Ammonia borane as an efficient and lightweight hydrogen storage medium
Bo
Peng
and
Jun
Chen
*
Institute of New Energy Material Chemistry, Key Laboratory of Energy Material Chemistry (Tianjin) and Engineering Research Center of High-Energy Storage and Conversion (Ministry of Education), Chemistry College, Nankai University, Tianjin, 300071, People's Republic of China. E-mail: chenabc@nankai.edu.cn
First published on 31st July 2008
Abstract
Ammonia borane (NH3BH3, AB) containing 19.6 wt% hydrogen has been considered as a promising candidate for on-board hydrogen storage applications on the way to the ideal “hydrogen economy”. Whereas, how to control the energy of the hydrogen releasing and recycling of AB efficiently is the present challenge for its wide use. In this mini review, we highlight the preparation, dehydrogenation and regeneration of AB and briefly discuss the current developments, problems and feasible solutions in AB hydrogen storage chemistry.
 B. Peng | Bo Peng was born in Nanchang, China, in 1983. He received his BSc in Chemistry in 2005 from Nankai University, P.R. China. He subsequently moved to the Institute of New Energy Material Chemistry, Nankai University, as a PhD candidate under the supervision of Professor J. Chen. His research interests are hydrogen storage, material simulation and parallel cluster management. |
 J. Chen | Dr Jun Chen became a Professor in the Institute of New Energy Material Chemistry at Nankai University in 2002. He graduated from Nankai University with a BSc in 1989 and a MSc in 1992. He obtained a PhD from Wollongong University (Australia) in 1999. He held the NEDO fellowship at National Institute of AIST Kansai Center (Japan) from 1999 to 2002. He has made contributions to research in energy storage and conversion in the fields of nanomaterials for batteries, fuel cells, and solar cells. |
The “hydrogen economy” as an energy solution has received worldwide attention.1–3 However, despite decades of extensive effort, there are still many hurdles on the path to achieving a “hydrogen economy”.4,5 A major technical challenge is the development of on-board hydrogen storage materials with the combination of high gravimetric/volumetric hydrogen density, adequate kinetics, reversibility, low cost and low toxicity. It is impractical for the current high-pressure and cryogenic hydrogen storage being widely applied in electric vehicles with fuel cells systems, because the low gravimetric/volumetric densities of the former (40 gL−1 at 700 bar) and the low storage temperatures of the latter (70 gL−1 at 20 K) have to be considered.6,7 Instead, the situation has prompted an extensive effort to develop solid hydrogen storage materials, such as metal hydrides and complex hydrides. To promote the research, the US Department of Energy (DOE) has set aggressive goals, e.g. by 2015, a gravimetric density of ≥90 (g H2) kg−1 (i.e. 9.0 wt% H2) and a volumetric density of ≥82 (g H2) L−1 should be reached with a transportation temperature between −20 °C and 85 °C for the whole system, including not only the hydrogen storage medium but also the tank, pumps, heaters, valves, etc. to deliver H2 to the fuel cells.8,9 To meet these targets, many kinds of solid materials have been investigated and found to be promising, typically: (1) metal–complex hydrides (e.g. MgH2, NaAlH4 and NH3BH3),10–12 (2) metal nitrides and imides (e.g. Li3N and LiNH2),13,14 (3) carbon materials (e.g. carbon nanofibers and single-wall carbon nanotubes),15,16 (4) inorganic nanostructures (e.g. TiS2 and MoS2 nanotubes),17,18 and (5) metal–organic frameworks (MOFs).19,20 Among these solid materials, ammonia borane (NH3BH3, AB), whose structure is schematically shown in Fig. 1, has attracted increasing attention as an efficient and lightweight storage medium for hydrogen because of its potential to store a significant percent of hydrogen chemically (19.6 wt% H2) and low molecular weight (30.7 g mol−1). Indeed, both Marder and Baker's group have published two excellent reviews on this subject in recent years.21,22 As this is still an area of intense investigation, many new publications and achievements should be noted, thus in this mini review we focus on the preparation, dehydrogenation and regeneration of AB.
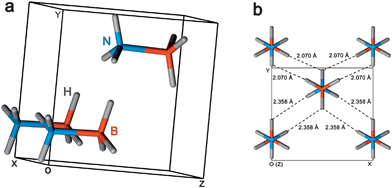 | ||
| Fig. 1 Schematic diagram of the crystal structure of NH3BH3 (a) and a view along z axis (b) at room temperature (298 K). | ||
The primary elements that compose AB are nitrogen, hydrogen and boron. The elements nitrogen and hydrogen exist widely in the nature, e.g. in air and water. The total world reserves of boron are, 369 million tons proven and 807 million tons possible on the basis of B2O3.23 Although, these element resources are abundant, it was not until 1955, that for the first time, Shore et al. synthesized the monomeric compound AB.24 Then in 1973, Mayer reported that a solid AB with high purity could be held at high temperatures (50 °C for 13 days).25 However, the cost of an AB complex is obviously expensive for practical application even now, (according to the Aldrich catalog, the price for 90% Tech. grade is $122 per 10 g and for 97% Tech. grade is $170 per 10 g), therefore, it is critical to find an efficient and economical synthesis procedure before its on-board application. Usually, either a salt metathesis of ammonium salts (e.g. sulfate or chloride) or a direct reaction of NH3 (g) with B2H6 (or BH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ·THF) is used to prepare AB on a laboratory-scale. In a recent paper, Ramachandran and Gargare reported an optimal preparative-scale (10 mol) preparation of AB using sodium borohydride and ammonium formate in dioxane (1 M) in high yield and purity,26 revealing a promising method for large-scale production of AB.
·THF) is used to prepare AB on a laboratory-scale. In a recent paper, Ramachandran and Gargare reported an optimal preparative-scale (10 mol) preparation of AB using sodium borohydride and ammonium formate in dioxane (1 M) in high yield and purity,26 revealing a promising method for large-scale production of AB.
There are various methods of dehydrogenation of AB and the simplest one would be direct heating. Neat AB thermally decomposes initially at 70 °C, it reaches a maximum at 112 °C with the observed melting of AB to yield 1.1 ± 0.1 equiv. H2 and mostly by-product polyaminoborane, [NH2BH2]n.27,28 The polyaminoborane obtained can decompose over a broad temperature range between 110 °C and 200 °C to lose 1 equiv. H2 and form polyiminoborane [NHBH]n at the heating rate of 1 °C min−1. When the rate of temperature is increased to 10 °C min−1, a small fraction of borazine, [N3B3H6], would also be detected by differential scanning calorimetry (DSC) and volumetric analyses.29–31 It should be noted that the main hurdle in the dehydrogenation of AB would be the long induction period for release of hydrogen.30 In the report by Bluhm et al., for the thermolysis of solid state AB carried out at 85 °C, negligible H2 was detected in the first 3 h, but after 17 h, 0.9 equiv. H2 was produced. After that, no further H2 was released even after prolonged heating to 67 h (similar results were obtained at 95 °C).32 In a recent mechanism work, Stowe et al. explained that the long induction period yielded a mobile phase of AB caused by disruption of the dihydrogen bonds.27 In order to reduce the induction period, the thermolysis of AB can be carried out in ionic liquid solvents to lower the reaction onset temperature (or speed up the kinetics)32 or in organic solvents, which are also used to synthesize AB derivants. Sneddon et al. reports that the thermolysis of AB in tetraglyme at 140–160 °C can prepare 10–20 g borazine, N3B3H6, in 65–70 % yields.33 In addition, Autrey's group inserted AB into a subtrate of mesoporous silica (SBA-15) to enhance the kinetics of the thermolysis of AB.34 The thermolysis onset temperature of AB within the template was ca. 15 °C less than that of neat AB and the enthalpy of thermolysis was ca. −2 ∼ 0 kJ mol−1 compared to ca. −20 kJ mol−1 of neat AB. This result indicates that the rate increasing may be benefited from either a templating effect or defect sites. Similarly, another example has been reported recently by Chen's group, showing that the Ni1 − xPtx (x = 0∼0.12) alloy hollow spheres of submicrometer sizes exhibited favorable catalytic activities for both the thermolysis and the hydrolysis of AB, which is mainly due to the high surface areas (e.g. 105 m2g−1 for Ni0.88Pt0.12) and a large surface-to-volume atomic ratio.35
The hydrogen releasing step of the previously mentioned hydrolysis of AB can usually be controlled by adjusting the pH of the system or using transition metal complex catalysts. The former case mainly occurs in acidic conditions, as the neat AB is relative stable in neutral or weak basic aqueous environments, except for some strong basic conditions (e.g. pyridine).36 When AB is added to an acidic solution, a rapid acid-catalyzed hydrolysis of AB occurs.37 It has been reported that the reaction is first order in AB and proton.38 Through analyzing the isotope effects and reaction rates for the interaction mechanism between AB and H+, Kelly and Marriott concluded that the H+ was more likely to interact with –NH3 rather than –BH3 to form NH4+ and dissociated BH3, which would rapidly release hydrogen when hydrolysis.39 It has also been found that the former step, i.e. the H+ interacting with –NH3 is the rate-limiting step, but when a strong Lewis acidic environment is encountered, the situation is slightly different, it becomes one in which hydrogen release is accelerated.40 The reaction path can be described as follows: a boronium cation [(solvent)NH3BH3]+ is formed at first, then the cation interacts with another 1 equiv. of AB to generate H2 and polyaminoborane verified by the 11B NMR experiment.32 Chandra and Xu have also extended the acid-catalyzed hydrolysis of AB from liquid acid cases to solid acid cases (such as some sulfonic acids, Si–OH functional groups, carbonic acid etc.) with the aim of solving the problem of intermittent use of H2 fuel in a vehicle. In such system, only when AB(aq) contacted with the solid acid will the H2 be produced.41
Alternatively, transition metal complex catalysts are another main strategy that controls hydrogen release to a much wider extent. Up to now, the “fastest” thermolysis catalyst known for releasing 1 equiv. H2 from AB is an iridium pincer complex, (POCOP)Ir(H)2 (POCOP = [η3-1,3-(OP-tert-Bu2)2C6H3]), reported by Goldberg, and with a 0.5 mol % catalyst loading, the releasing time of 1 equiv. H2 is approx. 14 min at ambient temperature.42 The inspired use of the iridium pincer complex for the dehydrogenation of AB results from the fact that the iridium pincer complex is a kind of traditional catalysts for alkane dehydrogenation and AB is isoelectronic with its similar structure C2H6. Besides, the reaction rate stays the same in the presence or absence of mercury, indicating the iridium catalyst remains homogeneous during the dehydrogenation. With further analysis by NMR spectroscopy, two intermediate complexes are detected, (POCOP)Ir(H)4 and (POCOP)Ir(H)2·BH3, from which the clean catalyst can be regenerated under H2 pressure, showing a promising initial practical step. In a subsequent theoretical study, Paul and Musgrave examined two possible catalysis mechanisms using density functional theory (DFT) calculations, and supported an energy favorable direct attack of AB on the iridium(III) dihydride species.43 In addition, many other metal complexes have also been investigated for AB analogue, e.g. Manners et al. reported that the metal Rh, Pd and Ru catalysts could dehydrogenate amine borane at room temperature with a 0.5 mol % catalyst loading.44 By combining XAFS (or Operando EXAFS) and 11B NMR spectroscopies, Fulton and coworkers proved that homogeneous Rh complexes, i.e. small Rhn (n = 4–6) clusters bound to AB, are catalytically active species for the dehydrogenation of a series of amine boranes.45,46 Further, with the knowledge of strongly donating ligands and electron-rich metal centers promoting B–H bond activation, Baker used a trialkylphosphine and N-heterocyclic carbine (NHC) combination with cheaper and more abundant first row metals, e.g. nickel, to develop long-lived catalysts with unprecedented ability of hydrogen release (>18 wt%).47 Remarkably, during the process, the main volatile intermediate product, borazine, which is poisonous for the fuel cell, can be further dehydrogenated to form a soluble cross-linker borazine and the reaction rate is in the same order as the consumption of AB starting material.
Besides metal-catalyzed thermolysis, we can also make use of metal–catalysis to promote the hydrolysis of AB, which is relative stable in neutral or weak basic solution. There have been many noble metal catalysts (such as Pt-related, [Rh(1,5-COD)(μ-Cl)]2 and Pd black) reported and the best performances are obtained with Pt-related materials (such as Pt–C, PtO2, Pt black and K2PtCl4etc.),41,48,49 by which 3 equiv. H2 can be released in between 2 and 19 min. Concluded from those experimental results, it has been found that the combination of smaller particle sizes with higher dispersion plays a role in the active catalyst. With this in mind, supported Co and Ni catalysts have shown high activity for AB hydrolysis with a metal–AB ratio of 0.018.49 Subsequently, Xu's group reported that Fe nanoparticles with no protective shell show excellent catalytic activity for the hydrolysis dehydrogenation of aqueous AB under argon, and even in air at room temperature.50 Approximately, 134 mL H2 is released by hydrolysis of aqueous AB (0.16 M, 10 mL, Fe–AB = 0.12) in ca. 8.5 min. The in situ prepared Fe nanoparticles with amorphous phase and zero-valence form exist as a perfect suspension in aqueous solution and contain aggregates with an average diameter of 60 nm, in which each aggregate is also an assembly of many smaller Fe nanoparticles. For more detailed information on this subject, readers are invited to refer to the review written by Xu and Chandra, in which the authors mainly focus on the recent development, catalytic hydrolysis of AB, by their group.51
Several other strategies for the dehydrogenation of AB have also been reported, such as methanolysis,26 strong oxidation,52 and metal amine activation,53etc. With the driving force of the high potential between the Hδ+ in –NH3 and Hδ− in metal hydrides, Diyabalanage et al. recently prepared calcium amidotrihydroborate, Ca(NH2BH3)2, and 2 equiv. H2 by the reaction of AB with calcium hydride in THF. The generated Ca(NH2BH3)2 has a lower propensity for borazine release (<0.1% of the weight change below 170 °C) and does not suffer from the long induction period for release of hydrogen that is present in AB.53 For example, for AB, any temperature above 120 °C results in the release of about two equiv. H2 because of the exothermic procedure (ca. −20 kJ mol−1), while for Ca(NH2BH3)2, the endothermic reaction (3.5 kJ mol−1) makes 1.1 equiv. H2 released from Ca(NH2BH3)2 at 120 °C, 2.4 equiv. at 150 °C and ∼4 equiv. at 170 °C. One more recent example is the reaction between AB and alkali-metal hydride reported by Xiong et al.54 1 equiv. H2 can be obtained by ball-milling 1:1 equiv. ratios of AB and the corresponding alkali-metal hydrides. Furthermore, a high storage capacity is easily accessible for alkali-metal aminoboranes (e.g. 10.9 wt% for LiNH2BH3 and 7.5 wt% for NaNH2BH3) and the obtained alkali-metal amidoboranes show substantially enhanced dehydrogenation characteristics with respect to AB itself (e.g. the enthalpies of dehydrogenation from LiNH2BH3 and NaNH2BH3 are −3 and −5 kJ mol−1vs. −20 kJ mol−1 of neat AB). At a dehydrogenation temperature of approximately 90 °C, 8 wt% and 6 wt% of hydrogen are released within the first hour without the unwanted by-product borazine. At present, the main problem for these alkali-metal aminoboranes is the lack of facile reversibility. The discussion of thermolysis and hydrolysis, it is not complete without mentioning the recent important development to the research of AB by Zhang et al.55 As the H2 (H0) obtained by dehydrogenation or hydrolysis of AB (as a source of H−) will be used to feed the fuel cell and further transforms into water (H+), the authors proposed a direct AB fuel cell and developed a direct electrochemical oxidation of AB in a fuel cell expecting to aid the search for other attractive alternative fuel. The experiment showed that in a 25 cm2 lab cell, a power density of >14 mW cm−2 could be reached at ambient temperature. Further open circuit potential and cyclic voltammetry results confirmed the direct electron transfer from H− in AB to H+, which opens the possibility of improvement in the performance of a direct AB fuel cell.
As mentioned above, an economically viable method for the regeneration of AB-decomposed by-product may be the ultimate barrier to its on-board application in hydrogen storage. Indeed, the methods for the efficient regeneration depend on the products, whilst the wide range of products show greatly differing reactivities.32 Calculations of the enthalpy of hydrolysis of AB indicate that the direct regeneration under H2 pressure is not energy favorable in thermodynamics (e.g. −227 kJ mol−1 would be required for AB + H+ + 3H2O → B(OH)3 + 3H2 + NH4+) and the recycling of AB from hydrolysis product was considered to be a multi-step process.56,57 Similarly, Tumas et al. pointed that there must be several steps to regenerate AB from the product BNHx and each step must be optimized to identify an energy efficient system.58 In the recent report, Sneddon and coworkers firstly used a superacidic system, HBr–AlBr3, in CS2 to digest the spent-AB fuel (at 23 °C for 4 h), then from the reaction mixture, BBr3 could be distilled, and finally, via a one-pot reduction followed by NH3 exchange, AB was regenerated in 84% yield, which was reported to be dependent on the reaction conditions and the type of spent fuels.59 In another independent report, Hausdorf et al. presented a general concept of the energy-efficient regeneration of AB from a wide spectrum of BNH-products, which is induced by the thermal dehydrogenation of AB. BNHx is initially treated with a solution of HCl (Brønsted acid) in organic solvents to form BCl3·base and NH4Cl, and then, followed by a hydrodechlorination process of the as-formed BCl3·base, B2H6 would be formed, which is finally reacted with NH3 (from NH4Cl) to regenerate AB.60 In this concept, the hydrodechlorination with H2 can be performed repeatedly via BCl3NR3 + 3H2 + 3NR2 → BH3NR3 + 3NR3HCl (R = alkyl). However, for the methanolysis of AB, Ramachandran and Gagare recently reported that AB can be formed from its methanolysis intermediate, ammonium tetramethoxyborate, through an efficient one-pot regeneration (NH4B(OMe)4 + NH4Cl + LiAlH4 → NH3BH3 + Al(OMe)3 + MeOH + H2 + LiCl + NH3).26
The clue to the preparation, dehydrogenation and regeneration of AB mentioned in this mini review has been demonstrated in Fig. 2 in a flow diagram. In conclusion, as a hydrogen storage medium, AB is a promising material not only because of its high hydrogen capacity and light weight, but also for the abundant AB chemistry, which offers various dehydrogenation methods from hydrolysis to thermolysis, from acid-catalyzed to metal-catalyzed and from homogenous to heterogeneous etc. Thus, considerable progress has been made in investigating the AB system. Undoubtedly, both an acceptable rate/extent of dehydrogenation and the reversibility of ammonia borane are still the main challenges to its practical use. For the former, it is expected that nanostructured materials will play a more important part as high surface areas and large surface-to-volume atomic ratios make an unprecedented difference between nano, meso, micro, and bulk materials. For the latter, Marder recently highlighted that finding new uses for the dehydrogenation by-products of AB may be as important as developing economically viable methods for regeneration of AB.22 Up to now, because high energy input is required to complete the energy cycle of AB and most of the energy is powered by non-renewable energy sources (e.g. fossil fuels), on the one hand we must enhance the utility of the non-renewable energy source with cleanliness and high efficiency, and on the other hand, we must consider using more renewable energy sources (such as solar photo-to-electricity conversion, etc.) to substitute for the non-renewable energy source. All in all, a comprehensive solution from current hydrogen storage materials to engineering systems is a reasonable way get close to the targets needed for transportation applications.
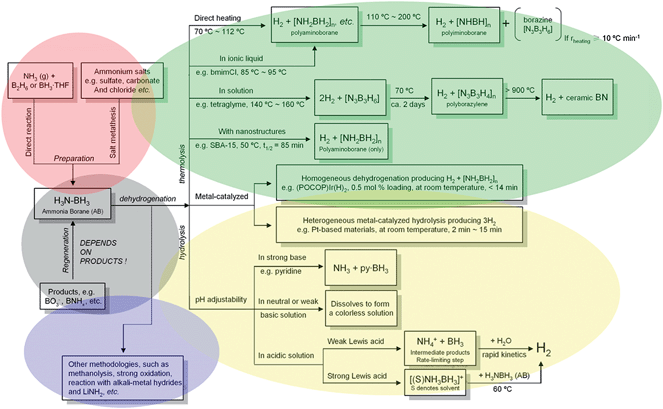 | ||
| Fig. 2 Schematic diagram of the preparation, dehydrogenation and regeneration of AB. | ||
Acknowledgements
The authors make a grateful acknowledgement for the reviewers' comments. This work was supported by the National NSFC (50631020 and 50771056) and MOST Programs (2007AA05Z108 and 2007AA05Z149) from P.R. China and FP-6 Hydrogen Solid Storage Activities (038941) from the European Community.References
- P. Grant, Nature, 2003, 424, 129 CrossRef CAS.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332 CrossRef CAS.
- L. de Palacio and P. Busquin, Report from Europa Commission, “Hydrogen Energy and Fuels Cells: A Vision for Our Future” from http://europa.eu.int/comm/research/energy/ Search PubMed.
- R. Coontz and B. Hanson, Science, 2004, 305, 957 CrossRef.
- G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, Phys. Today, 2004, 57, 39 CrossRef CAS.
- M. Gutowski and T. Autrey, Chem. World, 2006, 3, 44 Search PubMed.
- R. von Helmolt and U. Eberle, J. Power Sources, 2007, 165, 833 CrossRef CAS.
- Recent information can be found at: http://www.eere.energy.gov/hydrogenandfuelcells/pdfs/freedomcar_targets_explanations.pdf.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246 CrossRef CAS.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283 CrossRef CAS.
- D. K. Ross, Vacuum, 2006, 80, 1084 CrossRef CAS.
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111 CrossRef CAS.
- J. J. Hu, G. T. Wu, Y. F. Liu, Z. T. Xiong, P. Chen, K. Murata, K. Sakata and G. Wolf, J. Phys. Chem. B, 2006, 110, 14688 CrossRef CAS.
- P. Chen, Z. T. Xiong, J. Z. Luo, J. Y. Lin and K. L. Tan, Nature, 2002, 420, 302 CrossRef CAS.
- D. J. Browning, M. L. Gerrard, J. B. Lakeman, I. M. Mellor, R. J. Mortimer and M. C. Turpin, Nano Lett., 2002, 2, 201 CrossRef CAS.
- A. C. Dillion, K. M. Jones, T. A. Bekkedahl, C. H. Kiang, D. S. Bethune and M. Heben, Nature, 1997, 386, 377 CrossRef CAS.
- J. Chen, S. L. Li, Z. L. Tao, Y. T. Shen and C. X. Cui, J. Am. Chem. Soc., 2003, 125, 5284 CrossRef CAS.
- J. Chen, N. Kuriyama, H. T. Yuan, H. T. Takeshita and T. Sakai, J. Am. Chem. Soc., 2001, 123, 11813 CrossRef CAS.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS.
- X. B. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M J. Rosseinsky, Science, 2004, 306, 1012 CrossRef CAS.
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 25, 2613 Search PubMed.
- T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116 CrossRef CAS.
- Data collected from http://www.etimaden.gov.tr/.
- S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 8 CrossRef CAS.
- E. Mayer, Inorg. Chem., 1973, 12, 1954 CrossRef CAS.
- P. V. Ramachandran and P. D. Gargare, Inorg. Chem., 2007, 46, 7810 CrossRef CAS.
- A. C. Stowe, W. J. Shaw, J. C. Linehan, B. Schmid and T. Autrey, Phys. Chem. Chem. Phys., 2007, 9, 1831 RSC.
- V. Sit, R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1987, 113, 379 CrossRef CAS.
- M. G. Hu, R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1978, 23, 249 CrossRef CAS.
- G. Wolf, J. Baumann, F. Baitalow and F. P. Hoffmann, Thermochim. Acta, 2000, 343, 19 CrossRef CAS.
- F. Baitalow, J. Baumann, G. Wolf, K. Jaenicke-Röβler and G. Leitner, Thermochim. Acta, 2002, 391, 159 CrossRef CAS.
- M. E. Bluhm, M. G. Bradley, R. Butterick, III, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748 CrossRef CAS.
- T. Wideman and L. G. Sneddon, Inorg. Chem., 1995, 34, 1002 CrossRef CAS.
- A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578 CrossRef CAS.
- F. Y. Cheng, H. Ma, Y. M. Li and J. Chen, Inorg. Chem., 2007, 46, 788 CrossRef CAS.
- J. S. Wang and R. A. Geanangel, Inorg. Chim. Acta, 1988, 148, 185 CrossRef CAS.
- G. E. Ryschkewitsch, J. Am. Chem. Soc., 1960, 82, 3290 CrossRef CAS.
- A. D'Ulivo, M. Onor and E. Pitzalis, Anal. Chem., 2004, 76, 6342 CrossRef CAS.
- H. C. Kelly and V. B. Marriott, Inorg. Chem., 1979, 18, 2875 CrossRef CAS.
- F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., Int. Ed., 2007, 46, 746 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 159, 855 CrossRef CAS.
- M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048 CrossRef CAS.
- A. Paul and C. B. Musgrave, Angew. Chem., Int. Ed., 2007, 46, 8153 CrossRef CAS.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS.
- Y. S. Chen, J. L. Fulton, J. C. Linehan and T. Autrey, J. Am. Chem. Soc., 2005, 127, 3254 CrossRef CAS.
- J. L. Fulton, J. C. Linehan, T. Autrey, M. Balasubramanian, Y. S. Chen and N. K. Szymczak, J. Am. Chem. Soc., 2007, 129, 11936 CrossRef CAS.
- R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 156, 190 CrossRef CAS.
- Q. Xu and M. Chandra, J. Power Sources, 2006, 163, 364 CrossRef CAS.
- J. M. Yan, X. B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 1 CrossRef.
- Q. Xu and M. Chandra, J. Alloys Compd., 2007, 446–447, 729 CrossRef CAS.
- G. A. Olah and S. J. Kuhn, US Pat., 3103782, 1963 Search PubMed.
- H. V. K. Diyabalanage, R. P. Shrestha, T. A. Semelsberger, B. L. Scott, M. E. Bowden, B. L. Davis and A. K. Burell, Angew. Chem., Int. Ed., 2007, 47, 8995 CrossRef.
- Z. T. Xiong, C. K. Yong, G. T. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138 CrossRef CAS.
- X. B. Zhang, S. Han, J. M. Yan, M. Chandra, H. Shioyama, K. Yasuda, N. Kuriyama, T. Kobayashi and Q. Xu, J. Power Sources, 2007, 168, 167 CrossRef CAS.
- D. A. Dixon and M. Gutowski, J. Phys. Chem. A, 2005, 109, 5129 CrossRef CAS.
- C. R. Miranda and G. Ceder, J. Chem. Phys., 2007, 126, 184703 CrossRef.
- W. Tumas, R. T. Baker, A. Burrell and D. Thorn, DOE Chemical Hydrogen Storage Center of Excellence, DOE Hydrogen Program FY 2006 Annual Progress Report, 2006, http://www.hydrogen.energy.gov/pdfs/progress06/iv_b_4_tumas.pdf Search PubMed.
- L. G. Sneddon, Amineborane-Based Chemical Hydrogen Storage, DOE Hydrogen Program Review, 2007, http://www.hydrogen.energy.gov/pdfs/review07/st_27_sneddon.pdf Search PubMed.
- S. Hausdorf, F. Baitalow, G. Wolf and F. O. R. L. Mertens, Int. J. Hydrogen Energy, 2008, 33, 608 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |