), the closely related tetrabuytylammonium salt, (Bu4N)2[Ru(4-carboxy,4′-carboxylato-2,2′-bipyridine)2(NCS)2] (N719), or the black dye, (Bu4N)[Ru(4,4′,4″-tricarboxy-2,2′:6′,2″-terpyridine)(NCS)3] (Fig. 3b), on high surface area TiO2 nanocrystalline electrodes.12–14,24Ruthenium polypyridyl dyes have been reviewed extensively recently.25,26 These dyes show strong coupling to the underlying semiconductor and minimal recombination as evidenced by nearly quantitative APCE. For optimized DSSCs employing N3/N719, the IPCE is ∼80% for wavelengths < 650 nm, then tails off at longer wavelengths (750 nm) due to minimal (∼10%) LHE (see Fig. 4b).15,16
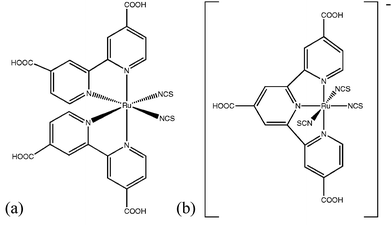 | ||
| Fig. 3 (a) Structure of the ruthenium polypyridyl dye N3. The closely related dye, N719, has tetrabutylammonium cations associated with two of the four carboxylate groups. (b) Structure of the “black dye”. | ||
Fig. 4a shows that there are a lot of photons in the 750–900 nm region of the solar spectrum that are not absorbed by N3. Therefore Jsc, and the overall efficiency, can be improved by decreasing the dye's optical gap, to extend the range of light that can be absorbed. Fig. 4a shows the increased overlap with the solar spectrum expected by shifting the ground state potential in 50 mV increments, with the simplifying assumption that the absorption spectra shape remains the same. This is a reasonable approximation since as long as the absorbance is over 1 (over 90% of light absorbed) for the majority of the spectrum, the overall light harvesting is insensitive to minor variations in extinction, and it is just the tail in the red/near-infrared region that matters. Fig. 4b shows the projected IPCEs resulting from shifting the ground state potential in 50 mV increments, assuming the APCE remains unity. The maximum short-circuit current density can be calculated by integration of the IPCE (Fig. 4b) with the solar spectrum (Fig. 4a). This results in an estimated Jsc of 16 mA cm−2 for N3/N719, in very good agreement with literature reports of optimized devices.14,15 Shifting the ground-state potential negatively by 350 mV, yielding an optical gap of ∼1.4 eV, would enable Jsc to reach 27 mA cm−2. This analysis demonstrates the large increase in current density that is potentially available through modest variations in the dye's electronics, without necessarily sacrificing voltage (or FF).
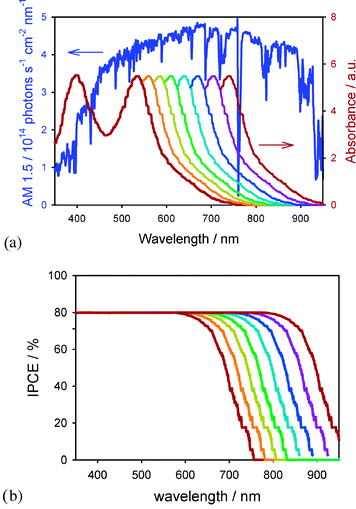 | ||
| Fig. 4 (a) AM 1.5 solar radiation spectrum (left axis). Absorption spectrum of N3 (right axis) and simulated spectra of dyes with extended absorption by decreasing ground state potential by 50 mV increments. (b) Simulated IPCEs for dyes in Fig. 3a assuming a maximum of 80%. | ||
A fundamental limitation of ruthenium polypyridyl dyes is their low molar absorptivity (εmax ∼1.4 × 104 M−1 cm−1).21 Hundreds of adsorbed dye monolayers are required to collect at least 90% of the light at the most strongly absorbing wavelength. In the most efficient cells, light harvesting is enhanced by using a nanocrystalline TiO2electrode with an effective surface area roughly 1200 times the geometric area and a capping light-scattering layer. While the high-electrode-area strategy works, a better strategy, all else being equal, may be to increase the molar absorptivity of the dye. For absorption of a given fraction of incident light, higher dye molar absorptivities allow lower semiconductor effective surface areas per unit of geometric area. At constant light harvesting, the overall cell efficiency will increase with decreasing surface area, since the potential-dependent rate of interception of electrons by the electrolyte scales with the effective surface area. It is generally assumed that the scaling is linear; however, one interesting example showed that interception scales super-linearly with surface area when different sized particles of TiO2 were studied, which would enhance even further the benefits of increased absorptivity.27 For example, in the ideal limit of a 1000-fold increase in molar absorptivity, a flat electrode would suffice to maintain excellent light harvesting. The corresponding 1000-fold decrease in interception rate would translate, via the diode equation, into significantly larger photovoltages.
Alternatively, the 1000-fold decrease in photoanode surface area would permit the use of redox shuttles with 1000-fold larger interception rate constants without a change in overall interception rate. To the extent it can be implemented, this strategy (lower surface area plus faster and more reactive redox shuttle) overcomes a problem associated with many alternative dyes: the inability of regeneration (viaiodide) to compete effectively with recombination.21,28–30 Faster redox couples (such as ferrocenium/ferrocene) can efficiently regenerate dyes with much less expenditure of overpotential than can I3−/I−.31 The unspent overpotential then becomes available to contribute to the cell photovolatge, thereby enhancing conversion efficiency.
B. Route to improvement
Attempts to extend the absorbance of N3/N719 analogues to longer wavelengths have included adjusting the orientation of thiocyanate ligands,32 changing the bipyridyl ligands,24,33 and substituting osmium for ruthenium as the central metal.21,33 The large spin–orbit coupling constant of osmium imparts substantial intensity to an otherwise forbidden singlet–triplet MLCT transition at low energy. Changing the dyes in such fashion in order to increase the spectral response, however, has been problematic, since the APCE generally decreases significantly, yielding lower overall efficiency.21,33 In order to maintain APCEs close to unity, the kinetics for recombination of the injected electron with oxidized dye must be slow compared to the regeneration of the neutral dye by the electrolyte. The commonly observed decreased APCEs (with modified dyes) are likely due to the concomitant problems of slower regeneration and faster recombination. A more negative (less positive) dye+/0 potential is expected to engender slower regeneration kinetics due, at least in part, to the lower driving force for the reaction.29 Further compounding the problem, recombination to oxidized ruthenium and osmium dyes has been shown (at least at shorter timescales) to be subject to Marcus “inverted region” kinetics.28,34–39 Therefore, shifting the ground state potential of the dye negative results in a lower driving force and hence faster recombination. These findings demonstrate the difficulty in changing just one component of the DSSC. Effective use of dyes with enhanced spectral response will likely also require new redox shuttles and new photoanodes (see below).Dyes that address both spectral coverage and absorptivity issues include indolines, coumarins, porphyrins and phthalocyanines (Fig. 5). The best performing organic dye to date is an indoline.40 This metal-free dye boasts an extinction coefficient roughly five times that of the ruthenium polypyridyl dyes and exhibits uniformly high IPCE values exceeding 80% throughout the range of 410 to 670 nm.40Coumarin dyes with thiophene extensions offer a seven-fold enhancement of extinction relative to the ruthenium polypyridyl dyes plus modest red enhancement: even without a scattering layer, IPCEs exceed 75% throughout the range from 400 to 700 nm.41
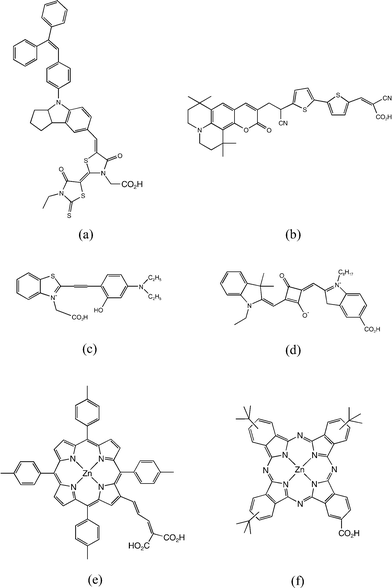 | ||
| Fig. 5 Organic dyes utilized in (a) 9% efficient indoline,40 (b) 6.5% efficient coumarin,41 (c) 5.2% efficient hemicyanine,42 (d) 4.5% efficient squarine,43 (e) 7.1% efficient porphyrin,44 and (f) 3.5% efficient phthalocyanine-based DSSCs.45 | ||
Hemicyanine dyes are another class of metal-free organic photosensitizers that incorporate both donor and acceptor molecular segments that effectively produce a photoinduced charge-separated state and demonstrate strong (ε is typically 0.5–1 × 105 M−1 cm−1), broad light absorbance in the visible and NIR. Investigations into the compatibility with nanocyrstalline TiO2 DSSCs have shown that the most efficient sensitizers are those that appear to use a dual surface-anchoring based on a carboxylate linker and a hydroxyl functionality. A cathodic shift of the excited state potential has been observed for dyes featuring the hydroxyl group, which could explain the enhanced cell performance by considering the increased driving force for electron injection. Fig. 5c gives the structure of a hemicyanine dye that has achieved a conversion efficiency of 5.2% and a nearly 75% IPCE between 450 and 650 nm utilizing an I3−/I−redox shuttle.42
A highly stable class of dyes, squarines absorb strongly (typical εmax around 105 M−1 cm−1) within the visible and NIR regions. Early attempts to incorporate these dyes into DSSCs relied mostly on symmetrical dye structures and resulted in poor conversion efficiencies. Recent efforts in dye design, however, have focused on the synthesis of unsymmetrical squarines and have lead to substantial improvements in cell performance. As opposed to symmetrical architectures that exhibit centralized charge density enhancement of the cyclobutane ring upon photoexcitation, unsymmetrical structures have been shown to facilitate unidirectional electron transfer, enhancing charge separation. Breaking symmetry also considerably broadens dye absorbance, improving spectral coverage. Also significant to dye design are the inclusion of solubilizing alkyl chains to prevent aggregation and conjugated surface-anchoring acid moieties to enhance electronic coupling between the dye and semiconductor and expedite electron injection. The best squaraine dye to date (Fig. 5d) displays all these traits, giving an IPCEmax of 85% at 650 nm and 4.5% conversion efficiency in optimized TiO2nanoparticle Grätzel cells.43
Nonetheless, indolines, coumarins, hemicyanines, and squaraines do not offer a straightforward pathway toward additional red enhancement, and they likely suffer the drawback common to many organic dyes with respect to enhanced interception when paired with the triiodide electrolyte (see below).
Porphyrins and phthalocyanines are structurally similar, exhibiting two major absorption bands: short wavelength Soret (B) bands and longer wavelength Q bands. Symmetrical porphyrin monomers show especially intense B bands (ε ∼ 4 × 105 M−1 cm−1). Though these bands are narrow, IPCEs for porphyrin monomers can exceed 60% out to 650 nm.44 The electronic configuration is readily manipulated through adjustments to the dye structure.44,46–49 Reducing the symmetry of a porphyrin monomer significantly enhances the extinction and red absorbance of the low energy Q bands and broadens the B bands, utilizing a greater fraction of the solar spectrum. In porphyrin oligomers, for example, where care is taken not to break conjugation, the Q bands can eventually become nearly as intense as the B bands as the length of the oligomer increases.50
The location of the acid linker on the porphyrin macrocycle used to affix the dye to the semiconductor surface significantly affects cell efficiencies. The most efficient sensitizers feature conjugated, carboxylate-terminated substituents attached at the beta-position (as illustrated in Fig. 5e) as opposed to methine bridging meso of the ring. This is likely due to the configurations of the respective HOMO and LUMO levels. The LUMO of the dye excited state must be in good electronic communication with the semiconductor surface for good electron injection.51,52 Experimentally, both porphyrins attached through meso and beta positions have been shown to do this well. The difference comes in the configuration of the electron-accepting HOMO level of the oxidized dye. Models of porphyrins linked to the surface through the meso position show a strong overlap and possibly easy route for electrons from the surface to recombine with the dye cation. The HOMO of the typical beta porphyrin, on the other hand, shows a node between the surface functionality and the ring, which likely inhibits geminate recombination from the semiconductor surface.53
The surface orientation of the dye and its molecular footprint must also be considered. Depending on the number of surface attachment functionalities incorporated onto the porphyrin ring, the macrocycle may lie flat, perpendicular, or at some angle to the surface. This effects the packing and therefore the loading of the dye, as well as the ultimate electronic coupling of the dye to the semiconductor.49 At present the most efficient porphyrins feature a beta-linked malonate group that tethers the chromophore in an edge-on geometry.44
Phthalocyanine monomers have enhanced near-infrared absorbance relative to porphyrin monomers. Although IPCEs for phthalocyanines can be relatively high in the Q band region (>60% from 600–725 nm), the B band typically absorbs only in the bluest part of the solar spectrum, leaving a large fraction of the incident sunlight unharvested. In addition, some portion of the relatively lower overall efficiency of these dyes appears to be due to dye aggregation and lack of directionality in the excited state.54 Successful phthalocyanine dyes, such as the one illustrated in Fig. 5f, employ a “push–pull” substitution pattern, wherein tetrabutyl groups enhance solubility, minimize aggregation, and tune the LUMO, while the carboxylate group promotes both physical attachment and electronic coupling to the underlying semiconductor.54 Co-adsorbents can be used as an additional means to minimize aggregation.45
An optimal DSSC dye would have a sufficiently high excited-state energy relative to the underlying semiconductor conduction band edge and would retain the strong coupling, slow recombination kinetics, and beneficial charge interception suppression of the ruthenium polypyridyl dyes while achieving a much higher molar absorptivity, especially in the red and near-infrared (NIR) regions. One way to engineer such “super chromophores” would be to develop highly conjugated porphyrin oligomers. Both the Soret and Q bands of such dyes broaden significantly in oligomers; in addition the Q bands red-shift and grow more intense with each additional unit, such that a tetramer would have a Q band onset at 880 nm with ε of 2 × 105 M−1 cm−1 at 800 nm.50 An array of these dye molecules attached perpendicularly to the semiconductor surface with their long axis perpendicular to the incident light would allow almost unity light harvesting efficiency from 880 through 580 and 520 through 400 nm, even with photoelectrodes having 5% of the effective surface area of typical photoelectrodes. The “hole” in the absorbance might be filled by an appropriate ligand coordinated to the metal center (typically zinc).
Photoelectrode surface areas could, in principle, be further reduced by constructing dye multilayers. Photonic energy harvested by the outer layers could be transported through intervening layers to the dye/electrode interface via Forster transfer (dipole–dipole energy transfer). Experimental implementation, however, is not straightforward. The dye multilayers must be porous enough to allow “potential determining” cations to reach the semiconductor surface.55–59 Additionally, aggregation must be avoided.60 Finally, the architecture of the photoelectrode itself may need to be altered from the standard sintered-nanoparticle motif in order to leave sufficient space to accommodate multilayers. Alternative architectures are considered in section 4, below.
3. Redox shuttles
A. Background
The function of the redox shuttle is to transfer electrons from the counter electrode to the oxidized dyes, formed via injection of photoexcited electrons into the photoanode, to complete the electrochemical circuit. There are two kinetic constraints for a successful redox shuttle: it must reduce the dye cation before the dye cation recombines with an electron in the photoanode, but not allow the oxidized form of the shuttle to intercept an electron from the photoanode. The dual criteria of fast dye regeneration and slow interception place a very challenging constraint on identifying effective redox shuttles.All reports of efficient (>4% at 1 sun illumination) DSSCs to date have utilized the I3−/I− couple as a redox shuttle. The uniquely good performance of I3−/I− in these cells can be attributed to efficient dye regeneration combined with exceedingly slow electron transfer from TiO2 to I3−. For example, when I3−/I− is employed with N3, the regeneration yield is quantitative.29 In addition, loss of electrons via interception by I3− is negligible (at short-circuit), allowing photo-injected electrons to be collected with near unity efficiency.
Despite the excellent performance of I3−/I− as a redox shuttle with N3, the regeneration of dyes with I− appears to be restricted to high driving-force reactions.29 Furthermore, the mechanism of the reduction of dye cations by iodide is still unclear. The overall yield of the regeneration reaction appears to be dependent upon interaction of a second iodide ion with a [dye+/I−] complex.29 Since the chemical nature, energetics and spatial configuration of this complex may be considerations for targeting the development of new sensitizer dyes for dye sensitized solar cells, there is no straightforward insight into which alternate dyes can be used effectively with I3−/I−. Attempts to use dyes with the potential to dramatically increase efficiency by extending the absorbance have been thwarted, at least in part, by inefficient regeneration with I−.21,28,30
Additionally, it has recently been demonstrated that electrodes sensitized with highly-polarizable organic dyes display much larger dark current densities than do electrodes sensitized with conventional ruthenium polypyridyl complexes when I3−/I− is used as the redox shuttle.61 An undesirable consequence of larger dark current densities is smaller open-circuit photovoltages. The enhanced dark current densities appear to reflect the formation of dye–I3− complexes and the resulting large local concentration of I3− at the semiconductor/dye interface.62 Formation of complexes can also lead to undesirable alternative photochemical reaction pathways, such as direct oxidative quenching of the dye excited state by I3−.61 In a semiconductor-based DSSC, the quenching sequence is manifest as a parasitic diminution of the expected anodic photocurrent density.61 These findings should provide impetus for researchers studying DSSC involving organic dyes to move away from I3−/I− electrolyte systems.
In addition to preventing efficient use of alternative dyes, reliance on I3−/I− also limits the photovoltage. Optimization of the photovoltage of DSSCs is critical to achieving improvements in device efficiencies. Typical DSSCs exhibit open circuit voltages in the range 0.75–0.85 V: only 50% of the ∼1.8 eV optical gap of N3. In other words, half of the energy available in a DSSC is lost in the electron injection and regeneration steps. Some of the energy lost in the electron-injection process could be recovered by shifting the dye+/* potential positive; however, the overpotential required for electron injection into the metal oxide at a rate sufficient to compete with radiative and nonradiative decay is already essentially optimized for a given photoanode material and dye. The other major energy loss is the ∼500 mV overpotential of I3−/I− that is used to regenerate N3. This large potential difference appears to be necessary in order to drive regeneration at an acceptable rate, and is likely a consequence of both mechanistic complexity and the unusually large reorganization energy for I3−/I−. In order to extract all the voltage available in a DSSC, alternative redox couples with potentials more closely matched to the dye oxidation potential need to be identified (Fig. 2). Even assuming 200 mV is necessary to drive regeneration, an increase in Voc of over 300 mV is available.
Perhaps most important is the scientific reason for employing alternate redox shuttles. The electrochemistry of iodine is generally very complicated.63 As noted above, it was only recently determined that the regeneration reaction appears to depend on a pre-association of iodide with the dye.29 The mechanism of the interception reaction remains uncertain despite over 15 years of research.64–66 Suffice it to say, however, that interception by I3− is a complicated, multi-electron-transfer process. Furthermore, it is not clear that what is learned about the I3−/I− shuttle can be applied to the development of alternative shuttles. Studies employing carefully prepared single crystal ZnO electrodes as a model system in contact with non-adsorbing, one-electron, outer-sphere redox couples were able to establish the dependence of the interfacial electron-transfer rate constant on driving force, reorganization energy, electrolyte composition and coupling.67–70 These measurements indicated that the tert-butyl groups are able to act as spacers to slow electron interception, which may explain why DSSCs using [Co(t-Bu2bpy)3]3+/2+ as a redox shuttle exhibited excellent efficiencies, while [Co(Me2bpy)3]3+/2+, systems showed poor performance (see below).71 Use of such non-adsorbing, one-electron, outer-sphere redox shuttles in DSSCs, in conjunction with measurements with single crystal model systems, should allow for more complete understanding of interception and guide the design of future redox shuttles.
B. Route to improvement
The most successful alternative redox couples to date consist of substituted cobalt polypyridyl complexes.71–73 A salient feature is relatively slow electron-exchange kinetics (albeit significantly faster than triiodide/iodide). The slow kinetics, which result from cobalt spin changes that in turn create large inner-sphere reorganization energies, likely inhibit electron interception. While the cobalt systems are one-electron outer-sphere couples, their potentials are slightly negative of I3−/ I−; consequently, they don't offer an opportunity for increasing the voltages of DSSCs. In general, fast one-electron, outer-sphere redox reagents such as ferrocenes have not been useful mediators in DSSCs.74,75 Although such species typically rapidly reduce the oxidized dye, they also undergo facile reduction of their oxidized form by electrons in the TiO2 conduction band. This rapid interception of electrons in the photoanode creates two distinct problems in the operation of a DSSC: loss of photovoltage (due to large dark current densities) and diminished charge collection efficiency (loss of photocurrent density).There have been several studies of alternative redox couples with potentials positive of I3−/ I−, which should in principle lead to increased Voc. In one interesting example Br3−/Br−, which is an obvious analogue of I3−/I− but with a ∼500 mV more positive redox potential, was employed as a redox shutle with a variety of sensitizers.76 In all cases the Voc was somewhat higher with Br3−/Br− compared to I3−/I−, as expected. The photocurrent density was generally low for the bromine systems, however, due at least in part, to slow regeneration kinetics. The low photocurrent densities resulted in low overall power conversion efficiencies. For example, when employed with N719, the Jsc and overall efficiency for the bromine system were only ∼20% of the comparable iodine system. The observed inefficient regeneration of N719 was likely due to the very minimal overpotential for the reaction. When a dye with more positive ground-state potential was employed, Eosin Y, Jsc values were similar for the iodine and bromine systems; but, Voc was 813 mV with Br3−/Br−, compared to 451 mV with I3−/I−.76 Overall, the highest efficiency employing the Br3−/Br−redox couple was the bromine/Eosin Y system (2.6%).76 While no overall improvement in performance was observed with Br−/Br3−, it proved superior to I3−/I− in a few cases and thus warrants further consideration.
Two pseudohalogen redox couples, (SeCN)2/SeCN− and (SCN)2/SCN−, with potentials 190 and 430 mV more positive than I3−/I−, were investigated as redox shuttles in DSSCs.77 With the N3 sensitizer, the maximum IPCE was 80%, 20% and 4% for the I3−/I−, (SeCN)2/SeCN− and (SCN)2/SCN− couples, respectively.77 This trend is attributed to the decreasing rates of dye regeneration in the order I− > SeCN− > SCN−. As a consequence, a greater fraction of the injected electrons recombine with the oxidized dye, N3+. No increase in voltage was observed with the pseudo halogen couples, however. Indeed, the couple with most positive potential, (SCN)2/SCN−, delivered the lowest Voc. These results illustrate that selecting a redox couple with a more positive potential does not necessarily translate into an increase of the open circuit potential; effective shuttles additionally need to meet the challenging criteria of fast regeneration and slow interception.
In an interesting report, a one-electron outer-sphere redox couple, Cu(dmp)22+/+ (dmp = 2,9-dimethy-1,10-phenanthroline), with a potential 430 mV positive of I3−/I−, was successfully employed as a redox shuttle in a DSSC sensitized with N719.78 The Voc with Cu(dmp)22+/+ was ∼700 mV, slightly greater than with I3−/I− (600 mV), possibly due to the more positive shuttle potential. The maximum IPCE, as well as the energy conversion efficiency, however, was ∼50% lower for [Cu(dmp)2]2+/+ (2.2%) compared to I3−/I− (4.3%).78 Despite its limitations, the I3−/I− shuttle has some properties that may be difficult to replicate with shuttles based on coordination complexes. Among them are extraordinarily high solubility for the reduced form of the shuttle (ensuring efficient dye regeneration, at least under low driving-force conditions) and efficient diffusion for the oxidized form of the shuttle (ensuring minimal concentration polarization (transport-based voltage loss) at the dark electrode).79,80 The use of alternative, higher-porosity architectures for photoelectrodes may help to alleviate the anticipated concentration polarization problems.
Recently there has also been much interest in employing solid organic redox couples (hole conductors), such as 2,2′–7,7′–tetrakis(N,N-di-p-methoxyphenylamine) 9,9′-spirobifluorene (spiro-OMeTAD), as shuttles in DSSCs.81–85 Solid-state electrolytes in DSSCs have been reviewed recently and thus won't be discussed extensively herein.85 Such solid-state electrolytes, however, directly address the challenge of achieving high shuttle concentrations. The redox potential for spiro-MeOTAD is reported to be about 450 mV positive of I3−/I−. While this is a very intriguing system, efficiencies have so far been limited to ∼4%. Poor filling of the nanoparticle TiO2pores has been suggested to be the major limitation of spiro-MeOTAD so far.85 Short charge diffusion lengths—a consequence of fast electron interception—may also be a limiting factor. Treatment of a pre-sensitized mesporous TiO2 film with amylose is a novel and potentially attractive route to minimizing interception losses in such solid state DSSCs without the loss of charge separation yields.86Amylose evidently serves to block physical access to interception sites. This strategy has resulted in an open circuit voltage of 1 V—to our knowledge the highest reported Voc to date for a TiO2 DSSC sensitized with ruthenium dyes.86
Finally, the amylose strategy suggests a related strategy for solution-based redox shuttles. The combination of relatively large redox shuttles (such as Cu(dmp)22+/+) and closely packed, but electrolyte porous, dyes of high aspect ratio (such as conjugated porphyrin oligomers) could serve to prevent shuttles from approaching the semiconductor surface. To the extent this can be achieved, rate constants for interception will diminish and photovoltages will increase.
4. Photoanode
A. Background
A good photoanode will facilitate light harvesting, electron injection and electron collection. Maximizing light harvesting requires: (a) transparency for the unsensitized semiconductor framework, and (b) sufficiently high internal area to enable absorptivities for surface-attached dyes to exceed 1 over the spectral region of interest. ηinj is maximized by having a large density of unpopulated states in the photoanode at potentials positive of the dye+/* potential (i.e. lower in energy than the thermalized dye excited-state on an absolute energy scale). When electron recombination with the oxidized dye can be neglected, the electron collection efficiency is determined by the kinetic competition between the effective rate of electron diffusion (the rate at which electrons are collected) and electron lifetimes (governed by rates of interception and recombination), τn, as described in detail elsewhere.87 The diffusion rate is dependent on both the apparent diffusion coefficient, Dn, and the diffusion distance, ln (rate ∝ Dnln2). The diffusion coefficient nominally varies with the mobility of the electron within the conduction band, according to the Einstein relation. In practice it is also governed to a large extent by the dynamics of trapping and thermal release of electrons from energetically distributed sub-bandedge states.64,88–90 Finally, τn, is related to the quasi-Fermi level of electrons in the film, and also shows trap density dependence. All else being equal, a photoanode that increases one of these independent variables will increase the efficiency of a DSSC, with the exception of Dn, which offers diminishing returns in the limit of kinetic redundancy. In the best DSSCs, the LHE and APCE are both close to unity. Consequently, changing just the photoanode will not substantially increase efficiency. Instead, improved photoanodes will only facilitate improvements when used in conjunction with new dyes and redox couples—specifically, those that are incompatible with conventional photoanodes.By combining optical transparency with a large internal surface area for dye loading, the introduction of a sintered titania nanoparticle (NP) film was largely responsible for the first large successes with DSSCs, and NP films remain a key component of the most efficient DSSCs. Additionally, those nanoparticle films that are based on TiO2 offer simple fabrication, low materials cost, ready scalability and tremendous chemical stability. Optimized photoelectrode films consist, in part, of 10–20 nm spherical particles that have been sintered to form a high surface area, 12 μm thick transparent structure. The surface area enhancement is described by the roughness factor, defined as the ratio of actual surface area to the projected surface area. Roughness factors in excess of 1000 are necessary for good light harvesting with standard ruthenium based sensitizers. A ∼4 μm thick film of much larger (∼400 nm) particle diameter is subsequently deposited in order to scatter photons back into the transparent film and enhance red and near-IR light harvesting. Despite the good performance of nanoparticle films in conventional DSSCs, this photoanode geometry has disadvantages for next-generation cells. These include relatively low porosity,80,91,92 limited materials generality93 and tedious particle synthesis. Additionally, the films are poorly suited to large vertically-attached light harvesters, such as the porphyrin oligomers described above. The primary weakness of the NP photoanode design, however, is the extraordinarily small apparent diffusion coefficient, Dn. As noted above, slow electron transport greatly limits the choice of dye regenerator (i.e.redox shuttle)—which, in turn, limits photovoltages and constrains the choice of light absorber. In contrast, an ideal photoanode would exhibit fast electron transport (as well as high transparency, high and tunable surface area and high porosity). Additionally, the anode fabrication method ideally would be inexpensive, scalable and materials general.
B. Route to improvement
One of the first significant advances in developing an alternative DSSC photoanode was the use of an array of aligned ZnO nanorods.94 Typically, nanorods are prepared on a conducting glass substrate on which a layer of ZnO particles has been deposited to seed the rod growth. Preferential growth of the [0001] crystal face from solution affords moderately high aspect ratio single crystal rods perpendicular to the TCO (transparent conductive oxide).94,95 Inclusion of poly(ethylenimine) in the deposition solution has been shown to enhance anisotropic growth, enabling rods with aspect ratios in excess of 125 to be formed. Compared to nanoparticle films (roughness factors > 1000) the arrays are most notably lacking in roughness (<200), significantly limiting Jsc and thus also limiting η to 1.5%. In addition, the solution growth of rods has been restricted primarily to ZnO. This is unfortunate, as ZnO photoanodes show consistently lower performance than similar TiO2 devices, owing primarily to the instability of ZnO in acidic dye solutions.96–99 Despite their modest success in DSSCs to date, nanorod photoanodes possess several attractive features including low cost, scalability and fast electron transport.100,101To overcome the apparent limitations on nanorod array surface area, various routes to low dimensionality DSSC photoanodes of another kind—oriented nanotubes—have been explored. In contrast to solution-phase nanorod growth, the electrochemical anodization of select metal films affords an array of metal oxide nanotubes with tunable roughness over 1000. When a suitable metal such as Ti is employed, the resulting membranes may be employed directly as photoanodes. For DSSCs with these electrodes, the best conversion efficiencies currently approach 7%. Surprisingly, nanotube films of this kind display similar electron transport kinetics to nanoparticle films; however, interception has been found to be slower in the nanotube films.102
The fabrication of nanotubes can be accomplished in a much more materials-diverse fashion by enlisting atomic layer deposition (ALD) as the synthesis technique and employing high-area anodic alumina oxide (AAO) membranes as templates for deposition. A key feature of ALD is its ability to coat templates conformally—even those presenting intricate and complicated geometries. The technique makes use of alternate exposures to reactive gas precursors (separated by inert gas purging) to deposit films of metal oxides, sulfides or nitrides. The self-limiting nature of the layer-by-layer growth allows for angstrom-level control over film thickness and facilitates the construction of pinhole-free coatings. As an example, ZnO tubes grown within and upon commercially available AAO afford DSSCs with up to 1.6% efficiency, limited primarily by the modest surface areas of commercial membranes (∼450 × geometric area). The fill factor and Voc of the ZnO nanotube devices, however, exceed those of any other ZnO photoanode reported to date.
The most obvious advantage of templated growth of photoanodes by ALD is the diversity of metal oxides that may now be employed in the study of DSSCs. The hydrothermal growth of nanorod arrays has been reported for only a small subset of metal oxides. Likewise, fabricating high surface area NP photoelectrodes of different metal oxides is a technical challenge that has yet to be overcome for several oxides of interest (e.g.NiO).93 In contrast, the list of transition-metal oxides accessible viaALD is long, suggesting many possibilities for high-surface-area photoelectrode fabrication. ALD also opens wide the possibility of mixed metal oxide DSSC devices as well as sophisticated multilayer DSSC devices (e.g. precisely defined TCO/semiconductor/blocking-layer devices).
We have recently extended the ALD strategy by using low-density, high surface area, mesoporous aerogels of silica as templates.103–107 Aerogel films are readily prepared with high and tunable surface area and porosity, with roughness factors > 1500, i.e. equal to or exceeding NP films. Thus, light harvesting and ion transport are not limiting factors. Like the fabrication of NP photoelectrodes, the fabrication of aerogel films is inexpensive and scalable. Because the aerogels are pseudo-one dimensional, they should exhibit faster electron transport than three-dimensional NP films. The greater porosity of the films should facilitate movement of shuttle molecules as well, thereby minimizing concentration polarization. Finally, both hydrothermal growth and ALD are expected to yield materials with larger polycrystalline domains compared to NP films, resulting in fewer surface trap states and grain boundaries or particle–particle junctions; these factors are important for achieving good electron transport dynamics. (We define “good” as sufficient to achieve essentially quantitative photocurrent collection within a given anode with a given redox electrolyte. As emphasized above, NP photoanodes are “good enough” in conventional cells featuring triiodide/iodide. For advanced cells employing alternative redox electrolytes, however, better electron transport dynamics clearly are required. Thus, we expect new photoanode architectures to offer no appreciable improvement in the performance of DSSCs based on conventional components, but very substantial improvement for DSSCs based on alternative components (especially alternative (fast) redox shuttles).)
 | ||
| Fig. 6 Cross-sectional SEM image of ZnO tubes grown on commercial AAOviaALD (scale bar = 100 nm). | ||
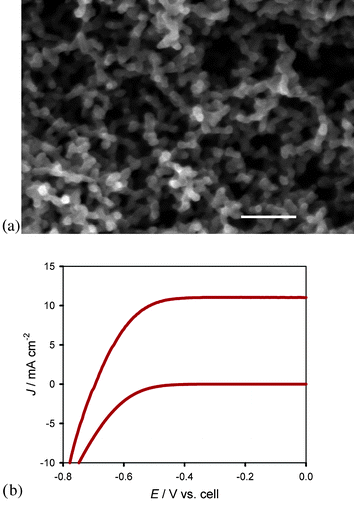 | ||
| Fig. 7 (a) SEM image of aerogel film coated with ∼9 nm TiO2viaALD (scale bar = 100 nm) (b) Jvs.E curve of TiO2 coated (∼9 nm) aerogel photoanode employed in a DSSC with N3 and I3−/I− under AM 1.5 illumination: Jsc = 11 mA cm−2, Voc = 0.7 V, FF = 0.7, η = 5.4% (lower plot = no illumination). | ||
Both ZnO and TiO2 have been conformally deposited with controlled variable thicknesses on the aerogel templates by ALD.103–107 This points to the materials flexibility of this approach. Electrodes incorporated into DSSCs have displayed excellent light harvesting and power efficiencies. Initial devices exhibited power conversion efficiencies of over 5% under 100 mW cm−2 light intensity.
5. Overall improvement strategies
In the above discussion, we have considered ways to optimize the individual components of a DSSC in order to increase the overall power conversion efficiency. Here, we consider the potential efficiency increase available by changing multiple components simultaneously. Fig. 8 shows the effect on efficiency due to varying the dye ground state potential and the redox shuttle potential. The typical DSSC employing N3 and I3−/I− is shown as the base case. In all cases the FF was assumed to be 0.8. Likewise, a constant quasi-Fermi level in the semiconductor was assumed. Each data point on a given colored line represents a 50 mV shift in dye potential. Shifting the dye's potential results in an increased Jsc, and thus efficiency, determined by integrating the IPCE's shown in Fig. 3b with the solar spectrum, but a constant Voc. The dye's potential is only varied to a maximum of 200 mV positive of the redox couple, which is the minimum overpotential assumed necessary for efficient dye regeneration. The different colored data points and lines represent the effect of shifting the redox shuttle potential positive, which produces an increase in voltage for any given dye (Jsc). Again, the potential is only shifted by a maximum of 350 mV, leaving 200 mV of overpotential for regeneration.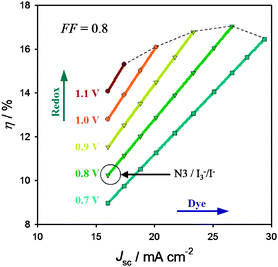 | ||
| Fig. 8 Estimated efficiency, η, of DSSCs employing dyes with increased spectral coverage in conjunction with redox shuttles with varying solution potentials. Efficiencies > 15% are, in principle, achievable in many configurations when there is minimal overpotential (ca. 200 mV) for dye regeneration (dotted line). | ||
It is important to note that the projections displayed in Fig. 8 are almost certainly not possible with I3−/I− as a redox shuttle. While the solution potential can be made more positive by increasing the concentration of I3−, the rate of interception also increases, resulting in a decreased APCE and overall efficiency. A higher concentration of I3− will also increase the absorption of incident light, further reducing the efficiency. Alternatively, as mentioned above, I− has not been successful regenerating dyes with enhanced spectral coverage efficiently, resulting in diminished APCEs.
The plot in Fig. 8 has several interesting features. Efficiencies exceeding 16% are reasonably achievable, and there are many combinations of dyes and redox shuttles to get there. Whatever combination is employed, the important criterion for achieving high efficiencies is a minimal overpotential for dye regeneration. Also shown is that while extending the dye absorption into the near IR will harvest more of the available photons producing more current density, there is an ultimate limitation imposed by the decreased voltage available for a single electrode DSSC. Finally, this analysis does not produce the maximum possible efficiencies available. For example, the effects of increasing the electrode's quasi-Fermi level (which will result in even larger increases in voltage) or fill factor (which can, in principle, reach 0.89 at 1.1 V) are excluded.
6. Conclusions
In achieving 11% overall energy conversion efficiency, the standard configuration of current generation DSSCs has been amazingly well optimized. Variation of any one of the three major photo-related components of a DSSC—the dye, redox shuttle and photoanode—has, thus far, led to worse overall photovoltaic performance. In order to realize the full promise of DSSCs as high efficiency energy-conversion devices, it will be necessary to alter at least two of the three major components simultaneously.Fortunately, as outlined above, there are multiple routes to improving device performance when components are varied simultaneously. Furthermore, there is now sufficient fundamental-level understanding of the performance of DSSCs to allow multi-component optimization strategies to be pursued rationally. We suggest that the simultaneous development of new dyes, shuttles and photoanodes, combined with further investigation of transport dynamics, will lead to DSSCs with efficiencies exceeding 16%.
Acknowledgements
The SEM work was performed in the EPIC facility of the NUANCE Center at Northwestern University. The NUANCE Center is supported by NSF-NSEC, NSF-MRSEC, Keck Foundation, the State of Illinois and Northwestern University. We gratefully acknowledge the contributions of collaborators and co-workers whose work is cited herein. We also gratefully acknowledge financial support from BP Solar, Argonne National Lab (Lab-Grad Fellowship for ABFM), and the U.S. Department of Energy, Basic Energy Sciences Program (Grant DE-FG02-87ER13808). Work at Argonne is supported by the U.S. Department of Energy, BES-Materials Sciences under Contract W-31-109-ENG-38.References
- In Basic Research Needs for Solar Energy Utilization, U.S. Department of Energy, 2005 Search PubMed.
- Clean-Soil Air Water, 2008, 36, 140.
- R. L. Hirsch, Energy Policy, 2008, 36, 881–889 CrossRef.
- J. R. Hughes, Oil Gas J., 2008, 106, 12–12 Search PubMed.
- A. Witze, Nature, 2007, 445, 14–17 CrossRef CAS.
- R. A. Kerr, Science, 2007, 318, 1230–1231 CrossRef CAS.
- N. S. Lewis, MRS Bull., 2007, 32, 808–820.
- G. W. Crabtree and N. S. Lewis, Phys. Today, 2007, 60, 37–42 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Muller, P. Liska, N. Vlachopoulos and M. Gratzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Y. Han, Jpn. J. Appl. Phys., Part 2, 2006, 45, L638–L640 CrossRef CAS.
- J. M. Kroon, N. J. Bakker, H. J. P. Smit, P. Liska, K. R. Thampi, P. Wang, S. M. Zakeeruddin, M. Gratzel, A. Hinsch, S. Hore, U. Wurfel, R. Sastrawan, J. R. Durrant, E. Palomares, H. Pettersson, T. Gruszecki, J. Walter, K. Skupien and G. E. Tulloch, Prog. Photovoltaics, 2007, 15, 1–18 CAS.
- M. K. Nazeeruddin, F. DeAngelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Gratzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS.
- M. Gratzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef.
- M. Gratzel, MRS Bull., 2005, 30, 23–27 CAS.
- Y. Chiba, A. Islam, R. Komiya, N. Koide and L. Y. Han, Appl. Phys. Lett., 2006, 88 Search PubMed.
- Z. S. Wang, M. Yanagida, K. Sayama and H. Sugihara, Chem. Mater., 2006, 18, 2912–2916 CrossRef CAS.
- M. D. Wei, Y. Konishi, H. S. Zhou, M. Yanagida, H. Sugihara and H. Arakawa, J. Mater. Chem., 2006, 16, 1287–1293 RSC.
- M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. Shklover, C. H. Fischer and M. Gratzel, Inorg. Chem., 1999, 38, 6298–6305 CrossRef CAS.
- G. Sauve, M. E. Cass, S. J. Doig, I. Lauermann, K. Pomykal and N. S. Lewis, J. Phys. Chem. B, 2000, 104, 3488–3491 CrossRef CAS.
- S. A. Haque, E. Palomares, B. M. Cho, A. N. M. Green, N. Hirata, D. R. Klug and J. R. Durrant, J. Am. Chem. Soc., 2005, 127, 3456–3462 CrossRef CAS.
- L. M. Peter, Phys. Chem. Chem. Phys., 2007, 9, 2630–2642 RSC.
- M. K. Nazeeruddin, P. Pechy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Gratzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338–2345 CrossRef CAS.
- R. Argazzi, N. Y. M. Iha, H. Zabri, F. Odobel and C. A. Bignozzi, Coord. Chem. Rev., 2004, 248, 1299–1316 CrossRef CAS.
- K. Zhu, N. Kopidakis, N. R. Neale, J. van de Lagemaat and A. J. Frank, J. Phys. Chem. B, 2006, 110, 25174–25180 CrossRef CAS.
- D. Kuciauskas, M. S. Freund, H. B. Gray, J. R. Winkler and N. S. Lewis, J. Phys. Chem. B, 2001, 105, 392–403 CrossRef CAS.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Gratzel and J. R. Durrant, J. Phys. Chem. C, 2007, 111, 6561–6567 CrossRef CAS.
- M. Alebbi, C. A. Bignozzi, T. A. Heimer, G. M. Hasselmann and G. J. Meyer, J. Phys. Chem. B, 1998, 102, 7577–7581 CrossRef CAS.
- S. Cazzanti, S. Caramori, R. Argazzi, C. M. Elliott and C. A. Bignozzi, J. Am. Chem. Soc., 2006, 128, 9996–9997 CrossRef CAS.
- T. Renouard, R. A. Fallahpour, M. K. Nazeeruddin, R. Humphry-Baker, S. I. Gorelsky, A. B. P. Lever and M. Graetzel, Inorg. Chem., 2002, 41, 367–378 CrossRef CAS.
- S. Altobello, R. Argazzi, S. Caramori, C. Contado, S. Da Fre, P. Rubino, C. Chone, G. Larramona and C. A. Bignozzi, J. Am. Chem. Soc., 2005, 127, 15342–15343 CrossRef CAS.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Gratzel, J. Nelson, X. Li, N. J. Long and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5225–5233 CrossRef CAS.
- H. Lu, J. N. Prieskorn and J. T. Hupp, J. Am. Chem. Soc., 1993, 115, 4927–4928 CrossRef CAS.
- J. E. Moser and M. Gratzel, Chem. Phys., 1993, 176, 493–500 CrossRef CAS.
- Y. X. Weng, Y. Q. Wang, J. B. Asbury, H. N. Ghosh and T. Q. Lian, J. Phys. Chem. B, 2000, 104, 93–104 CrossRef CAS.
- D. Kuciauskas, J. E. Monat, R. Villahermosa, H. B. Gray, N. S. Lewis and J. K. McCusker, J. Phys. Chem. B, 2002, 106, 9347–9358 CrossRef CAS.
- D. A. Gaal, J. E. McGarrah, F. Liu, J. E. Cook and J. T. Hupp, Photochem. Photobiol. Sci., 2004, 3, 240–245 RSC.
- S. Ito, S. M. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvet, P. Comte, M. K. Nazeeruddin, P. Pechy, M. Takata, H. Miura, S. Uchida and M. Gratzel, Adv. Mater., 2006, 18, 1202 CrossRef CAS.
- Z. S. Wang, Y. Cui, K. Hara, Y. Dan-Oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138–1141 CrossRef CAS.
- Y. S. Chen, C. Li, Z. H. Zeng, W. B. Wang, X. S. Wang and B. W. Zhang, J. Mater. Chem., 2005, 15, 1654–1661 RSC.
- J. H. Yum, P. Walter, S. Huber, D. Rentsch, T. Geiger, F. Nuesch, F. De Angelis, M. Gratzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2007, 129, 10320 CrossRef CAS.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Gratzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CrossRef CAS.
- J. J. Cid, J. H. Yum, S. R. Jang, M. K. Nazeeruddin, E. M. Ferrero, E. Palomares, J. Ko, M. Gratzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358–8362 CrossRef CAS.
- J. R. Stromberg, A. Marton, H. L. Kee, C. Kirmaier, J. R. Diers, C. Muthiah, M. Taniguchi, J. S. Lindsey, D. F. Bocian, G. J. Meyer and D. Holten, J. Phys. Chem. C, 2007, 111, 15464–15478 CrossRef CAS.
- Q. Wang, W. M. Carnpbell, E. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Gratzel, J. Phys. Chem. B, 2005, 109, 15397–15409 CrossRef CAS.
- W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 1363–1379 CrossRef CAS.
- J. Rochford, D. Chu, A. Hagfeldt and E. Galoppini, J. Am. Chem. Soc., 2007, 129, 4655–4665 CrossRef CAS.
- S. J. Lee, K. L. Mulfort, J. L. O'Donnell, X. B. Zuo, A. J. Goshe, P. J. Wesson, S. T. Nguyen, J. T. Hupp and D. M. Tiede, Chem. Commun., 2006, 4581–4583 RSC.
- P. Persson and M. J. Lundqvist, J. Phys. Chem. B, 2005, 109, 11918–11924 CrossRef CAS.
- J. Snook, L. Samuelson, J. Kumar, Y. Kim and J. Whitten, Org. Electron., 2005, 6, 55–64 CrossRef CAS.
- S. I. Yang, J. Seth, T. Balasubramanian, D. Kim, J. S. Lindsey, D. Holten and D. F. Bocian, J. Am. Chem. Soc., 1999, 121, 4008–4018 CrossRef CAS.
- P. Y. Reddy, L. Giribabu, C. Lyness, H. J. Snaith, C. Vijaykumar, M. Chandrasekharam, M. Lakshmikantam, J. H. Yum, K. Kalyanasundaram, M. Graetzel and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2007, 46, 373–376 CrossRef CAS.
- A. M. Massari, R. W. Gurney, C. P. Schwartz, S. T. Nguyen and J. T. Hupp, Langmuir, 2004, 20, 4422–4429 CrossRef CAS.
- A. M. Stux and G. J. Meyer, J. Fluoresc., 2002, 12, 419–423 CrossRef CAS.
- L. A. Lyon and J. T. Hupp, J. Phys. Chem., 1995, 99, 15718–15720 CrossRef CAS.
- G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1993, 97, 1426–1430 CrossRef CAS.
- C. A. Kelly, F. Farzad, D. W. Thompson, J. M. Stipkala and G. J. Meyer, Langmuir, 1999, 15, 7047–7054 CrossRef CAS.
- K. E. Splan and J. T. Hupp, Langmuir, 2004, 20, 10560–10566 CrossRef CAS.
- B. C. O'Regan, I. Lopez-Duarte, M. V. Martinez-Diaz, A. Forneli, J. Albero, A. Morandeira, E. Palomares, T. Torres and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 2906–2907 CrossRef CAS.
- K. E. Splan, A. M. Massari and J. T. Hupp, J. Phys. Chem. B, 2004, 108, 4111–4115 CrossRef CAS.
- Y. A. Yaraliev, Uspekhi Khimii, 1982, 51, 990–1016 CAS.
- L. M. Peter, J. Phys. Chem. C, 2007, 111, 6601–6612 CrossRef CAS.
- J. Bisquert, A. Zaban, M. Greenshtein and I. Mora-Sero, J. Am. Chem. Soc., 2004, 126, 13550–13559 CrossRef CAS.
- A. J. Frank, N. Kopidakis and J. van de Lagemaat, Coord. Chem. Rev., 2004, 248, 1165–1179 CrossRef CAS.
- T. W. Hamann, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. B, 2006, 110, 25514–25520 CrossRef CAS.
- T. W. Hamann, F. Gstrein, B. S. Brunschwig and N. S. Lewis, J. Am. Chem. Soc., 2005, 127, 13949–13954 CrossRef CAS.
- T. W. Hamann, F. Gstrein, B. S. Brunschwig and N. S. Lewis, J. Am. Chem. Soc., 2005, 127, 7815–7824 CrossRef CAS.
- T. W. Hamann, F. Gstrein, B. S. Brunschwig and N. S. Lewis, Chem. Phys., 2006, 326, 15–23 CrossRef CAS.
- S. A. Sapp, C. M. Elliott, C. Contado, S. Caramori and C. A. Bignozzi, J. Am. Chem. Soc., 2002, 124, 11215–11222 CrossRef CAS.
- H. Nusbaumer, J. E. Moser, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Gratzel, J. Phys. Chem. B, 2001, 105, 10461–10464 CrossRef CAS.
- H. Nusbaumer, S. M. Zakeeruddin, J. E. Moser and M. Gratzel, Chem.–Eur. J., 2003, 9, 3756–3763 CrossRef CAS.
- B. A. Gregg, Coord. Chem. Rev., 2004, 248, 1215–1224 CrossRef CAS.
- B. A. Gregg, F. Pichot, S. Ferrere and C. L. Fields, J. Phys. Chem. B, 2001, 105, 1422–1429 CrossRef CAS.
- Z. S. Wang, K. Sayama and H. Sugihara, J. Phys. Chem. B, 2005, 109, 22449–22455 CrossRef CAS.
- G. Oskam, B. V. Bergeron, G. J. Meyer and P. C. Searson, J. Phys. Chem. B, 2001, 105, 6867–6873 CrossRef CAS.
- S. Hattori, Y. Wada, S. Yanagida and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 9648–9654 CrossRef CAS.
- P. J. Cameron, L. M. Peter, S. M. Zakeeruddin and M. Gratzel, Coord. Chem. Rev., 2004, 248, 1447–1453 CrossRef CAS.
- N. Papageorgiou, M. Gratzel and P. P. Infelta, Sol. Energy Mater. Sol. Cells, 1996, 44, 405–438 CrossRef CAS.
- W. H. Howie, F. Claeyssens, H. Miura and L. M. Peter, J. Am. Chem. Soc., 2008, 130, 1367–1375 CrossRef CAS.
- H. J. Snaith, A. J. Moule, C. Klein, K. Meerholz, R. H. Friend and M. Gratzel, Nano Lett., 2007, 7, 3372–3376 CrossRef CAS.
- H. J. Snaith and M. Gratzel, Adv. Mater., 2007, 19, 3643 CrossRef CAS.
- W. H. Howie, J. E. Harris, J. R. Jennings and L. M. Peter, Sol. Energy Mater. Sol. Cells, 2007, 91, 424–426 CrossRef CAS.
- H. J. Snaith and L. Schmidt-Mende, Adv. Mater., 2007, 19, 3187–3200 CrossRef CAS.
- S. Handa, S. A. Haque and J. R. Durrant, Adv. Funct. Mater., 2007, 17, 2878–2883 CrossRef CAS.
- A. B. F. Martinson, T. W. Hamann, M. J. Pellin and J. T. Hupp, Chem.–Eur. J., 2008, 14, 4458–4467 CrossRef CAS.
- J. Kruger, R. Plass, M. Gratzel, P. J. Cameron and L. M. Peter, J. Phys. Chem. B, 2003, 107, 7536–7539 CrossRef.
- J. Bisquert, J. Phys. Chem. C, 2007, 111, 17163–17168 CrossRef CAS.
- J. A. Anta, I. Mora-Sero, T. Dittrich and J. Bisquert, J. Phys. Chem. C, 2007, 111, 13997–14000 CrossRef CAS.
- N. Papageorgiou, P. Liska, A. Kay and M. Gratzel, J. Electrochem. Soc., 1999, 146, 898–907 CrossRef CAS.
- N. Papageorgiou, C. Barbe and M. Gratzel, J. Phys. Chem. B, 1998, 102, 4156–4164 CrossRef CAS.
- J. J. He, H. Lindstrom, A. Hagfeldt and S. E. Lindquist, J. Phys. Chem. B, 1999, 103, 8940–8943 CrossRef CAS.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. D. Yang, Nat. Mater., 2005, 4, 455–459 CrossRef CAS.
- L. E. Greene, B. D. Yuhas, M. Law, D. Zitoun and P. Yang, Inorg. Chem., 2006, 45, 7535–7543 CrossRef.
- T. P. Chou, Q. F. Zhang and G. Z. Cao, J. Phys. Chem. C, 2007, 111, 18804–18811 CrossRef CAS.
- M. Quintana, T. Edvinsson, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2007, 111, 1035–1041 CrossRef CAS.
- H. Horiuchi, R. Katoh, K. Hara, M. Yanagida, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2003, 107, 2570–2574 CrossRef CAS.
- C. Bauer, G. Boschloo, E. Mukhtar and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 5585–5588 CrossRef CAS.
- A. B. F. Martinson, J. E. McGarrah, M. O. K. Parpia and J. T. Hupp, Phys. Chem. Chem. Phys., 2006, 8, 4655–4659 RSC.
- E. Galoppini, J. Rochford, H. H. Chen, G. Saraf, Y. C. Lu, A. Hagfeldt and G. Boschloo, J. Phys. Chem. B, 2006, 110, 16159–16161 CrossRef CAS.
- K. Zhu, N. R. Neale, A. Miedaner and A. J. Frank, Nano Lett., 2007, 7, 69–74 CrossRef CAS.
- S. O. Kucheyev, J. Biener, Y. M. Wang, T. F. Baumann, K. J. Wu, T. van Buuren, A. V. Hamza, J. H. Satcher, J. W. Elam and M. J. Pellin, Appl. Phys. Lett., 2005, 86 Search PubMed.
- T. F. Baumann, J. Biener, Y. M. M. Wang, S. O. Kucheyev, E. J. Nelson, J. H. Satcher, J. W. Elam, M. J. Pellin and A. V. Hamza, Chem. Mater., 2006, 18, 6106–6108 CrossRef CAS.
- J. W. Elam, J. A. Libera, M. J. Pellin, A. V. Zinovev, J. P. Greene and J. A. Nolen, Appl. Phys. Lett., 2006, 89 Search PubMed.
- T. W. Hamann, A. B. F. Martinson, J. W. Elam, M. J. Pellin and J. T. Hupp, Adv. Mater., 2008, 20, 1560–1564 CrossRef.
- T. W. Hamann, A. B. F. Martinson, J. W. Elam, M. J. Pellin and J. T. Hupp, J. Phys. Chem. C, 2008, 112, 10303–10307 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |