Organic liquid CO2 capture agents with high gravimetric CO2 capacity
David J.
Heldebrant
*a,
Clement R.
Yonker
b,
Philip G.
Jessop
c and
Lam
Phan
c
aPacific Northwest National Laboratory, Materials Chemistry and Surface Research Group, Energy and Efficiency Division, Richland, WA 99352, USA. E-mail: david.heldebrant@pnl.gov; Fax: +1-509-375-2186; Tel: +1-509-372-6359
bPacific Northwest National Laboratory, Molecular Interactions & Transformations Group, Fundamental and Computational Sciences Directorate, Richland, WA 99352, USA. E-mail: clem.yonker@pnl.gov; Fax: +1-509-376-6660; Tel: +1-509-372-4748
cQueens University, Department of Chemistry, 90 Bader Lane, Kingston, ON, Canada K7L 3N6. E-mail: jessop@chem.queensu.ca; Fax: +1-613-533-3212; Tel: +1-613-533-6669
First published on 18th July 2008
Abstract
We report a new class of CO2 binding organic liquids that chemically capture and release CO2 much more efficiently than aqueous alkanolamine systems. Mixtures of organic alcohols and amidine/guanidine bases reversibly bind CO2 chemically as liquid amidinium/guanidinium alkylcarbonates. The free energy of CO2 binding in these organic systems is very small and dependent on the choice of base, approximately −9 kJ mol−1 for DBU and Barton's base and +2 kJ mol−1 for 1,1,3,3-tetramethylguanidine. These CO2 capturing agents do not require an added solvent because they are liquid, and therefore have high CO2 capacities of up to 19% by weight for neat systems, and slightly less when dissolved in acetonitrile. The rate of CO2 uptake and release by these organic systems is limited by the rate of dissolution of CO2 into and out of the liquid phase. Gas absorption is selective for CO2 in both concentrated and dilute gas streams. These organic systems have been shown to bind and release CO2 for five cycles without losing activity or selectivity.
Introduction
With global demands for energy from fossil fuels expected to rise, an efficient CO2 trapping system must be employed to minimize the greenhouse gas emissions from fossil fuel combustion. Chemical CO2 trapping agents, such as aqueous alkanolamines, rapidly bind CO2 (forming water-soluble carbamate and bicarbonate salts) and are generally effective in post-combustion systems where the CO2 concentrations are very low (5–15 vol%). However, the process has serious disadvantages.1 The concentration of ethanolamine rarely exceeds 30 wt% due to the corrosive nature of the solution,2 and this reduces the maximum CO2 volumetric (≤108 g L−1) and gravimetric capacity (≤7 wt%) of the CO2 scrubber.2 The ≤30 wt% loading of ethanolamine also means that a large excess of water must be pumped and heated during CO2 capture and release, and this greatly increases the energy requirements. One way of lowering the energy requirements and increase the volumetric and gravimetric capacity of CO2 scrubbers would be to remove the solvent. Herein, we present a novel class of CO2 binding organic liquids (CO2BOL) that do not require dilution with a solvent and chemically bind CO2 with high volumetric and gravimetric CO2 capacity.CO2BOLs are based on Jessop's ‘switchable solvents’, one version of which is a liquid mixture of an alcohol and an amidine or guanidine base that chemically bind CO2 to form an amidinium or guanidinium alkylcarbonate salt (Fig. 1).3–6 The switchable solvents were based on reactions of CO2 with amidine bases such as diazabicyclo[5.4.0]-undec-7-ene (DBU) and polymeric amidines.7–11 While polymer-bound amidines have been proposed in the literature as CO2 capturing agents,9,12 those studies assumed that amidines are capable of chemically binding CO2 as a stable zwitterionic adduct. However, there is no direct evidence of the existence of such an adduct of CO2 with any amidine, and we have shown that the product found by previous researchers from the reaction of CO2 with amidines is actually a bicarbonate salt and not a zwitterionic adduct.7 We therefore conclude that the ability of DBU or polymer-bound DBU to capture CO2 will be limited to the amount of CO2 that can physically adsorb or dissolve plus the amount that can be trapped as the bicarbonate by adventitious water. The CO2BOLs, because they include a stoichiometric amount of alcohol, should be superior because they can chemically bind CO2 without relying on adventitious water.

Conventional (i.e. non-switchable) ionic liquids functionalized with terminal amines have been shown to be good CO2 scrubbing agents with moderate weight capacities of CO2,13–15 Davis' group showed their amine tethered immidazolium IL absorbed 0.5 molar equivalents of CO2 (7.4% CO2 by weight) as a carbamate salt and took three hours to reach saturation.13 CO2BOLs differ from these ionic liquids because they are only ionic liquids after the CO2 is chemically bound. Furthermore, CO2BOLs do not contain CO2 trapping functional groups tethered to a charged but inert core, and subsequently have the potential for higher weight capacities of CO2.
In the studies of switchable solvents, CO2BOLs were shown to reversibly bind CO2 with a high gravimetric and volumetric capacity. CO2BOLs are liquids before and after reacting with CO2, eliminating the need for superfluous inert solvents that reduce the weight and volumetric capacity of the trapping agent. Gravimetric measurements showed that DBU:1-hexanol CO2BOL is capable of capturing 1.3 mol of CO2 per mol DBU, the additional 0.3 (4 wt%) being presumably due to physical rather than chemical absorption; the total CO2 absorbed is 19% by weight and 147 g CO2 L−1 liquid. The combination of chemical and physical adsorption gives CO2BOLs potentially higher CO2 gravimetric capacity and volumetric capacity than aqueous ethanolamine systems (7 wt%, 108 g L−1 liquid) for 30% MEA in water. Using lower molecular weight alcohols and bases could increase the gravimetric CO2 capacities of CO2BOLs even further.
CO2 is chemically bound in CO2BOLs, as an alkylcarbonate salt rather than the bicarbonate or carbamate salts seen in conventional aqueous amine CO2 scrubbing systems.2 Carbamate and bicarbonate salts have strongly bound CO2 with high hydrogen bonding. CO2 is bound more weakly in an alkylcarbonate salt, at least partly because of decreased hydrogen bonding, so that less energy is required to thermally strip the CO2 from the liquid. CO2 release from some CO2BOLs has been shown to occur at temperatures as low as room temperature, although the reaction is slow under those conditions.3,4
Physical and chemical properties of CO2BOLs can be manipulated by changing alcohol/base pairs, as well as by chemically modifying the alcohol/base pairs. Almost any primary or secondary alkanol could be used. Appropriate bases include amidines, guanidines, phosphazines, and possibly some amines; four examples that we have studied are shown in Fig. 2. Due to the potential for dozens of CO2BOL systems, they can be tailor-made for a specific weight capacity, volumetric capacity, regeneration temperature and physical properties. We present here this novel class of liquid organic, high-capacity CO2 scrubbing agents.
![Bases investigated in this study. I, diazabicyclo[5.4.0]-undec-7-ene (DBU); II, 1,1,3,3 tetramethylguanidine (TMG); III, Barton's base; IV, Hünig's base.](/image/article/2008/EE/b809533g/b809533g-f2.gif) | ||
| Fig. 2 Bases investigated in this study. I, diazabicyclo[5.4.0]-undec-7-ene (DBU); II, 1,1,3,3 tetramethylguanidine (TMG); III, Barton's base; IV, Hünig's base. | ||
Experimental
All chemical reagents were purchased from the Aldrich Chemical Company. Bases were distilled over CaH2 under an inert atmosphere and then dried over 4 Å molecular sieves. Alcohols were distilled over Mg activated with I2 under an inert atmosphere and stored over 4 Å molecular sieves. All reagents were handled under a N2 atmosphere in a dry box. CO2 was purchased from Praxair, SFE grade (99.9995%), and pumped directly into the pressure cell. Water content measurements were performed on a Mettler Toledo DL-37 Karl Fischer Titrator. 1H NMR and 13C NMR spectra were acquired on a Varian 300 MHz and a Bruker 400 MHz spectrometers.All CO2 uptake measurements were performed in a Parr 160 mL pressure vessel. The pressure vessel incorporated an Omega CDCE-90-1 (10–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μS conductivity probe and Omega PX01CO-200 A5T 0–200 psi pressure transducer. CO2 uptake was measured by changes in conductivity and decrease in pressure. The pressure vessel was retrofitted to contain an internal sampling system to add liquid reagents to the system. The conductivity measurements were started when the base component was transferred into the reaction solution of alcohol in MeCN.
000 μS conductivity probe and Omega PX01CO-200 A5T 0–200 psi pressure transducer. CO2 uptake was measured by changes in conductivity and decrease in pressure. The pressure vessel was retrofitted to contain an internal sampling system to add liquid reagents to the system. The conductivity measurements were started when the base component was transferred into the reaction solution of alcohol in MeCN.
CO2 evolution experiments were performed in an automated burette system designed in-house.16 CO2BOL components were syringed inside an oven-dried round bottom flask and sealed. The reactor flask was attached to the burette system, opened to the burette system, and then plunged, up to its neck, in a preheated oil bath.
For competitive binding experiments, a 1 : 1 : 1 (by moles) mixture of DBU, MeOH, and another reagent (2-propanol, t-butanol, water or aniline) was prepared in a vial. 1H and 13C NMR spectra of each sample were acquired before and after CO2 treatment. The CO2 bubbling was continued until the NMR spectra showed no further change. The integration ratio of the methyl groups of methanol, methylcarbonate, and the unreacted and reacted forms of the other reagent were measured by 1H NMR spectroscopy (32 scans, delay time of 20 s). Analyses were performed in duplicate. For the experiment with water, quantitative 13C NMR spectroscopy was used to determine the concentrations of the bicarbonate and methylcarbonate anions. To obtain reliable integration values, the 13C NMR spectra were obtained in inverse-gated mode with a delay time of 50 s, which is 5 times the T1 relaxation time.
Thermodynamic measurements were performed with 0.1 M solutions of base and alcohol in d-MeCN. All liquid reagents were syringed under an N2 environment at room temperature to an NMR tube. CO2 was sparged through the solution for 10 min, and then the tube was capped and sealed with teflon tape. Equilibrium constants were calculated from the concentrations of species in solution plus the pressure of CO2. The CO2 pressure was 1 atm of CO2 at 24 °C (the temperature at which the sample was flushed with CO2) and was assumed to slightly rise at higher temperatures due to the release of CO2 from the salt. The amount of that pressure rise was calculated from the observed increase in the concentration of free alcohol.
Results and discussion
CO2 uptake
The rate of CO2 uptake by CO2BOLs (eqn (1)) was evaluated by measuring the change in conductivity of a solution of an amidine or guanidine base and an alcohol in acetonitrile as a function of time. As shown in previous studies,3–5 solutions of amidine and guanidine bases and alcohols are non-conductive until CO2 is present and the amidinium alkylcarbonate salt is produced (Fig. 1).| CO2(g) + DBU + ROH → [DBUH+][ROCO2−] | (1) |
The uptake of CO2 at 28 °C was complete within 20 seconds regardless of choice of base (DBU or TMG) or the choice of alcohol (ethanol, 1-propanol, 1-butanol, 1-pentanol, 1-hexanol). However, the rate was strongly dependent on the stirring rate, indicating that the reaction was limited by the rate of mass-transfer of CO2 from the gas phase into solution rather than the reaction of dissolved CO2 with the base and alcohol. We attempted to increase the rate of mass transfer of CO2 into solution by exclusively using liquids pre-saturated with CO2 and by increasing the stir-rate to 500 rpm but the rate remained dependent on the stir-rate. The process is as rapid as current mixing will allow, which is clearly promising for CO2 capture applications.
Selectivity of CO2 absorption
In order for a CO2 scrubbing system to be applicable for post and pre-combustion CO2 capture, it needs to have high selectivity towards CO2 in either pressurized (pre-combustion) or atmospheric (post-combustion) gas streams. The selectivity of CO2BOLs towards CO2 was demonstrated by reacting DBU and 1-hexanol with CO2 under a N2 atmosphere (Fig. 3). A 1 : 1 molar ratio of DBU and 1-hexanol was placed in a pressure vessel, exposed to gases, and monitored for changes in pressure and conductivity. At first, 50 psig of pure N2 was added; the N2 pressure did not decrease and there was no change in conductivity, indicating no absorption of N2. Under constant stirring, one equivalent of CO2 (50 psig) was added to the reactor vessel, bringing the total pressure to 100 psig. A subsequent pressure drop of 50 psig (Fig. 3) indicated that the CO2 was absorbed. The conductivity of the solution increased, indicating that [DBUH+][ROCO2−] was formed. Thus CO2BOLs are selective towards capture of CO2 in a pressurized 50% gas mixture of CO2 and N2, suggesting that CO2BOLs may be useable for pre-combustion CO2 capture. As shown in previous work, DBU: 1-hexanol are able to selectively capture CO2 from a mixture of CO2 and N2 at 1 atm,17 demonstrating its applicability towards post-combustion CO2 capture.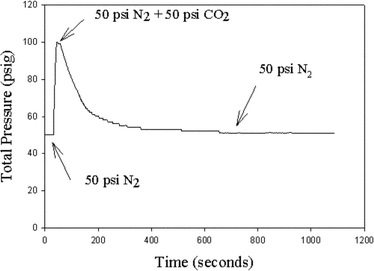
CO2 release
For measurements of the rate of CO2 evolution, CO2 saturated CO2BOLs were placed in the automatic burette system and plunged into a pre-heated oil bath while being stirred and monitored for CO2 evolution. On average, decarboxylations from [DBUH+][ROCO2−] (R = 1-hexyl, 1-pentyl, 1-butyl) and [TMGH+][ROCO2−] (ROH = 1-hexyl, 1-pentyl, 1-butyl) were achieved within 1 min of heating at 90 °C under a static atmosphere at 250 rpm stirring. The rate of CO2 evolution from the CO2BOLs (eqn (2)) was first order with respect to the concentration of CO2BOL salt in solution. Ethanol, 1-propanol, 1-butanol, 1-pentanol and 1-hexanol showed similar rates of CO2 evolution when paired with DBU and heated at the same temperature and solution stir-rate. Changing the solution stir-rate altered the rate at which CO2 evolved from the CO2BOL, suggesting that the rate-limiting step was mass-transfer of CO2 from solution, rather than chemical release of CO2 from the alkylcarbonate anion. The Eact for the process was determined to be 23–33 kJ mol−1 from an Arrhenius plot of the first order rate constant.| [BaseH+][ROCO2−] → CO2 + Base + ROH | (2) |
The total amount of released CO2 detected by the gas burette system was highly dependent on the temperature. CO2BOLs do not decompose or evolve CO2 at an appreciable rate at room temperature under a static atmosphere.17 CO2 evolution from CO2BOLs at a reasonable rate requires either mild heating or continuous flushing of an inert gas or air through the liquid.3 In the burette experiments without flushing of a gas through the liquid, the [DBUH+][ROCO2−] (R = 1-hexyl, 1-pentyl, 1-butyl) and [TMGH+][ROCO2−] (ROH = 1-hexyl, 1-pentyl, 1-butyl) CO2BOLs, on average 0.25 equivalents of CO2 are evolved when heated to 50 °C, 0.50 equivalents at 70 °C and up to 0.65 equivalents at 90 °C. This is attributed to the established thermodynamic equilibrium between the gaseous and dissolved CO2. Elevating the temperature of the CO2BOL shifts the equilibrium, forcing more CO2 evolution.
In comparison with currently employed aqueous alkanolamine systems, CO2BOLs have the potential to be much more energy efficient for CO2 release. 60% of the energy penalty for CO2 capture from power plants is attributed to the thermal stripping of CO2 from solution.2 The large inefficiency of MEA systems is caused by the high specific heat of water (4.18 J g−1 deg−1).18 The specific heat of CO2BOLs to date have not been measured, however the specific heat of other ionic liquids such as 3-ethyl-1-methyl-imidazolium tetrafluoroborate and 3-butyl-1-methyl-imidazolium tetrafluoroborate have been shown to be 1.28 J g−1 deg−1and 1.66 J g−1 deg−1, respectively.19 It is predicted that the specific heat of CO2BOLs would fall between 1.2–2.0 J g−1 deg−1, analogous to other ionic liquids, and therefore at least 50% less energy would be required to thermally strip CO2 from CO2BOLs than from aqueous systems at comparable temperatures.
Another problem with the aqueous ethanolamine system is solvent loss arising from the requirement for heating the liquid above the boiling point of the solvent.2 This is less likely to be a problem because the temperature required for CO2 release from CO2BOLs is well below the boiling point of the components (159 °C for 1-hexanol and 256 °C for DBU).
Recycling
For industrial applications, a CO2 capture agent must be recyclable. To assess the ability of CO2BOLs to be used repeatedly, we performed a series of capture and release cycles with DBU:1-hexanol using our automated gas burette system. The CO2BOL was isolated from the burette system and then carboxylated by sparging the solution with CO2. The CO2BOL was connected to the burette system and then decarboxylated by plunging the stirred flask into a pre-heated oil bath at 90 °C. For each cycle, the mixture of DBU and 1-hexanol was sparged with CO2 for five minutes to form the alkylcarbonate salt [BaseH+][ROCO2−] and then subsequently plunged into a hot oil bath and measured for CO2 release (Fig. 4). After CO2 release, the flask was cooled to room temperature and CO2 was sparged into the solution for another five minutes, this process was repeated for five cycles. DBU and 1-hexanol were carboxylated and decarboxylated five times with no visible loss of activity. We anticipate that the DBU and 1-hexanol CO2BOL could be cycled indefinitely as long as the gas stream is anhydrous and the evaporative losses remain low. | ||
| Fig. 4 Lifetime/repeated CO2 release from DBU and HexOH at 90 °C. Heating begins at one minute. | ||
If water is present, the system selectively forms the much more thermally stable bicarbonate salt, [DBUH+][HCO3−], rather than the CO2BOL alkylcarbonate [DBUH+][ROCO2−]. While gas streams used in this study have very low water content, industrial flue gas streams can have water content as high as 15%,20 so there is a high probability that CO2BOLs would form significant amounts of the stable bicarbonate salt unless steps are taken to exclude water. While the [DBUH+][HCO3−] salt may therefore appear to be problematic, it can be stripped of CO2 at 121 °C, the same temperature used for MEA systems. The stripping of CO2 from CO2BOLs, even with small conversion to [DBUH+][HCO3−], will still be more energy efficient than MEA systems because the specific heat of [DBUH+][HCO3−] should be lower than that of water and the enthalpy for releasing CO2 from that salt should not be significantly more than that from MEA.
Competitive binding experiments
A series of competitive experiments were performed to determine the preference of DBU for certain alcohols. Methanol was chosen as the standard. The competitive binding experiments were performed by bubbling CO2 through 1 : 1 : 1 mixtures of DBU, methanol and another reagent ROH (R = n-propyl, iso-propyl, t-butyl, or H, eqn (3)–(6)). The ratio of equilibrium constants KROH/KMeOH was calculated from the NMR integration ratios. | (3) |
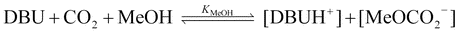 | (4) |
| DBU + CO2 + MeOH + ROH ⇌ [DBUH+] + [MeOCO2−] + [ROCO2−] | (5) |
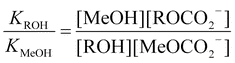 | (6) |
The relative equilibrium constants (Table 1) showed that DBU and CO2 react more favourably with water (forming the bicarbonate salt) than methanol, although the preference for H2O over alcohols is not strong. This result suggests that any water present in the gas stream would be in direct competition with alcohols, and that the formation of the HCO3− salt could be mitigated by competition in solution. Using hydrophobic reagents, or ensuring that there is always significantly more alcohol than water could potentially inhibit formation of significant amounts of the CO2BOL bicarbonate salt. The shortest/most acidic species, water, binds stronger than longer and less acidic species such as methanol and propanol. There was little preference for methanol over a longer primary alcohol (1-propanol), but both of those primary alcohols were preferred over the secondary alcohol iso-propanol and very strongly preferred over the tertiary alcohol t-butanol. In a competition between methanol and t-butanol, there was no detectable conversion of t-butanol to t-butylcarbonate anion. However, in the absence of methanol, a small amount of t-butylcarbonate anion was detectable at 158.0 ppm in the 13C NMR spectrum. These experiments show that for optimum binding of CO2, CO2BOLs should be made using primary alcohols or aniline, with secondary and especially tertiary alcohols being poorer choices.
| ROH | K ROH/KMeOH |
|---|---|
| H2O | 1.43 |
| MeOH | 1.0 |
| PhNH2 | 0.97 |
| PrOH | 0.96 |
| iso-PrOH | 0.80 |
| t-BuOH | 0 |
Aromatic amines such as aniline can serve the same role as an alcohol in this type of reaction, forming a carbamate salt [DBUH+][PhNCO2−] rather than an alkylcarbonate salt. A competitive binding experiment showed that aniline has roughly the same affinity for CO2 and DBU as methanol. Alkylamines were not tested because, unlike anilines, they can serve as bases and thereby could complicate the competition experiments.
Thermodynamics
The thermodynamics of binding are critical for designing a system that can efficiently capture CO2 without requiring too much energy input for the subsequent CO2 release. Approximate thermodynamic data for select CO2BOLs were obtained by 1H NMR spectroscopic measurements of the CO2 binding equilibrium (eqn (1), Table 2). The experiments were performed in d-MeCN to prevent the viscosity of the CO2BOLs from broadening the 1H NMR spectrum. The CO2 absorption has been reported to be slightly lower in solvents21 than in neat systems.5 The concentrations of all relevant species were determined spectroscopically after flushing of CO2 through dilute solutions of base and alcohol in deuterated MeCN. The 1H NMR integrations of the alpha-hydrogen on the bound (RCH2OCO2−) and free alcohol (RCH2OH) were measured over a temperature range of 24–60 °C. The reaction enthalpy was determined from the slope of the Van't Hoff plot of the equilibrium data. Such plots are known to exhibit curvature if the temperature range is too large; significant curvature was observed with Barton's base, so a shorter temperature range of 25–50 °C was used for the tests with that base.| Base/alcohol pair | ΔH/kJ mol−1a | ΔS/J mol−1Kb | ΔG/kJ mol−1c | CO2 absorption in MeCN at 25 °C (%) |
|---|---|---|---|---|
| a Data rounded to two significant figures. b Calculated at 25 °C from NMR integrations using ΔG = −RTlnKeq, Keq = [BaseH+][ROCO2−]/PCO2[Base][ROH]. c Calculated at 25 °C using ΔG = ΔH – TΔS. d Average of the unrounded values for PrOH, BuOH, PentOH, HexOH. | ||||
| DBU/HexOH | −140 | −440 | −9.4 | 87 |
| DBU/PentOH | −120 | −390 | −7.5 | 82 |
| DBU/BuOH | −140 | −450 | −9.7 | 88 |
| DBU/ PrOH | −130 | −420 | −7.8 | 83 |
| DBU/i-PrOH | −140 | −450 | −5.7 | 76 |
| DBU/linear alcohold | −136 | −425 | −8.6 | — |
| TMG/HexOH | −160 | −530 | 1.6 | 42 |
| TMG/PentOH | −210 | −710 | 0.7 | 47 |
| TMG/BuOH | −180 | −590 | 2.4 | 38 |
| TMG/PrOH | −170 | −590 | 2.3 | 39 |
| TMG/i-PrOH | −160 | −550 | 5.5 | 25 |
| TMG/linear alcohold | −180 | −610 | 1.7 | — |
| Barton's/HexOH | −83 | −250 | −11 | 90 |
| Barton's/PentOH | −52 | −150 | −8.7 | 85 |
| Barton's/BuOH | −60 | −180 | −8.0 | 83 |
| Barton's/PrOH | −53 | −160 | −9.0 | 86 |
| Barton's/i-PrOH | −76 | −240 | −7.7 | 82 |
| Barton's/linear alcohold | −72 | −210 | −9.2 | — |
While there is significant scatter in the data, there are very clear trends. The ΔH and ΔG values are almost independent of the choice of alcohol, which is not surprising because the linear alcohols have almost identical pKa values in MeCN. The less favourable ΔG values for iso-propanol could be due to the steric bulkiness of the secondary alcohol destabilizing the alkylcarbonate anion. Tertiary alcohols such as t-butanol were unable to form [BaseH+][ROCO2−] salts in significant quantities.
The reaction energetics depend strongly on the choice of base. Looking at the average values for each base combined with linear alcohols (Table 2), the enthalpy varies in the following order of decreasing exothermicity: TMG > DBU > Barton's base. The weakest base of the three, TMG, (pKaH of the conjugate acid, meaning the pKa of the protonated base, in MeCN is 23.3)22,23 had the least favourable ΔG of reaction and therefore the weakest ability to capture CO2. Surprisingly, however, this was not due to reaction enthalpy; CO2 capture by TMG and alcohol had the most favourable reaction enthalpy, presumably because the salt [TMGH+][ROCO2−] is capable of more hydrogen bonding interactions than the corresponding DBU and Barton's salts (see discussion below and Fig. 5). It is the very negative ΔS that is responsible for the positive ΔG and therefore the weak capture of CO2 by TMG/alcohol mixtures. Extensive hydrogen bonding would be expected to lower the entropy. DBU, the intermediate base (pKaH in MeCN is 24.3)16,18 and Barton's base, the strongest of the three bases (estimated pKaH in MeCN is 25.3)24 had almost identical abilities to bind CO2 but DBU had a much more favourable reaction enthalpy. Barton's base, on the other hand, has a better reaction entropy (−390 J mol−1 K−1 for DBU vs. −185 J mol−1 K−1 for Barton's. The enthalpy of Barton's base is also comperable to that for a 30 wt% of MEA solution in water at 40 °C (−80 kJ mol−1 CO2).25 The pKaH of the base is not a sufficient predictor of the ability of a base to react with CO2 and an alcohol.
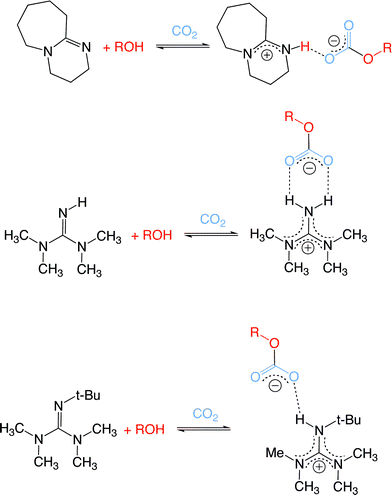 | ||
| Fig. 5 Proposed hydrogen bonding of cation with anion for salts made from DBU, TMG and Barton's base with ROH and CO2. | ||
Hydrogen bonding is believed to be important in the stabilization of the salts. The TMG data suggest that too much hydrogen bonding can lower the ΔG of CO2 binding by negatively impacting the ΔS. Therefore the design of a CO2BOL must take into consideration the number of hydrogen bonding interactions in the salt as well as the strength of those interactions. The alkylcarbonate salts of DBU and Barton's base are likely to have fewer hydrogen-bonding interactions than the salt of TMG because [TMGH+] has two hydrogen-bond donor sites. Additionally, hydrogen-bonding in the TMG salt is likely to be stronger due to the formation of an entropically-favoured 6-member ring (Fig. 5). Similar 6 member rings of carboxylates and amidines have been reported previously by Kraft et al.26 In general, however, the strength of hydrogen-bonding in the alkylcarbonate salts of all three bases is likely weaker than would be expected in salts that lack delocalization in the cation. DBU is reported to be a weak H-bond donor because of its highly delocalized cation.27 Galezowski et al. claim that highly delocalized charges make amidines really weak H-bond donors; as guanidines are more delocalized than amidines, they should therefore be poorer H-bond donors. Hydrogen bonding may also be responsible for the preference for DBU and CO2 to bind water rather than alcohol. Crystallographic structures of [DBUH+][HOCO2−]10 and [DBUH+][CH3OCO2−]5 show that the proton on the anion gives the former salt more extensive hydrogen bonding than the latter.
The reaction entropies can also be explained by considering the hydrogen-bonding of the salts. The ΔS term is least negative for Barton's base, perhaps because the bulky t-butyl group forces the alkylcarbonate anion farther away from the BartonH+ cation. The ΔS value for DBU is more negative due to the decreased steric bulk around the protonated nitrogen. The ΔS value is most negative for TMG, because of the proposed 6-member H-bonding ring being the most ordered and entropically unfavorable. From this data, we can conclude that the ability of a base/alcohol combination to trap CO2 can not be predicted or explained by the pKaH of the protonated base; it is necessary to include hydrogen-bonding arguments and the entropy of reaction.
The thermodynamic results correspond well with the relative equilibrium constants of DBU paired with methanol and other alcohols as discussed previously. The relative thermodynamics show that alcohol choice (>C2) plays a minimal role in the binding energies of CO2 compared to the choice of base. As discussed in previous work,3,4 the alcohol chain length does affect physical properties such as melting point and viscosity of the CO2BOL. While the alcohol may play a minimal role in thermodynamic properties, alcohols can be chosen to alter the physical properties of the CO2BOLs, making them molecularly tunable.
Tertiary amines such as triethylamine and Hünig's base do not form CO2BOLs. Whether Hünig's base should be basic enough (pKaHs of trialkylamines in MeCN are 18.1–18.8)28,29 to accept a proton from the produced alkylcarbonic acid is unclear because the pKas of alkylcarbonic acids are unknown. It is known that applying high pressures of CO2 over a solution of NEt3 in methanol causes the production of [NEt3H+][CH3OCO2−], but reducing the pressure to ambient causes the salt to fall apart into the amine, alcohol, and CO2.30
The amidine and guanidine bases and alkylcarbonate salts that comprise CO2BOLs have charges that are highly delocalized, which weakens the attractions between the ions. We suggest that the delocalization inherent to protonated amidines and guanidines plays a vital role in determining not only the chemical properties (reversibility of CO2 binding and release) of CO2BOLs, but also the physical properties (e.g. melting point or viscosity). A more in-depth investigation of CO2BOLs is underway in our laboratory to confirm the effect of the highly delocalized charges of the amidine and guanidine bases on the binding and release of CO2.
Conclusions
CO2BOLs have been shown to repeatedly bind and release CO2 with a high gravimetric and volumetric CO2 capacity (19% by weight, 147 g L−1 liquid) for neat mixtures because they require no solvent to dissolve the CO2 carrier. The binding and release of CO2 from CO2BOLs appears to be mass-transfer limited, with the dissolution of CO2 in and out of the liquid phase being the rate-limiting step. Binding of CO2 under dilute and concentrated streams was selective, making CO2BOLs applicable in post or pre-combustion CO2 capture. CO2BOLs chemically bind CO2 weakly as alkylcarbonate salts, the binding energies for CO2 is no more than 10 kJ mol−1, which is lower than CO2 release from bicarbonate salts observed in aqueous systems. CO2BOLs organic composition and lower specific heat mean that far less energy is required for stripping CO2 from these agents than from conventional MEA aqueous systems, which are constrained by the high specific heat of water as well as the large volumes of water needed to keep the system liquid. CO2BOLs can be competitively formed in the presence of water, by dilution in a large excess of alcohol. If water is in significant quantities, undesirable bicarbonate salts will be formed, however they can be broken down more efficiently than in MEA systems due to the anticipated lower specific heat of CO2BOLs than water. The alcohol was shown to have a limited role in the energetics of binding CO2, while the base showed some influence on ΔH and ΔG. The low energetics of CO2 binding and release is due to the weak ion-pairs of the CO2BOLs and is not a simple function of the pKa of the base. Calorimetric measurements of the heat capacity and ΔH of reaction are being pursued. With multiple alcohols and bases to choose from, CO2BOLs can be tuned at the molecular level to further enhance their physical and chemical properties. Ultimately, CO2BOLs have tremendous potential to be energy efficient industrial liquid CO2 capture agents.Acknowledgements
We would like to thank Richard Zheng for the design and assembly of the automated gas burette system and John C. Linehan for helpful discussions regarding the NMR measurements. This work was funded by the Department of Energy's Energy Conversion Initiative (ECI), providing internal Pacific Northwest National Laboratory (PNNL) Laboratory Directed Research and Development. PNNL is operated by Battelle for the U. S. Department of Energy.References
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321–348 CrossRef CAS.
- A. N. M. Peeters, A. P. C. Faaij and W. C. Turkenburg, Int. J. Greenhouse Gas Control, 2007, 1, 396–417 CrossRef CAS.
- P. G. Jessop, D. J. Heldebrant, X. W. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102–1102 CrossRef CAS.
- Y. X. Liu, P. G. Jessop, M. Cunningham, C. A. Eckert and C. L. Liotta, Science, 2006, 313, 958–960 CrossRef CAS.
- L. Phan, D. Chiu, D. J. Heldebrant, H. Huttenhower, E. John, X. W. Li, P. Pollet, R. Y. Wang, C. A. Eckert, C. L. Liotta and P. G. Jessop, Ind. Eng. Chem. Res., 2008, 47, 539–545 CrossRef CAS.
- L. Phan, J. R. Andreatta, L. K. Horvey, C. F. Edie, A. L. Luco, A. Mirchandani, D. J. Darensbourg and P. G. Jessop, J. Org. Chem., 2008, 73, 127–132 CrossRef CAS.
- D. J. Heldebrant, P. G. Jessop, C. A. Thomas, C. A. Eckert and C. L. Liotta, J. Org. Chem., 2005, 70, 5335–5338 CrossRef CAS.
- E. R. Perez, M. O. da Silva, V. C. Costa, U. P. Rodrigues and D. W. Franco, Tetrahedron Lett., 2002, 43, 4091–4093 CrossRef CAS.
- T. Endo, D. Nagai, T. Monma, H. Yamaguchi and B. Ochiai, Macromolecules, 2004, 37, 2007–2009 CrossRef CAS.
- E. R. Perez, R. H. A. Santos, M. T. P. Gambardella, L. G. M. de Macedo, U. P. Rodrigues, J. C. Launay and D. W. Franco, J. Org. Chem., 2004, 69, 8005–8011 CrossRef CAS.
- E. Haruki, Organic and Bio-organic Chemistry of Carbon Dioxide, Kodansha Ltd., Tokyo, Japan, 1982 Search PubMed.
- B. Ochiai, K. Yokota, A. Fujii, D. Nagai and T. Endo, Macromolecules, 2008, 41, 1229–1236 CrossRef CAS.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS.
- L. M. G. Sanchez, G. W. Meindersma and A. B. de Haan, Chem. Eng. Res. Des., 2007, 85, 31–39 CrossRef.
- J. B. Tang, W. L. Sun, H. D. Tang, M. Radosz and Y. Q. Shen, Macromolecules, 2005, 38, 2037–2039 CrossRef CAS.
- S. R. F. Zheng, D. J. Heldebrant, D. D. Caldwell, C. L. Aardahl, T. S. Autrey and J. C. Linehan, Rev. Sci. Instrum., 2008 Search PubMed , in press.
- P. G. Jessop, 2005, unpublished results.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, Florida, 84th edn, 2003 Search PubMed.
- J. S. Wilkes, J. Mol. Catal. A: Chem., 2004, 214, 11–17 CrossRef CAS.
- H. Schurmann, P. B. Monkhouse, S. Unterberger and K. R. G. Hein, Proc. Combust. Inst., 2007, 31, 1913–1920 CrossRef.
- Y. N. Y. Hori, J. Nakau and J. Nakau, Chem. Express, 1986, 1, 173–176 Search PubMed.
- I. Kaljurand, T. Rodima, I. Leito, I. A. Koppel and R. Schwesinger, J. Org. Chem., 2000, 65, 6202–6208 CrossRef CAS.
- K. Izutsu, Acid–Base Dissociation Constants in Dipolar Aprotic Solvents, Blackwell Science, Oxford, UK, 1990 Search PubMed.
- E. D. Raczynska, P. C. Maria, J. F. Gal and M. Decouzon, J. Phys. Org. Chem., 1994, 7, 725–733 CrossRef CAS.
- H. S. I. Kim, Ind. Eng. Chem. Res., 2007, 46, 5803–5809 CrossRef.
- A. Kraft, L. Peters, S. Johann, A. Reichert, F. Osterod and R. Frohlich, Mater. Sci. Eng., C, 2001, 18, 9–13 CrossRef.
- W. Galezowski, A. Jarczewski, M. Stanczyk, B. Brzezinski, F. Bartl and G. Zundel, J. Chem. Soc., Faraday Trans., 1997, 93, 2515–2518 RSC.
- I. Kaljurand, A. Kutt, L. Soovali, T. Rodima, V. Maemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS.
- E. I. Room, A. Kutt, I. Kaljurand, I. Koppel, I. Leito, I. A. Koppel, M. Mishima, K. Goto and Y. Miyahara, Chem.–Eur. J., 2007, 13, 7631–7643 CrossRef CAS.
- P. Munshi, A. D. Main, J. C. Linehan, C. C. Tai and P. G. Jessop, J. Am. Chem. Soc., 2002, 124, 7963–7971 CrossRef.
| This journal is © The Royal Society of Chemistry 2008 |