On board catalytic NOx control: mechanistic aspects of the regeneration of Lean NOx Traps with H2
Pio Forzatti*, Luca Lietti and Isabella Nova
Politecnico di Milano, Dipartimento di Energia, Laboratory of Catalysis and Catalytic Processes, P.zza L. Da Vinci 32, 20133, Milano, Italy. E-mail: pio.forzatti@polimi.it; Fax: +390223993318; Tel: +390223993238
First published on 2nd July 2008
Abstract
In this paper the results obtained in our previous studies on the reduction of nitrates by H2 over Pt–Ba/Al2O3 catalyst samples are presented and discussed along with new data recently collected in order to explain the chemistry that leads to nitrogen and ammonia evolution during the regeneration of Lean NOx Traps.
Pio Forzatti is a Full Professor of Chemical Engineering at the Department of Energy, Politecnico di Milano, Italy. His main research interests are in the field of energy-related and environmental catalysis, including Catalytic Combustion, Catalytic Partial Oxidation of HCs, Lean NOx removal, Fischer Tropsch synthesis. |
Luca Lietti is a Full Professor of Chemical Engineering at the Department of Energy, Politecnico di Milano, Italy. His research covers energy-related and environmental catalysis, including Fischer Tropsch synthesis, catalytic removal of soot and Lean NOx Trap systems. |
Isabella Nova is an Associate Professor of Chemical Engineering at the Department of Energy, Politecnico di Milano, Italy. Her research activities focus on environmental catalytic processes, including Selective Catalytic Reduction of NOx by NH3 and Lean NOx Traps. |
A. Introduction
Nitrogen oxides (NOx) are blamed for the production of acid rain, the formation of ozone in the troposphere and the production of secondary particulate matter. Although some NOx emissions are produced from natural sources such as lightning and microbiological activities, a considerable amount of nitrogen oxides emissions have a human origin. In particular NOx is generated during combustion processes in the energy and transportation sectors. The transportation sector and in particular diesel-equipped vehicles have become, in recent years, one of the primary sources of NOx emissions. For this reason, regulations to control NOx emissions in industrialised countries are becoming very severe: in Europe, Euro 5 (2009) and Euro 6 (2014) regulations for diesel passenger cars will require a three-fold decrease of NOx emissions from 0.25 g km−1 (Euro 4) to 0.08 g km−1 (Euro 6).Gasoline-powered engines are characterized by lower NOx emissions than diesel engines (0.08 g km−1 was the limit set for gasoline engines by Euro 4), but due to their higher fuel consumption and CO2 emissions their market share has decreased in these last years. Indeed, in the European Community vehicles equipped with diesel engines have increased their market share up to nearly 60%.
Contrary to stoichiometry, in gasoline engines three-way catalysts are not effective in the reduction of NOx under lean conditions, as is the case for diesel engines. A viable solution for the control of NOx in the exhausts from lean burn engines is the urea–SCR technique, which accomplishes NOx reduction by injecting urea (a precursor of NH3) into the flue gases, or the NOx Storage Reduction (NSR) or Lean NOx Trap (LNT) system.1 In such a system the removal of NOx is realized by adsorbing nitrogen oxides in the form of nitrates and nitrites under lean conditions, and then reducing the stored NOx to N2 by short excursions under rich conditions: accordingly, these systems operate under cyclic conditions alternating a long lean period with a short rich phase.
A typical NSR catalyst consists of a NOx storage component, such as an alkaline earth metal oxide (e.g. Ba), and of a noble metal (Pt), which catalyses the oxidation of NO, CO and unburned hydrocarbons (UHC) during the lean phase, and reduction of stored NOx during the rich phase. These elements are dispersed on a high surface area support, such as γ-alumina. Commercial catalyst formulations may also include: potassium, as a substitute to or in addition to Ba; TiO2, which is reported to decrease SOx adsorption; Rh, which favours NOx reduction with H2/CO/HCs and H2 generation via steam reforming of hydrocarbons; ZrO2 or CeO2–ZrO2, which are used to promote the formation of hydrogen through steam reforming and WGS reactions.1
Although urea–SCR is preferred for heavy trucks and mini-vans, LNTs are cheaper for small engines.2 Accordingly, in the last few years the potential of LNT technology has motivated extensive investigation by both the academic and industrial communities. Great efforts have been devoted to the analysis of the storage of NOx and to the identification of the nature and the role of the adsorption sites.3–24 In subsequent regeneration of the trap, adsorbed NOx species are reduced to N2 but the formation of other by-products (NH3, NO and N2O) may be observed.
In spite of the fact that the regeneration step plays a crucial role in the overall process, relatively few studies have been dedicated to the analysis of this phase. There is a general consensus that the regeneration of NSR catalysts includes, at first, the release of NOx from the catalyst surface upon decomposition of nitrite–nitrates ad-species, followed by the reduction of the released NOx.1 However, the mechanistic details of these steps and the precise role of the catalyst components are still unclear.
In recent years, an extensive investigation of the mechanisms involved in the storage of NOx was carried out in our labs.18–24 By the combined use of transient reactivity techniques and FTIR spectroscopy, the major routes involved in the adsorption of NOx were clarified. More recently, the regeneration of model Pt–Ba/Al2O3Lean NOx Trap systems was also investigated.18,25–30 In this review the results obtained in our studies of the reduction phase will be reported, along with new data recently collected which better explain the chemistry involved in the reduction by H2 of the stored NOx.
B. NOx storage–reduction over model Pt–Ba/Al2O3 catalyst under isothermal conditions
A typical result obtained upon NO adsorption in He + O2 at 200 °C over a model Pt–Ba/Al2O3 LNT catalyst (Pt–Ba/Al2O3 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:100), followed by reduction with H2 at the same temperature, is presented in Fig. 1 (A) (storage) and Fig. 1 (B) (reduction).
:20:100), followed by reduction with H2 at the same temperature, is presented in Fig. 1 (A) (storage) and Fig. 1 (B) (reduction).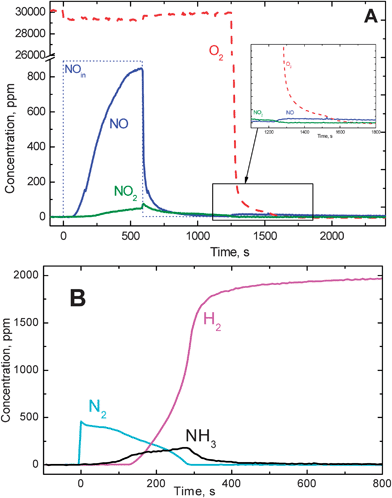 | ||
| Fig. 1 (A) Temporal evolution of NO, NO2 and O2 outlet concentrations during adsorption of NO (1000 ppm) in He + O2 3% (v/v) at 200 °C over the Pt–Ba/Al2O3 catalyst. (B) Temporal evolution of H2, N2 and NH3 outlet concentrations during reduction with H2 (2000 ppm in He) over the Pt–Ba/Al2O3 catalyst. | ||
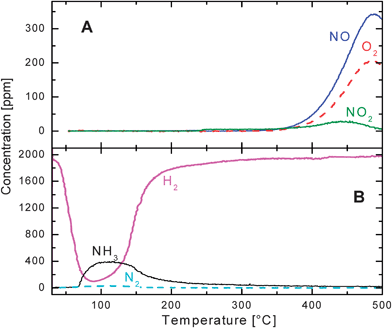
NO (1000 ppm) was admitted to the reactor at t = 0 s under He + O2 (3% v/v) flow; after 100 s NO breakthrough was observed, while NO2 evolution was seen starting from 200 s. Both the NO and NO2 outlet concentrations increased with time until the NO feed flow was switched off (t = 600 s). NOx was stored primarily in the form of nitrates, as shown by FTIR experiments,22 although the presence of nitrites cannot be excluded. After the NO switch off a tail was observed in the NO and NO2 profiles, due to desorption of weakly adsorbed species. The amounts of NOx stored over the catalyst after the He purge was close to 3 × 10−4 mol gcat−1, corresponding to an overall Ba utilization (i.e. the fraction of Ba involved in the storage assuming the formation of Ba(NO3)2) close to 12%. The desorption process was very slow since, after several minutes the NO and NO2 concentrations had not yet decreased to zero. An additional minor NO release was observed at t ∼ 1200 s, upon switching off the oxygen flow (He purge), in line with the effect of oxygen partial pressure on the stability of the nitrate species formed on the catalyst surface.25,31 It is worth noticing that these data indicate that the rate of nitrate decomposition under an inert atmosphere is rather slow, and that substantial amounts of NOx are retained on the catalyst surface after a He purge at the adsorption temperature. Similar results were obtained at higher temperatures.25
Part (B) of Fig. 1 shows the results obtained following stepwise addition of H2 (2000 ppm) in He after NOx storage at the same temperature (200 °C). H2 was immediately and completely consumed and N2 was the main product along with traces of NO (not shown in the figure); the reaction was very fast and was limited by the concentration of H2. Upon hydrogen admission a very small temperature increase of 3–5 °C was observed, so that the reduction was accomplished under nearly isothermal conditions. The concentration of nitrogen was rather constant (roughly 400 ppm) for 100 s, and then progressively decreased to zero while ammonia formation was observed. Hydrogen breakthrough was also detected after ammonia formation. Accordingly, the reduction process was initially very selective to nitrogen (almost quantitative); then a switch of the selectivity to ammonia was measured. NH3 was by far the most important by-product of the reduction of stored NOx, as is also reported by several authors.5,21,32,33
The following reactions can be invoked to explain the formation of N2 and NH3 from nitrites and nitrates:
| Ba(NO2)2 + 3H2 → BaO + N2 + 3H2O | (1) |
| Ba(NO3)2 + 5H2 → BaO + N2 + 5H2O | (2) |
| Ba(NO2)2 + 3H2 → BaO + 2NH3 + 3H2O | (3) |
| Ba(NO3)2 + 8H2 → BaO + 2NH3 + 5H2O | (4) |
The N2 concentration of 400 ppm in Fig. 1 (B) was close to that expected from reaction (2) (400 ppm) and was significantly lower than that expected from reaction (1)(660 ppm). This eventually confirms that nitrates are the most abundant ad-species when the storage of NOx is extended up to saturation.22
In reactions (1)–(4)the formation of BaO is envisaged upon regeneration of Ba(NO2)2 and Ba(NO3)2. However, due to the formation of water upon reduction of the stored nitrites/nitrates, Ba(OH)2 may also be formed due to the reaction:
| BaO + H2O → Ba(OH)2 | (5) |
Accordingly, on the basis of the results shown in Fig. 1 (B), the reduction of the stored NOx:
• is a very fast process that is limited already at 200 °C by the reductant concentration (similar effects were observed at higher H2 concentration, close to 1%);
• it occurs under isothermal conditions, due to the very small temperature excursion upon admission of the reductant;
• is initially very selective to N2; the selectivity changes with time since ammonia prevails at the end of the reduction phase.
In order to clarify the mechanistic aspects of nitrate reduction by H2 over model Pt–Ba/Al2O3 NSR catalyst samples, a detailed analysis of the thermal stability and of the reactivity of the stored nitrates in H2 was carried out in our laboratories using different experimental techniques, and the results are reported below.
C. Thermal stability—reactivity of nitrates stored on model Pt–Ba/Al2O3 catalysts.
It was suggested that the first step in the reduction of NOx stored over typical NSR catalysts is the decomposition of nitrates to gaseous NOx, which is then reduced at the noble metal.1,5 To provide further indication of the mechanisms operating during the reduction of the stored NOx, the thermal stability and reactivity with H2 of NOx stored over the model Pt–Ba/Al2O3 catalyst were investigated in our labs by Temperature Programmed Desorption (TPD), and Temperature Programmed Surface Reaction (TPSR).25–30 TPD experiments provided information on the thermal stability of nitrate species adsorbed onto the catalyst surface, whereas TPSR carried out in the presence of H2 (H2–TPSR) shed light on the stability and reactivity of the stored nitrate species under a reducing environment. Experiments were also performed at constant temperature by imposing stepwise changes in the H2 inlet concentration (Isothermal Transient Response Method, ITRM) to analyze the stability of nitrate species in the presence of H2 at constant temperature. All the experiments were carried out over the model Pt–Ba/Al2O3 catalyst, but the corresponding binary Ba/Al2O3 and Pt/Al2O3 samples and a Ba/Al2O3–Pt/Al2O3 physical mixture were also considered for comparison purposes. Accordingly, the role of the various catalyst components on the thermal stability/reactivity of the stored NOx could be elucidated.Notably, in all cases near isothermal conditions in the catalyst bed were maintained, i.e. the runs were performed in the absence of any significant thermal effects upon temperature programming and/or lean–rich switches due to the dilute conditions employed in the experiments.
If not otherwise stated, NOx was stored on the catalyst surface starting from He/NO/O2 mixture at 350 °C, i.e. under conditions of practical interest.22 After adsorption, the catalyst was purged with He at the adsorption temperature and cooled down to RT while flowing He: this prevented any further contact of the catalyst with NO at temperatures different from that of adsorption. This saturation procedure was adopted for all the investigated samples except for the binary Ba/Al2O3 catalyst, for which the NOx storage was carried out starting from NO2 instead of NO, due to the poor NO adsorption ability of this sample.22
TPD experiments
Fig. 2 shows the results of the TPD experiments performed over the different catalytic systems after adsorption of NOx at 350 °C: the concentrations of NOx (obtained as the sum of measured NO and NO2 outlet concentrations) are plotted as a function of the catalyst temperature.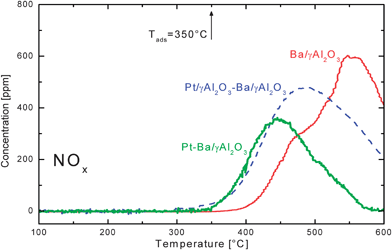 | ||
| Fig. 2 NOx outlet concentrations versus temperature during He–TPD runs after NOx adsorption at 350 °C over the Ba/Al2O3, Pt–Ba/Al2O3 catalysts and over the Pt/Al2O3–Ba/Al2O3 physical mixture. | ||
In the case of the Pt–Ba/Al2O3 catalyst no desorption peak was observed below the adsorption temperature; only above this temperature was the detection of gaseous NOx observed due to the decomposition of the stored nitrates. Oxygen (not shown) was also present in the reactor exit gases. The decomposition process ended below 600 °C, since at such a temperature the gas-phase NOx concentration was negligible.
For the Ba/γ-Al2O3 sample, nitrates decomposition started above 400 °C and the process was not yet completed at 600 °C; the same was found in the case of the Pt/Al2O3–Ba/Al2O3 mixture.
The results presented above indicate that Pt favours the decomposition of nitrates, but only when dispersed over the same support particle where the nitrates are stored. Several authors observed that Pt does affect the decomposition of stored nitrates.5,10,19,22,25,31,34–37 In particular Coronado et al.,36 upon comparing the results obtained after NO2 adsorption on a physical mixture of Pt/SiO2 and Ba/SiO2 and on Pt–Ba/SiO2 catalyst, suggested that the decomposition of nitrates takes place at the interface between the Ba component and the noble metal. These findings are in line with our data where nitrate decomposition is not completed at 600 °C in the case of both Ba/Al2O3 sample (i.e., in the absence of Pt) and of the Pt/Al2O3–Ba/Al2O3 physical mixture, for which the NOx spill-over process from Ba to Pt cannot occur.
TPD experiments were also performed over the Pt–Ba/Al2O3 catalyst after NOx adsorption at different temperatures, namely 300 °C and 400 °C.25 The temperature threshold for nitrate decomposition was always close to the adsorption temperature, thus indicating that the thermal stability of the NOx adsorbed species over all the investigated catalyst samples is determined by the temperature at which the nitrates were stored.
H2–TPSR experiments
The reactivity of nitrates stored at 350 °C with gaseous H2 (H2–TPSR experiment) was also addressed, and results are shown in Fig. 3.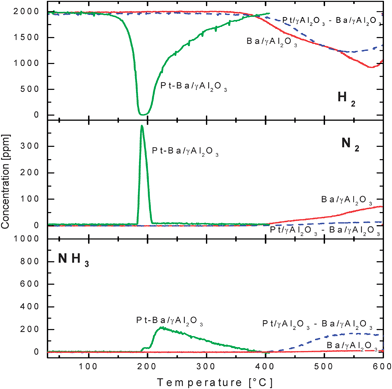 | ||
| Fig. 3 Temporal evolution of H2, N2 and NH3 outlet concentrations during H2–TPSR (H2 = 2000 ppm) over the Ba/γ-Al2O3, Pt–Ba/γ-Al2O3 catalysts and over the Pt/γ-Al2O3–Ba/γ-Al2O3 physical mixture after NOx adsorption at 350 °C. | ||
In the case of the Ba/Al2O3 catalyst and of the physical Pt/Al2O3–Ba/Al2O3 mixture, H2 consumption was apparent only above 350 °C, i.e. above the NOx adsorption temperature; the evolution of N2 and of minor amounts of NO2 and NO (not shown) were seen over the binary Ba/Al2O3 sample, while over the physical mixture significant production of ammonia was observed. The comparison with the TPD data previously shown (Fig. 2) indicates that the presence of hydrogen does not significantly affect the temperature threshold for nitrate decomposition (i.e. their stability), but leads to a different product distribution. In particular the appearance of N2 and of NH3 and the corresponding H2 consumption indicate that the NOx evolved upon nitrate decomposition were reduced to N2 and to NH3 by H2 present in the gas stream.
A different picture was apparent in the case of the Pt–Ba/Al2O3 catalyst: in this case the reduction of the stored NOx was observed at much lower temperatures (180 °C) with production of N2 and NH3. Notably, H2 consumption was apparent at a temperature slightly below that corresponding to the evolution of the reaction products (140 °C vs. 180 °C).
The overall H2 consumption and the corresponding N2 and NH3 formation observed during the TPSR run are in line with the stoichiometry of reactions (2) and (4), respectively.
TPSR experiments were also performed over the same Pt–Ba/Al2O3 catalyst after NOx adsorption at different temperatures (300 and 400 °C):25 H2 consumption was always observed starting near 140 °C, in spite of the fact that different adsorption temperatures were used. This indicates that the temperature onset for nitrate decomposition is not affected by the temperature at which the NOx has been stored.
H2–ITRM experiments
The stability/reactivity in a reducing environment of the nitrates stored over Pt–Ba/Al2O3, Ba/Al2O3 and Pt/Al2O3–Ba/Al2O3 physical mixture was also investigated under isothermal conditions at different temperatures, in the range 100–350 °C (H2–ITRM). In these experiments NOx was adsorbed on the catalysts at 350 °C; then, after a He purge at 350 °C the catalyst temperature was set at the selected value under He flow and eventually H2 was fed to the reactor.In the case of both the Ba/Al2O3 sample and the Pt/Al2O3–Ba/Al2O3 physical mixture, no reactivity was observed at any investigated temperature, in line with the H2–TPSR data as previously shown (Fig. 3). In fact, over these samples decomposition of the stored NOx was observed only above 350 °C.
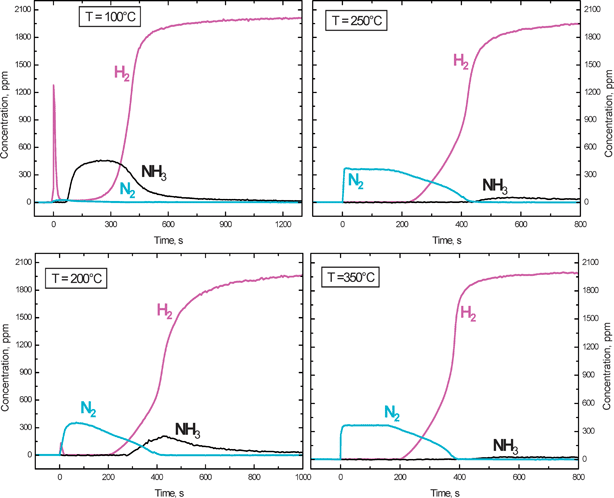
A different picture was apparent in the case of the Pt–Ba/Al2O3 catalyst. Fig. 4 shows the results collected upon H2 addition to the reactor (at t = 0 s) at different temperatures, after NOx adsorption at 350 °C followed by a He purge at the same temperature. At 150 °C (a temperature which was close to the temperature onset of the reaction as indicated by TPSR experiments, see Fig. 3) an induction period in the reaction was observed; indeed hydrogen was not consumed until ∼150 s. Thereafter, H2 was consumed and evolution of NH3 and of minor amounts of N2 was observed.
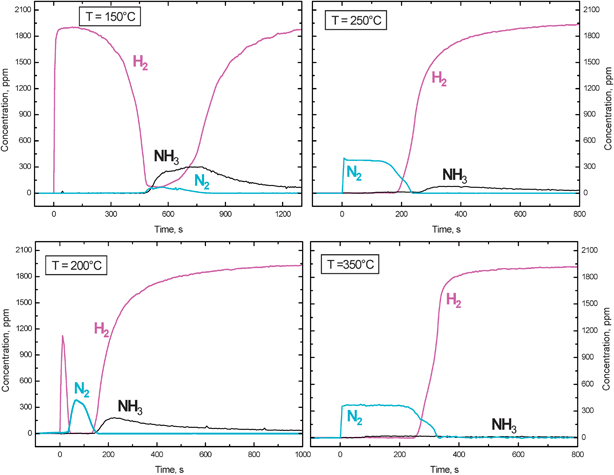 | ||
| Fig. 4 Temporal evolution of H2, N2 and NH3 outlet concentrations during reduction with H2 (2000 ppm) over the Pt–Ba/γ-Al2O3 catalyst at 150, 200, 250, 350 °C (H2–ITRM experiment) after NOx adsorption at 350 °C. | ||
Upon increasing the temperature at which H2 was fed to the reactor (the NOx storage was always carried out at 350 °C) the induction period for H2 consumption decreased so that it was still visible at 200 °C but disappeared at higher temperatures. A switch in the product selectivity (from NH3 to N2) was also observed upon increasing the temperature, so that at 350 °C the reaction was almost completely selective to N2.
Notably, at 250 °C and above, upon H2 admission the N2 outlet concentration immediately increased to the level of 370 ppm: this concentration level corresponds well to the stoichiometry of reaction (2), i.e. to the reduction of nitrates by H2, being reaction limited by the H2 concentration.
Fig. 5 shows the efficiency in the removal of the stored NOx (i.e., the fraction of NOx stored at 350 °C which was reduced in the reaction with H2) and the N2 selectivity of the reduction process, calculated from the experiments shown in Fig. 4 (full square symbols) and in other runs that will be discussed later on. The N2 selectivity (i.e. the time weighted average selectivity of the reduction during the entire reduction phase) was estimated according to the following equation:
 | (6) |
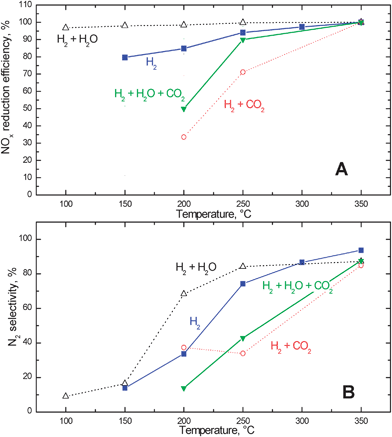 | ||
| Fig. 5 NOx reduction efficiency (A) and N2 selectivity (B) upon reduction at different temperatures with hydrogen in the presence and in the absence of 1% v/v water and/or 3000 ppm of CO2 after NOx adsorption at 350 °C over the Pt–Ba/γ-Al2O3 catalyst. | ||
N2O concentration was always found to be negligible in the experiments and therefore this species was not included in eqn (6).
Fig. 6 (full square symbols) shows that complete NOx removal efficiency was achieved at temperatures higher than 250 °C and that the selectivity to N2 increases significantly with temperature. This point will be addressed in the following.
 | ||
| Fig. 6 He–TPD run (A) and H2–TPSR run (B) with 2000 ppm hydrogen in the presence of 1% v/v water over the Pt–Ba/γ-Al2O3 catalyst after NOx adsorption at 350 °C. | ||
D. A Pt–catalyzed route for nitrate decomposition–reduction
As previously reported, it is generally believed that the regeneration of NSR catalysts includes, at first, the release of NOx from the catalyst surface upon decomposition of nitrite–nitrates ad-species, followed by the reduction of the released NOx.5 It was suggested that the initial NOx release may be driven by several factors4 including the establishment of a net reducing environment which shifts towards desorption the adsorption–desorption equilibrium of stored nitrates,6,23,31,34,37 or by heat generated upon the reducing switch (thermal release).38 This last mechanism can be ruled out in our experiments performed under nearly-isothermal conditions, and on the basis of the results of TPD experiments (see Fig. 2).Poulston and Rajaram34 investigated the stability of the nitrates stored on Pt–Ba/Al2O3 catalysts by heating the catalyst samples under a different atmosphere (He, CO, H2). They observed that under a reducing environment (i.e. in the presence of H2 or CO) the decomposition temperature of the nitrate was significantly reduced with respect to pure He; besides, H2 was more effective at regenerating the NOx storage activity.
Along similar lines, Liu and Anderson,31 by a combined use of in-situ DRIFT spectroscopy and TPD experiments, discussed potential mechanisms involved in the initial release of stored NOx. These include: (i) destabilization of nitrates due to the decrease of oxygen concentration in the gas-phase; (ii) release of NOx into the gas phase during conversion of barium nitrate into barium hydroxide or carbonate; (iii) spill-over of nitrates onto available Pt sites. However, these mechanisms have been considered unlikely by the authors, who instead suggested that the reductant molecule (or an activated reductant molecule spilled over from Pt) directly reduce stored nitrates to nitrites, which, being substantially less stable than nitrates, decompose to NO and gaseous oxygen.
Our data are consistent with this latter mechanism, since over the investigated model Pt–Ba/Al2O3 catalyst the reduction of NOx ad-species was initiated at temperatures well below that of thermal decomposition. Notably, the temperature onset for the reaction was not affected by the adsorption temperature, i.e. by the thermal stability of the stored NOx. These findings provide clear evidence of the fact that under isothermal conditions the reduction of the stored NOx is not initiated through the thermal decomposition of nitrates. Since, in the absence of Pt no activity was observed in the reduction of the stored nitrates below the NOx adsorption temperature, it is concluded that the first step in the nitrate reduction by H2 involves a Pt–catalyzed process, already active at low temperatures. This process is likely to be favoured when NOx is adsorbed in close proximity to Pt and requires that Pt and the NOx adsorption sites (Ba) are dispersed over the same support: in fact for the Pt/Al2O3–Ba/Al2O3 physical mixture the stored nitrates were not reduced below the adsorption temperature, i.e. the temperature of thermal decomposition of stored NOx.
Scheme 1 shows possible pathways for the reduction mechanism consistent with the data previously shown. The Pt–catalyzed route may involve the activation of H2 on Pt sites, followed by spill-over on the alumina support towards nitrate ad-species (Scheme 1 (A)). The reduction of stored nitrates is hence operating because H ad-species promote the decomposition of nitrates to gaseous NO and/or NO2, possibly via the formation of nitrites as intermediates.31 NO/NO2 are then reduced on Pt as discussed later on. Alternatively, the reduction of the Pt sites by H2 creates a driving force for O atoms migration from the Ba sites to Pt, resulting in the reduction of stored nitrates and leading to their destabilization/decomposition. The role of the reductant in this mechanism is, hence, to keep Pt in a reduced state.
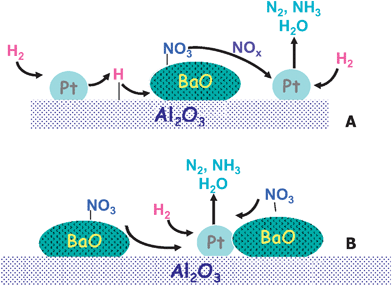
Also, a mechanism involving the surface diffusion of NOx ad-species towards reduced Pt sites cannot be excluded (Scheme 1 (B)):5 in this case, NOx spills over the surface and is decomposed at reduced Pt sites. This mechanism is in line with the observed effect of Pt on the thermal decomposition of stored nitrates; it also implies a high mobility of NOx species adsorbed onto Ba, which is likely to be poor at low temperature. In this respect, it is expected that NOx ad-species present on Ba sites neighbouring Pt are readily involved in the reduction step. Hence, the proximity between Pt and Ba should play an important role in the reduction process, similar to what is already proposed for the storage process.22 It is worth noticing that the reduction of the stored nitrates requires either, a proximity between the adsorption sites and the Pt sites, or implies spill-over phenomena involving the reductant and/or the nitrates. As a matter of fact, in the absence of Pt or when spill-over effects cannot occur (like in the case of the Pt/Al2O3–Ba/Al2O3 physical mixture) the suggested Pt-catalyzed route does not operate.
E. Effect of water and CO2 on the Pt-catalyzed route for nitrate reduction.
Since water and CO2 are always present in flue gases, their effect on the stability/reactivity of nitrate ad-species was also addressed.Fig. 6 shows the results obtained during TPD and H2–TPSR runs over the Pt–Ba/Al2O3 catalyst carried out in the presence of 1% v/v water after storage of NOx at 350 °C.
Results collected during the TPD run (Fig. 6 (A)) showed that nitrate decomposition occurs above 350 °C leading to the evolution of NO and O2 as the major decomposition products, along with minor amounts of NO2. As was the case in the TPD experiment carried out in a dry environment (see Fig. 2), no desorption peak was observed below 350 °C, i.e. the NOx adsorption temperature. The overall picture is indeed very similar to that obtained in the case of TPD performed in the absence of water, indicating that the thermal decomposition of nitrates is not affected by the presence of water.
The stability/reactivity of nitrates in He/H2/H2O was also investigated by running a H2–TPSR experiment in the presence of water (Fig. 6 (B)). H2 was consumed starting from roughly 50 °C; a minimum in the H2 concentration was seen near 100 °C. H2 consumption is accompanied by the formation of NH3 and small amounts of N2. The stored nitrates were completely reduced at temperatures below 200 °C.
A comparison with the results of the same experiment carried out in dry conditions (Fig. 3) clearly indicates that water, contrary to the TPD experiment, has a dramatic effect on the stability of the stored nitrates in the presence of hydrogen, since nitrate decomposition (followed by reduction) occurred at temperatures well below those observed in the absence of water. Besides, the product selectivity was also changed because in the presence of water the reduction of the stored nitrates was almost completely selective towards the formation of ammonia.
The reactivity of nitrates with hydrogen in the presence of water was also investigated under isothermal conditions at different temperatures, in the range 100–350 °C (H2–ITRM). Fig. 7 shows the results collected at 100, 200, 250 and 350 °C. The reaction was already very fast at 100 °C, and indeed was limited by the H2 concentration. At variance with the experiments carried out in a dry environment (Fig. 4), H2 was immediately consumed and the formation of NH3 and of N2 was observed. Besides, almost complete removal of the stored nitrates could be achieved at low temperatures (see Fig. 5 (A), empty triangles).
 | ||
| Fig. 7 Temporal evolution of H2, N2 and NH3 and NO outlet concentrations during reduction with H2 (2000 ppm) in the presence of 1% v/v water over the Pt–Ba/γ-Al2O3 catalyst at 100, 200, 250, 350 °C (H2–ITRM experiment) after NOx adsorption at 350 °C. | ||
Accordingly, the data obtained in the presence of water shows, in line with the results of TPSR experiments, that water favours the decomposition/reactivity of the stored nitrates. It is worth noticing that also in the presence of water the amount of nitrogen formed upon reduction increased with temperature at the expense of ammonia, so that almost complete N2 selectivity was attained at high temperatures (see Fig. 5 (B), empty triangles).
The reasons why the decomposition/reduction of the stored nitrates is greatly enhanced in the presence of water have not been completely clarified so far. It can be speculated that water favours the H2 spill-over, which was considered an important step in the reduction of the stored nitrates.39,42 As a matter of fact, several literature reports indicate that water increases the rate of H2 spill-over over oxide surfaces.43 A significant enhancement is expected in the reduction of nitrates stored some distance from Pt sites because the reaction implies the mobility of H ad-species on the surface. Specific effects of water on the spill-over of adsorbed nitrates is a factor which may play a role in the reaction.
In addition, water has an impact on the nature of the adsorbed NOx species as well. In a FTIR investigation carried out over the same catalyst samples used in this study, it was found that the presence of water affects the nature of the stored nitrates adsorbed over Ba, namely bidentate nitrate ad-species are transformed into ionic species upon addition of water vapour.40 Changes in the nature of adsorbed nitrate species induced by water were also reported by Kim et al.41 over Ba/Al2O3 samples with different Ba loadings. After NO2 adsorption, formation of nano-sized Ba(NO3)2 particles was observed by XRD; however, when water was applied to the NO2 pre-adsorbed samples at room temperature, a crystalline Ba(NO3)2 phase was formed. Still it is questionable whether a crystalline phase could be more active than the nano-sized particles.
In spite of the fact that structural changes are induced by water on the adsorbed nitrate species, their thermal stability is apparently not affected by water, as shown by TPD experiments (see Fig. 6). However, their reactivity may be modified and this may play a role in the reduction of the stored nitrates in the presence of water.
The effect of CO2 on the reduction of the stored nitrates was also investigated, and results are presented in Fig. 8 (TPD and H2–TPSR) and 9 (H2–ITRM).
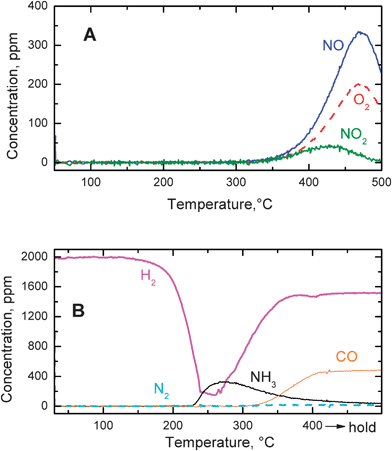 | ||
| Fig. 8 He–TPD run (A) and H2–TPSR run (B) with 2000 ppm hydrogen in the presence of 3000 ppm CO2 over the Pt–Ba/γ-Al2O3 catalyst after NOx adsorption at 350 °C. | ||
Fig. 8 (A) and (B) show the results obtained during TPD and H2–TPSR runs carried out in the presence of 3000 ppm of CO2 after storage of NOx at 350 °C, respectively. In the TPD run the decomposition of nitrates started above 350 °C and resulted in the evolution of NO and O2 along with minor amounts of NO2. Desorption peaks were observed only above the NOx adsorption temperature (350 °C), thus indicating that the thermal decomposition of nitrates is not affected by the presence of CO2, as in the case of the TPD experiments carried out in dry (see Fig. 2) and wet (see Fig. 6) environments.
H2–TPSR experiments in the presence of CO2 were carried out to investigate the stability/reactivity of nitrates in He/H2/CO2, and the results are shown in Fig. 8 (B). H2 was consumed starting from roughly 150 °C, with production of NH3 and of negligible amounts of N2. Formation of CO was observed above 320 °C, due to the occurrence of the reverse water gas shift reaction (WGSR):
| CO2 + H2 → CO + H2O | (7) |
Comparing these results with those collected in the absence of CO2 (Fig. 3) it appears that CO2 has a significant inhibiting effect on the reduction of the stored nitrates by H2. Indeed the minimum in H2 consumption which was observed near 180 °C in the absence of CO2, was shifted towards 240–260 °C when CO2 was present in the feed.
It may be argued that the inhibiting effect of CO2 on the reduction of the stored nitrates is related to the poisoning of the Pt sites by carbonyls, formed upon CO2 reduction by H2 (i.e. according to reverse WGSR). This reaction can occur to a limited extent at low temperatures, as shown by dedicated FTIR experiments40 carried out over the same catalyst sample showing that Pt–carbonyls are formed at temperatures as low as 150 °C in the presence of a CO2–H2 mixture. At these temperatures, CO was not detected in significant amounts in the gas-phase.
The reactivity with hydrogen of nitrates stored at 350 °C over the Pt–Ba/γ-Al2O3 catalyst in the presence of CO2 was also investigated under isothermal conditions in the range 200–350 °C, and results are shown in Fig. 9 where the concentration profiles of hydrogen, nitrogen and ammonia measured at the reactor outlet are shown as a function of time.
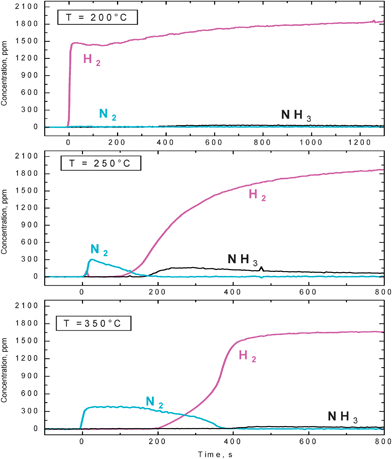 | ||
| Fig. 9 Temporal evolution of H2, N2 and NH3 outlet concentrations during reduction with H2 (2000 ppm) in the presence of 3000 ppm CO2 over the Pt–Ba/γ-Al2O3 catalyst at 200, 250, 350 °C (H2–ITRM experiment) after NOx adsorption at 350 °C. | ||
In line with TPSR data, at 200 °C no reaction was observed. H2 was immediately detected at the reactor outlet upon admission, and no ammonia or nitrogen formation was observed.
By increasing the reaction temperature up to 250 °C and further to 350 °C, H2 was completely consumed and N2 formation was observed, followed by NH3 evolution.
As shown in Fig. 5 (empty circles), at 200 and 250 °C both the NOx removal efficiency and the nitrogen selectivity were lower than in the absence of CO2, whereas at 350 °C no significant differences were apparent.
Finally, the combined effect of the presence of water and of carbon dioxide on the reduction of the stored nitrates was investigated by performing TPSR and ITRM runs in the presence of water (1% v/v) and CO2 (3000 ppm).29 CO2 inhibition on the reduction by H2 of the stored nitrates was apparent also in the presence of water, although in a wet environment the effect of CO2 inhibition was somehow reduced. Accordingly, a slightly higher NOx removal efficiency was observed at low temperatures in the presence of H2O and CO2 when compared to the presence of CO2 only (full triangles and empty circles in Fig. 5 (A), respectively). The presence of water likely drives the reverse WGSR reaction (7)from right to left hence limiting the extent of Pt–carbonyls formation. As in the absence of water, the effect CO2 tended to vanish at high temperature.
F. Selectivity and role of ammonia in the reduction of stored nitrates
It has been previously shown that decomposition of the stored nitrates in a reducing environment leads to formation of N2 and other products, particularly ammonia. As a matter of fact, formation of N2 and NH3 was observed upon nitrate reduction during the H2–TPSR and H2–ITRM experiments previously discussed. The products distribution strongly depended on the type of experiment (TPSR vs. ITRM) and on the operating conditions, i.e. temperature, presence/absence of water and CO2, etc. No significant N2O formation was observed during our experiments, although several authors reported its formation upon regeneration of LNT catalysts.45,46According to literature indications, nitrates are at first decomposed to gaseous NOx, which are then reduced upon reaction at the noble metal. Since the reduction of evolved NOx occurs in a rich environment, the three-way catalyst (TWC) mechanism formalisms were tentatively applied in the case of LNT catalysts as well, to explain the formation of the reaction products.5 Accordingly, in many studies the reactivity of gaseous NO with various reductants, including H2, was investigated.45,47,48
Based on in situ FTIR spectroscopy coupled with mass spectrometry and time-resolved XRD, Szailer et al.44 investigated the reduction of stored NOx with CO and/or H2. In the case of H2 only, a mechanism in which hydrogen initially cleaned the platinum sites from adsorbed oxygen left from the previous lean phase was proposed. Water produced from the reaction between the H and O adsorbed species destabilized the adsorbed nitrates, and desorbed NOx decomposes into –N and –O ad-species at the empty Pt site, Pt*:
| (x + 1)Pt* + NOx → xPt⋯Oad + Pt⋯Nad | (8) |
The dissociation of NOx species on the Pt particles was followed by the recombination of N–ad-species to form N2 or by reaction of Nad atoms with H2 to form NH3:
| 2Pt⋯Nad → 2Pt* + N2 | (9) |
| Pt⋯Nad + 3/2 H2 → NH3 + Pt* | (10) |
Ammonia could also react with NOx to form N2 and H2O:
| NH3 + NOx + H2O → [NH4NOx] → N2 + H2O | (11) |
Accordingly, (see also49–51), in this process the primary role of H2 is to keep the surface of Pt clean for the dissociation of NOx, by reacting with Oad to form H2O.
Therefore, the selectivity to N2/NH3/N2O formation depends on the surface concentration of adsorbed Pt⋯Nad, Pt⋯Hadetc. species, as also suggested.52 This was also proved by steady-state flow reactor experiments with NO + H2 mixtures showing that the selectivity of the reaction switched from mainly N2 when the feed was stoichiometric for N2 production (1:1), to almost complete selectivity to NH3 at high H2/NO ratios.45,47 N2O formation was also measured, generally at low H2/NO feed ratio and low temperature.47
The mechanisms of N2, NH3 and N2O formation on noble metals upon NO reduction by H2 over supported Pd catalysts were recently discussed by Dhainaut et al.47, and mechanisms similar to those reported by Szailer et al.44 were considered to account for N2, NH3 and N2O formation. However a different pathway was invoked, i.e. the H–assisted NO dissociation followed by reaction of NH fragments with adsorbed NO leading to N2 and NH3:
| Pt⋯NOad + Pt⋯H2ad → Pt⋯NHad + Pt⋯OHad | (12) |
| Pt⋯NOad + Pt⋯NHad → N2 + Pt⋯OHad | (13) |
| Pt⋯NHad + Pt⋯H2ad → NH3 + 2 Pt | (14) |
Since ammonia was generally observed among reaction products, the possibility of NH3 SCR-like mechanisms involving NO and adsorbed NHx fragments was also suggested. Mechanistic aspects of this reaction were recently investigated by Kondratenko and Baerns53 over Pt supported catalysts with NH3 + NO mixtures using the temporal analysis of product (TAP) reactor. Two routes for N2 formation were identified, i.e. (i) reaction between NO and NHx adsorbed species, and (ii) recombination of –N ad-atoms deriving from NO dissociation or NH3 complete dehydrogenation. N2O formation was ascribed to the recombination of two NO molecules or to the interaction of adsorbed NO with NHx species, with the participation of additional oxygen containing species. The selectivity to the various reaction products was found to depend strongly on the coverage of the catalyst by the various adsorbed species.
It is worth noticing that the above mentioned mechanisms can hardly explain both the very high N2 selectivity and the temporal sequence of products (with ammonia detection following that of nitrogen, see Fig. 1 (B)), which characterizes the regeneration by hydrogen of LNTs systems under isothermal conditions.
In a recent paper from Cumaranatunge et al.,45 a simplified scheme for the reduction of the stored NOx by H2 was proposed. In particular, the authors provided evidence for the development of a reaction front which travels along the catalyst bed during the regeneration stage: ammonia and N2 can be simultaneously formed in the H2–rich zone of the front, according to previously suggested mechanisms for three-way catalysts. However, NH3 may further react with the stored nitrates, leading to the formation of N2. This would explain the temporal sequence of products formation with NH3 breakthrough observed after N2 production when the stored NOx starts to deplete, and is insufficient to react with the NH3 formed upstream. A similar scheme was also proposed by Pihl et al.46
In order to get additional information on the mechanisms operating in the reduction of the stored nitrates, we investigated the reactivity of ammonia as well. The study was also motivated by the observation that during both H2–TPSR and H2–ITRM experiments, under specific conditions, ammonia was produced with very high selectivity. Notably, in the case of ITRM experiments, ammonia formation decreased with temperature whereas that of N2 showed an opposite trend (see Fig. 5 (B)): this may suggest that ammonia is an intermediate species in the formation of N2. Accordingly, the reactivity of ammonia with nitrates was analyzed under temperature programmed and isothermal conditions (NH3–TPSR and NH3–ITRM respectively).
Fig. 10 shows the results of NH3–TPSR experiments performed over the Pt–Ba/γ-Al2O3 catalyst in the presence (Fig. 10 (A)) and in the absence of water (Fig. 10 (B)), after having stored NOx species at 350 °C.
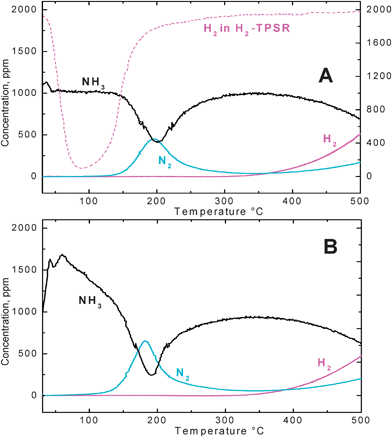 | ||
| Fig. 10 NH3–TPSR run with 1000 ppm ammonia in the presence (A) and in the absence (B) of 1% v/v H2O over the Pt–Ba/γ-Al2O3 catalyst after NOx adsorption at 350 °C. | ||
In the absence of water, desorption of weakly adsorbed ammonia species was observed as the catalyst was heated up. This peak was not observed in the presence of water, ammonia being displaced by water from the catalyst surface.
Then, both in the absence and in the presence of water, ammonia was consumed starting from roughly 130 °C and simultaneous formation of nitrogen only was observed, according to the following overall stoichiometry:
| 3Ba(NO3)2 + 10NH3 → 8N2 + 3BaO + 15H2O | (15) |
At temperatures higher than 350 °C the NH3 concentration decreased again and N2 and H2 evolution was observed, due to the occurrence of the ammonia decomposition reaction:
| 2NH3 → N2 + 3H2 | (16) |
At variance to what was observed when hydrogen was used as reducing agent, water inhibited (even if very slightly) the reactivity of ammonia in the reduction of the stored nitrates. This was also confirmed by NH3–ITRM experiments.29
For comparison purposes, Fig. 10 (A) also shows the hydrogen concentration profile measured during H2–TPSR under wet conditions (see Fig. 6 (B)): it clearly appears that, in the presence of water the reactivity of NH3 was markedly lower than that of H2, which was indeed capable of reducing the stored nitrates at 60 °C, with formation of NH3 only.
The bulk of data herein reported clearly indicates that in the presence of water the stored nitrates were easily reduced by H2 at very low temperatures, starting from 60 °C, leading to the formation of ammonia. Also, ammonia was active in the reduction of the stored nitrates, but at higher temperatures when compared to H2 (above 130–140 °C vs. 60 °C). Notably, complete selectivity to nitrogen was observed in this case. Accordingly, these data are consistent with the hypothesis that the formation of N2 during the regeneration of LNTs by H2 occurs via an in series 2-step pathway involving, at first, the fast formation of ammonia upon reaction of nitrates with H2reaction (4), followed by the slower reaction of ammonia with the stored nitrates leading to the selective formation of N2reaction (15). The sum of reactions (4) and (15) leads to the overall stoichiometry for the reduction of nitrates to N2, reaction (2).
The reduction of the stored nitrates with hydrogen to give ammonia reaction (4) is very fast; indeed it was observed at very low temperatures, slightly above room temperature. On the other hand, the subsequent reaction of ammonia with the stored nitrates to give nitrogen is much slower, and hence is rate determining in the formation of nitrogen. As a matter of fact, when considering the stoichiometry of reaction (2) (the reduction of nitrates to N2 with H2) and (15) (the reduction of nitrates to N2 with NH3), similar amounts of N2 are produced during both H2–ITRM and NH3–ITRM experiments, carried out at the same temperature, in line with the hypothesis that ammonia is an intermediate in the formation of N2.
The mechanism of ammonia formation upon reduction of the stored nitrates with H2 (step 1) is not completely clear. As shown above, it may be suggested that H2, activated over the Pt sites, provokes nitrates decomposition to gaseous NOx which are then reduced by H2 to NH3 over the Pt sites. The studies on the reduction of NO by H2 over Pt cited above45,47,48 showed that the selectivity of the process depends on the relative surface concentration of the –N, –H and –O adsorbed species. Accordingly, the very high selectivity to ammonia observed at low temperatures suggests that high H:N and H:O ratios are locally attained on the catalyst surface where the reaction takes place, thus driving the selectivity to ammonia and water. Notably, as shown in a previous paper the reduction of NO by H2 can be accomplished at temperatures as low as 60 °C over the same Pt–Ba/Al2O3 catalyst sample used in this study.28
Once ammonia has been formed, it may further react with adsorbed nitrates, and this reaction is very selective towards nitrogen. This was clearly demonstrated by both the NH3–TPSR and NH3–ITRM data. It is worth noticing that the reaction of ammonia with NOx obeys the stoichiometry of reaction (15), which does not involve gas-phase NOx. As a matter of fact, we analyzed by dedicated experiments the NO + NH3 reaction over the same catalyst sample used in this study:30 it was found that the NO + NH3 reaction leads to significant amounts of N2O, as opposed to the nitrate + NH3 reaction which was highly selective to N2 (see Fig. 10). Besides, it was found that Pt was involved in the reaction of ammonia with the stored nitrates since reduction of nitrates (and of nitrites) stored on Ba could not be accomplished by NH3 over a Pt-free sample.30 As previously discussed, in the case of H2, Pt could activate NH3 and/or could favour nitrate decomposition. The mechanistic features of this reaction are still not completely clear; however an intermediate species must be invoked that decomposes fast and selectively to N2.
G. The H2 front model for the regeneration of LNTs
The data discussed above provide clear evidence in favour of the existence of an in series two-step process for the reduction of nitrates with H2 to give N2. Step 1 is represented by the fast reaction of H2 with the stored nitrates to give ammonia; this step is followed by the subsequent slower reduction of nitrates to give nitrogen (step 2). It is worth noticing that under the experimental conditions adopted in the present study (i.e. nearly isothermal conditions with limited temperature changes upon the lean–rich switches) the reaction of ammonia with nitrates does represent the major route for nitrogen formation.A simplified sketch of the processes ongoing during the reduction of the trap is shown in Scheme 2. The occurrence of a fast reaction of the adsorbed nitrates with H2 to give ammonia and the integral “plug-flow” behaviour of the reactor imply the complete consumption of the reductant H2 and the formation of an H2 front travelling along the reactor axis. At a given instant of the regeneration phase, different zones are present in the reactor: (i) an initial zone, upstream of the H2 front (zone I), where the trap has already been regenerated with reduction of nitrates by H2 and the Ba storage sites have been restored; (ii) the zone corresponding to the development of the H2 front (zone II), where the concentration of hydrogen decreases from the inlet value to almost zero. Here, the formation of ammonia due to reduction of nitrates by H2 (fast reaction) is taking place; (iii) a zone immediately downstream of the H2 front (zone III) where nitrates, still available because they have not yet come in contact with H2, react with ammonia formed upstream (i.e. in zone II) if the temperature is high enough; (iv) the last zone (zone IV) where the nitrates initially stored have not yet been reduced. Notably, the spatiotemporal H2 concentration profile inside the reactor is complex, a result of H2 consumption that leads to the formation of ammonia and from the depletion of adsorbed nitrates downstream of the H2 front, due to their reaction with ammonia (at high temperatures).
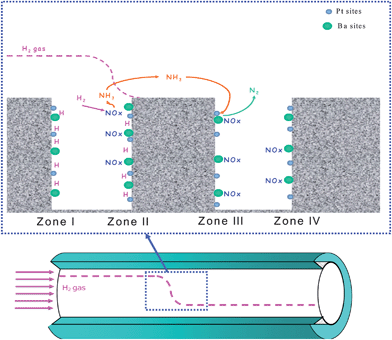 | ||
| Scheme 2 Sketch of the reduction mechanism for Pt–Ba/Al2O3 catalyst upon regeneration with H2. | ||
When the regeneration of the trap is carried out at low temperatures, e.g. 150 °C or below, H2 reacts with surface nitrates to give NH3, according to reaction (4), and an H2 front develops (zone II). Besides, due to the low temperature, ammonia hardly reacts with the stored nitrates to form N2 which, according to reaction (15), a temperature threshold of 130 °C has been measured. As a result, ammonia is by far the major reaction product and the nitrogen selectivity is very low. Upon increasing the reduction temperature, the reactivity of NH3 with nitrates reaction (15) becomes appreciable and this drives the selectivity to N2. Since H2 is by far more reactive than NH3 towards surface nitrates, NH3 reacts preferentially with nitrates located downstream of the H2 front (zone III). Accordingly, N2 is observed at the reactor outlet (Fig. 4) and the release of unreacted NH3 decreases.
Finally, at the highest investigated temperatures (e.g. 350 °C, Fig. 4), NH3 readily reacts with the adsorbed NOxreaction (15) even in the presence of H2. This has been pointed out by dedicated ITRM experiments with H2 + NH3 mixtures40 showing that at high temperatures NH3 effectively competes with H2 for the reduction of the stored nitrates. Accordingly, at high temperatures NH3 evolution at the reactor outlet becomes of minor relevance, being readily consumed by the reaction with stored nitrates. The in series 2-step pathway suggested above for nitrogen formation, together with the integral behaviour of the trap, can nicely explain the observed increase in the N2 selectivity with temperature (Fig. 5 (B)) in the reduction of the stored NOx by H2.
Conclusions
Mechanistic aspects of the reduction with H2 of NOx stored on Lean NOx Trap catalysts are critically reviewed. It was shown that, under nearly isothermal conditions nitrogen formation occurs via an in series two-step process involving the participation of ammonia as an intermediate. The first step of this process is ammonia formation through the reaction of H2 with stored nitrates; ammonia then reacts with the nitrates left on the catalysts’ surface leading to the formation of nitrogen. Over the investigated Ba-containing catalysts, the first step (i.e. NH3 formation) is much faster than the second one which, therefore, is rate determining in the formation of nitrogen. Both steps are catalyzed by Pt and, under nearly isothermal conditions, do not involve the occurrence of a thermal decomposition step of the stored nitrates.Due to the fast reaction of the adsorbed nitrates with H2 to give ammonia and to the integral behaviour of the trap, an H2 front develops in the trap which travels along the reactor axis. Ammonia formed upon reaction of nitrates with H2 reacts downstream of the H2 front with nitrates leading to N2 formation, if the temperature is high enough. This explains both the observed change in the selectivity of the process with time upon regeneration of the trap (with selectivity changing from N2 to NH3), and the increase in the N2 selectivity with temperature as well.
The identification of the pathway for the reduction of stored NOx, where ammonia is suggested as the intermediate product in the formation of nitrogen, may favour the improvement of the combined NSR + SCR technology that has been proposed by several car manufacturers to make NOx removal by NSR more effective and to simultaneously limit the ammonia slip.2
References
- S. Matsumoto, Catal. Technol., 2000, 4, 102 Search PubMed.
- T. Johnson, Platinum Met. Rev., 2008, 52, 23 CrossRef CAS.
- H. Shinjoh, N. Takahashi, K. Yokota and M. Sugiura, Appl. Catal., B, 1998, 15, 189 CrossRef CAS.
- S. Hodjati, C. Petit, V. Pitchon and A. Kiennemann, Appl. Catal., B, 2000, 27, 117 CrossRef CAS.
- W. S. Epling, L. E. Campbell, A. Yezerets, N. W. Currier and J. E. Park II, Catal. Rev., 2004, 46, 163 CrossRef.
- E. Fridell, M. Skoglundh, B. Westerberg, S. Johansson and G. Smedler, J. Catal., 1999, 183, 196 CrossRef CAS.
- E. Fridell, H. Persson, B. Westerberg, L. Olsson and M. Skoglundh, Catal. Lett., 2000, 66, 71 CrossRef CAS.
- P. Broqvist, I. Panas, E. Fridell and H. Persson, J. Phys. Chem. B, 2002, 106, 7 CrossRef CAS.
- B. Westerberg and E. Fridell, J. Mol. Catal. A: Chem., 2001, 165, 249 CrossRef CAS.
- L. Olsson, H. Persson, E. Fridell, M. Skoglundh and B. Andresson, J. Phys. Chem. B, 2001, 105, 6895 CrossRef CAS.
- J. A. Anderson, B. Bachiller-Baeza and M. Fernández-García, Phys. Chem. Chem. Phys., 2003, 5, 4418 RSC.
- C. Hess and J. H. Lunsford, J. Phys. Chem. B, 2002, 106, 6358 CrossRef CAS.
- C. Hess and J. H. Lunsford, J. Phys. Chem. B, 2003, 107, 1982 CrossRef CAS.
- H. Y. Huang, R. Q. Long and R. T. Yang, Energy Fuels, 2001, 15, 205 CrossRef CAS.
- H. Mahzoul, J. F. Brilhac and P. Gilot, Appl. Catal., B, 1999, 20, 47 CrossRef CAS.
- P. J. Schmitz and R. J. Baird, J. Phys. Chem. B, 2002, 106, 4172 CrossRef CAS.
- N. V. Cant and M. J. Patterson, Catal. Today, 2002, 73, 271 CrossRef CAS.
- L. Lietti, P. Forzatti, I. Nova and E. Tronconi, J. Catal., 2001, 204, 175 CrossRef CAS.
- F. Prinetto, G. Ghiotti, I. Nova, L. Lietti, E. Tronconi and P. Forzatti, J. Phys. Chem. B, 2001, 105, 12732 CrossRef CAS.
- I. Nova, L. Castoldi, F. Prinetto, V. Dal Santo, L. Lietti, E. Tronconi, P. Forzatti, G. Ghiotti, R. Psaro and S. Recchia, Top. Catal., 2004, 30/31, 181 CrossRef.
- L. Castoldi, I. Nova, L. Lietti, E. Tronconi and P. Forzatti, Catal. Today, 2004, 96, 43 CrossRef CAS.
- I. Nova, L. Castoldi, F. Prinetto, G. Ghiotti, L. Lietti, E. Tronconi and P. Forzatti, J. Catal., 2004, 222, 377 CrossRef CAS.
- I. Nova, L. Castoldi, L. Lietti, E. Tronconi and P. Forzatti, SAE [Tech. Pap.], 2005 Search PubMed 2005-01-1085.
- A. Scotti, I. Nova, E. Tronconi, L. Castoldi, L. Lietti and P. Forzatti, Ind. Eng. Chem. Res., 2004, 43, 4522 CrossRef CAS.
- I. Nova, L. Castoldi, L. Lietti, E. Tronconi and P. Forzatti, SAE [Tech. Pap.], 2006 Search PubMed 2006-01-1368.
- I. Nova, L. Lietti, L. Castoldi, E. Tronconi and P. Forzatti, J. Catal., 2006, 239, 244 CrossRef CAS.
- I. Nova, L. Castoldi, L. Lietti, E. Tronconi and P. Forzatti, Top. Catal., 2007, 42–43, 21 CrossRef.
- L. Castoldi, I. Nova, L. Lietti, E. Tronconi and P. Forzatti, Top. Catal., 2007, 42–43, 189 CrossRef.
- L. Lietti, I. Nova and P. Forzatti, Role of ammonia in the reduction by hydrogen of NOx stored over Pt-Ba/Al2O3 Lean NOx Trap catalysts, J. Catal., 2008 Search PubMed doi: 10.1016/j.jcat.2008.05.005.
- I. Nova, L. Lietti and P. Forzatti, Catal. Today, 2008, 136, 128 CrossRef CAS.
- Z. Liu and J. A. Anderson, J. Catal., 2004, 224, 18 CrossRef CAS.
- T. Lesage, C. Terrier, P. Bazin, J. Saussey and M. Daturi, Phys. Chem. Chem. Phys., 2003, 5, 4435 RSC.
- H. Abdulhamid, E. Fridell and M. Skoglundh, Top. Catal., 2004, 30/31, 161 CrossRef.
- S. Poulston and R. Rajaram, Catal. Today, 2003, 81, 603 CrossRef CAS.
- M. Sharma, M. P. Harold and V. Balakotaiah, Ind. Eng. Chem. Res., 2005, 44, 6264 CrossRef CAS.
- J. M. Coronado and J. A. Anderson, J. Mol. Catal. A: Chem., 1999, 138, 83 CrossRef CAS.
- N. W. Cant and M. J. Patterson, Catal. Lett., 2003, 85, 153 CrossRef CAS.
- K. S. Kabin, R. L. Muncrief and M. P. Harold, Catal. Today, 2004, 96, 79 CrossRef CAS.
- J. R. Theis, H. W. Jen, R. W. McCabe, M. Sharma, V. Balakotaiah and M. P. Harold, SAE [Tech. Pap.], 2006 Search PubMed 2006-01-1067.
- Unpublished results from our labs.
- D. H. Kim, J. H. Kwak, J. Szanyi, S. D. Burton and C. H. F. Peden, Appl. Catal., B, 2007, 72, 233 CrossRef CAS.
- M. Machida, D. Kurogi and T. Kijima, J. Phys. Chem. B, 2003, 107, 196 CrossRef CAS.
- M. Stoica, M. Caldararu, N. I. Ionescu and A. Auroux, Appl. Surf. Sci., 2000, 153, 218 CrossRef CAS.
- T. Szailer, J. H. Kwak, D. H. Kim, J. C. Hanson, C. H. F. Peden and J. Szanyi, J. Catal., 2006, 239, 51 CrossRef CAS.
- L. Cumaranatunge, S. S. Mulla, A. Yezerets, N. W. Currier, W. N. Delgass and F. H. Ribeiro, J. Catal., 2007, 246, 29 CrossRef CAS.
- J. A. Pihl, J. E. Parks II, C. S. Daw and T. W. Root, SAE [Tech. Pap.], 2006 Search PubMed 2006-01-3441.
- F. Dhainaut, S. Pietrzyk and P. Granger, Appl. Catal., B, 2007, 70, 100 CrossRef CAS.
- J. Xu, R. Clayton, V. Balakotaiah and M. P. Harold, Appl. Catal., B, 2008, 77, 395 CrossRef CAS.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283 CrossRef CAS.
- J. P. Breen, R. Burch and N. Lingaiah, Catal. Lett., 2002, 79, 171 CrossRef CAS.
- R. Burch, Catal. Rev. Sci. Eng., 2004, 46, 271 CrossRef CAS.
- T. Szailer, J. H. Kwak, D. H. Kim, J. Szanyi, C. Wang and C. H. F. Peden, Catal. Today, 2006, 114, 86 CrossRef CAS.
- V. A. Kondratenko and M. Baerns, Appl. Catal., B, 2007, 70, 111 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |