The current status of hydrogen storage in metal–organic frameworks†
Dan Zhao, Daqiang Yuan and Hong-Cai Zhou*
Department of Chemistry and Biochemistry, Miami University, Oxford, Ohio 45056, USA. E-mail: zhouh@muohio.edu; Fax: +1 513 529-0452; Tel: +1 513 529-8091
First published on 24th June 2008
Abstract
The theoretical and experimental hydrogen storage studies on metal–organic frameworks (MOFs) have been reviewed. Seven distinct factors influencing hydrogen uptake capacity in MOFs have been classified and discussed. Based on existing studies, some possible future developments have been proposed.
 Dan Zhao |
 Daqiang Yuan |
 Hong-Cai Zhou | Hongcai “Joe” Zhou obtained his BSc degree from Beijing Normal University and PhD from Texas A&M University. After a postdoctoral stint at Harvard, he joined the faculty of Miami University, Oxford in 2002. He will move back to Texas A&M as a full professor in the coming fall. His research focuses on hydrogen/methane storage and gas separation that are relevant to clean energy technologies. |
1. Introduction
The rapid consumption of petroleum deposits and the escalating air pollution problems caused by burning fossil fuels have driven the global research community to look for cleaner and renewable energy resources. Albeit not a primary energy source, hydrogen is an ideal energy carrier. It almost triples the gravimetric heat of combustion of gasoline (120 MJ kg−1vs. 44.5 MJ kg−1).1 More importantly, the energy-releasing procedure of hydrogen oxidation, in either an internal combustion engine or a fuel-cell stack, produces only water as a by-product.Energy spent on transportation accounts for a significant part of total energy consumption. It is estimated that in industrialized countries, one-third of all the energy generated annually is consumed in transportation.2 For a modern vehicle with a driving range of 400 km per tank of fuel, about 8 kg of hydrogen is needed for a combustion engine-driven automobile and 4 kg for a fuel-cell-driven one.3 Although these gravimetric requirements are far less demanding than that of gasoline (24 kg), hydrogen is notoriously difficult to compress for on-board storage. Volumetrically, even liquid hydrogen has a much smaller combustion heat than that of gasoline (8960 MJ m−3vs. 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 170 MJ m−3). In the gas phase, 4 kg of hydrogen occupy 45 m3 of space at room temperature and 1 atm.3
170 MJ m−3). In the gas phase, 4 kg of hydrogen occupy 45 m3 of space at room temperature and 1 atm.3
In order to facilitate the research and application of hydrogen as an energy carrier, the US Department of Energy (DOE) has set the targets for on-board hydrogen storage systems: 6.0 wt% and 45 g L−1 by the year 2010, and 9.0 wt% and 81 g L−1 by 2015.4 These targets should be reached at ambient temperature (from −40 to 85 °C) and applicable pressure (less than 100 atm). Note that these are the goals for the system including container and any necessary accessories, the hydrogen storage capacity of the material itself should be even higher.
A safe and effective hydrogen storage technique has become the bottleneck for a possible hydrogen economy. High pressure or cryogenic liquid hydrogen tanks have been certified worldwide and demonstrated in some prototype fuel-cell vehicles. However, their limited storage densities prevent them from reaching the DOE targets. For example, high pressure tanks can reach a pressure of 10000 psi (680 atm) with a 2.35 safety factor (23500 psi burst pressure).4 However, the heavy weight of the system offsets the gain in gravimetric storage density under pressurized conditions, and the volumetric density is far from that of liquid hydrogen (70.8 g L−1). Cryogenic liquid hydrogen tanks, on the other hand, can be used to improve the volumetric hydrogen storage capacity. However, about 20% of the recoverable energy is needed to liquefy the hydrogen and another 2% is spent to keep the tank cool.1 The hydrogen storage capacity of the aforementioned tanks is demonstrated to be between 3.4 to 4.7 wt% gravimetrically and 14 to 28 g L−1 volumetrically.4
In solid-state storage systems, a hydrogen atom/molecule either forms a strong chemical bond to a solid support (chemisorption) or interacts weakly with a sorbent (physisorption).
In chemisorption, dihydrogen molecules split into hydrogen atoms upon contacting the solid support. The highly reactive hydrogen atoms can form chemical bonds with the solid, leading to the formation of metal hydrides or chemical hydrides, depending on the nature of the solid support. Due to the short bonds between hydrogen and the solid, some hydride compounds can reach a relatively high hydrogen storage capacity. However, this strong bonding also leads to severe kinetic and thermodynamic problems during the charging and discharging procedures. Complete charging may take several hours, and the hydrogen-releasing temperature is typically very high (300 °C or higher).5 Alanates are among the most-explored hydride compounds for hydrogen storage. For example, an alanate treated with titanium doping and ball milling showed more than 5 wt% hydrogen storage capacity with a hydrogen-releasing temperature only modestly higher than ambient.6 In another example, Balde et al. showed that by reducing the particle size of NaAlH4, the hydrogen desorption temperature and activation energy can be decreased from 186 °C and 116 kJ mol−1 for the largest particles to 70 °C and 58 kJ mol−1 for the smallest particles, respectively.7 However, heat management and reversibility of these systems remain a concern.8
The physisorption method, on the other hand, stores hydrogen in the molecular form in a sorbent with a large surface area. The most frequently-studied sorbents are activated carbons, carbon nanostructures, zeolites, porous polymers, and metal–organic frameworks (MOFs). Because of the weak sorbent–sorbate interaction, physisorption-based hydrogen storage systems show fast kinetics with a charging time of minutes. However, the same weak interaction results in gravimetric hydrogen uptake of a sorbent at ambient temperature and applicable pressure of typically less than 2 wt%.
2. MOFs as physisorbent materials for hydrogen storage
In 2003, Rosi et al. reported the first MOF-based hydrogen storage result.9 Since then, the hydrogen storage capacities of about 150 MOFs have been reported (Table 1).| Materiala | SA/m2 g−1 | Pore volume/cm3 g−1 | H2 uptake at 77 K, 1 atm/wt% | Maximum H2 uptake/wt% (g L−1) | ∆Hads/kJ mol−1 | Reference | ||
|---|---|---|---|---|---|---|---|---|
| BET Langmiur | 77 K | 298 K | ||||||
| a 1,4-ndc = 1,4-naphthalenedicarboxylate; 2,6-ndc = 2,6-naphthalenedicarboxylate; 2,4-pdc = 2,4-pyridinedicarboxylate; 2-pmc = 2-pyrimidinecarboxylate; 2-pymo = 2-pyrimidinolate; 4,4′-bpe = 4,4′-trans-bis(4-pyridyl)-ethylene; 4,4′-bipy = 4,4′-bipyridine; 4,6-pmdc = 4,6-pyrimidicarboxylate; 4-pymo = 4-pyrimidinolate; 5-bbdc = 5-tert-butyl-1,3-benzenedicarboxylate; 6-mna = 6-mercapto-3-pyridinecarboxylate; abdc = 2-aminobenzene-1,4-dicarboxylate; abtc = azobenzene-3,3′,5,5′-tetracarboxylate; adc = 4,4′-azobenzenedicarboxylate; atdc = 9,10-anthracenedicarboxylate; bbdc = 2-bromobenzene-1,4-dicarboxylate; bdc = 1,4-benzenedicarboxylate; bdp = 1,4-benzenedi(4′-pyrazolyl); bdt = 1,4-benzeneditetrazolate; bhtc = biphenyl-3,4′,5-tricarboxylate; bpdc = 4,4′-biphenyldicarboxylate; bptc = 3,3′,5,5′-biphenyltetracarboxylate; bpydc = 2,2′-bipyridyl-5,5′-dicarboxylate; bpytc = 4,4′-bipyridine-2,6,2′,6′-tetracarboxylate; btc = 1,3,5-benzenetricarboxylate; btt = 1,3,5-benzenetristetrazolate; cbbdc = 1,2-dihydrocyclobutabenzene-3,6-dicarboxylate; cyclam = 1,4,8,11-tetraazacyclotetradecane; dabco = 1,4-diazabicyclo[2.2.2]octane; dbbd = 6,6′-dichloro-2,2′-dibenzyloxy-1,1′-binaphthyl-4,4′-dibenzoate; dccptp = 3,5-dicyano-4-(4-carboxyphenyl)-2,2′:6′4″-terpyridine; debd = 6,6′-dichloro-2,2′-diethoxy-1,1′-binaphthyl-4,4′-dibenzoate; detkb = 2,2′-diethoxylbiphenyl-3,3′,5,5′-tetra-kis(benzoate); dhbdc = 2,5-dihydroxyterephthalate; dipyni = N,N′-di-(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide; dpac = 4,4′-dipyridylacetylene; fma = fumarate; hfipbb = 4,4′-(hexafluoroisopropylidene)bis(benzoate); meim = 2-methylimidazole; ntb = 4,4′,4″-nitrilotrisbenzoic acid; ntc = naphthalene-1,4,5,8-tetracarboxylate; ox = C2O42−; oxdc = oxydiacetate; pbpc = pyridine-3,5-bis(phenyl-4-carboxylate); p-cdc = 1,12-dihydroxy-carbonyl-1,12-dicarba-closo-dodecaborane; pda = p-phenylenediacrylate; pdc = pyridine-3,5-dicarboxylate; phim = benzimidazolate; pic = 3-picoline; pydc = pyrene-2,7-dicarboxylate; pyen = 3,3′-(1E,1′E)-(ethane-1,2-diylbis(azan-1-yl-1-ylidene))bis(methan-1-yl-1-ylidene)dipyridin-4-olate; pyz = pyrazine; qptc = quaterphenyl-3,3‴,5,5‴-tetracarboxylate; sbtc = trans-stilbene-3,3′,5,5′-tetracarboxylate; sip = 5-sulfoisophthalate; tatb = 4,4′,4″-s-triazine-2,4,6-triyltribenzoate; tcppda = N,N,N′,N′-tetrakis(4-carboxyphenyl)-1,4-phenylenediamine; tfbdc = tetrafluoroterephthalate; tmbdc = tetramethylterephthalate; tpb-3tz = 1,3,5-tri-p-(tetrazol-5-yl)phenylbenzene; tptc = terphenyl-3,3″,5,5″-tetracarboxylate; trz = 1,2,4-triazole; ttdc = thieno[3,2-b]thiophene-2,5-dicarboxylate; tz = 3,5-bis(trifluoromethyl)-1,2,4-triazolate; tzi = 5-tetrazolylisophthalate; (refer to ESI1 for more details).b 1.2 bar.c 1.17 bar.d absolute hydrogen uptake. | ||||||||
| Co(bdp) | 2670 | 0.93 | 3.1, 30 bar | 10 | ||||
| Co(2,6-ndc)(4,4′-bipy)0.5 | 115 | 0.10 | 0.72 | 11 | ||||
| Co(ox)(4,4′-bipy) | 0.1 | 12 | ||||||
| Co(pyz)[Ni(CN)4] | 127 | 7.2 | 13 | |||||
| Co(pyz)[Pd(CN)4] | 122 | 7.8 | 13 | |||||
| Co(pyz)[Pt(CN)4] | 138 | 7.6 | 13 | |||||
| Co3(bdc)3(dabco) | 360 | 538 | 0.195 | 1 | 14 | |||
| Co3(bpdc)3(4,4′-bipy) | 922 | 0.38 | 1.98 | 6.8 | 15 | |||
| Co3(D2-Htcppda)2 | 504 | 16 | ||||||
| Co3(ndc)3(dabco) | 1502 | 2293 | 0.822 | 2.45 | 4.2 (35.3), 40 bar | 0.89 (4.48), 17.2 bar | 14 | |
| Cu(2-pymo)2 | 350 | 0.86b | 17 | |||||
| Cu(4-pymo)2 | 65 | 0.03b | 17 | |||||
| Cu(bdc)(dabco)1/2 | 1461 | 0.52 | 1.8 | 18 | ||||
| Cu(bdt) | 200 | 0.66c | 19 | |||||
| Cu(dccptp)(NO3) | 268 | 0.113 | 1.34 | 1.91 (23.0), 20 bar | 6.12 | 20 | ||
| Cu(fma)(4,4′-bpe)1/2 | 0.8 | 21 | ||||||
| Cu(hfipbb)(H2hfipbb)1/2 | 0.23 | 1.0 (16), 48 bar | 22 | |||||
| 12 | ||||||||
| Cu2(C2h-tcppda) | 504 | 1.2 | 23 | |||||
| Cu2(D2-tcppda) | 626 | 1.4 | 23 | |||||
| Cu2(detkb) | 733 | 0.42 | 0.8 | 24 | ||||
| Cu2(qptc) | 2932 | 1.138 | 2.24 | 6.07 (35.6), 20 bar | 25 | |||
| Cu2(tptc) | 2247 | 0.886 | 2.52 | 6.06 (39.4), 20 bar | 25 | |||
| Cu2.9Mn1.1Cl(btt)3/8 (Cu) | 1695 | 1778 | 2.02b | 6.0–8.5 | 26 | |||
| Cu3[(Cu4Cl)3(tpb-3tz)8]2 | 1120 | 1200 | 2.8 (11.4), 30 bar | 0.5 (2.03), 65 bar | 8.2 | 27 | ||
| Cu4Cl(btt)3/8 | 1710 | 1770 | 2.42b | 4.2 (38), 30 bar | 9.5 | 28 | ||
| 5.7 (53), 90 bard | ||||||||
| Cu6O(tzi)3(NO3) | 2847 | 3223 | 1.01 | 2.4 | 9.5 | 29 | ||
| CUK-1, Co3(2,4-pdc)2(OH)2 | 630 | 0.26 | 1.60 | 30 | ||||
| CUK-2, Co2(6-mna)2 | 420 | 0.17 | 0.66 | 30 | ||||
| Dy(btc) | 655 | 1.32 | 31 | |||||
| Er2(pdc)3 | 427 | 32 | ||||||
| Fe3(OH)(pbpc)3 | 1200 | 1.6 | 3.05(33.1), 20 bar | 33 | ||||
| Fe3O(tfbdc)3 | 635 | 0.9 | 34 | |||||
| Fe4O2(btb)8/3 | 1121 | 1835 | 0.69 | 2.1 | 35 | |||
| FMOF-1, Ag2[Ag4(tz)6] | 810.5 | 0.324 | 2.33 (41), 64 bar | 36 | ||||
| HKUST-1, Cu3(btc)2 | 1154 | 1958 | 3.6 (31.6), 50 bar | 0.35 (3.08), 65 bar | 37 | |||
| 2.27 | 3.6 (31.6), 10 bar | 38 | ||||||
| 1944 | 2260 | 3.3 (29), 77 bar | 39 | |||||
| 1507 | 2175 | 0.75 | 2.54 | 5.8–6.6 | 40 | |||
| 0.4 | 1.44 | 6.0–7.0 | 12 | |||||
| 1239 | 0.62 | 2.18 | 6.1 | 41 | ||||
| HKUST-1 + Pt/Ac | 1.12, 100 bar | 42 | ||||||
| In3O(abtc)3/2(NO3) | 1417 | 0.5 | 2.61b | 6.5 | 43 | |||
| IRMOF-1, MOF-5, Zn4O(bdc)3 | 3534 | 4170 | 5.2 (31), 45 bar | 39 | ||||
| 3080 | 4.3 (25.5), 30 bar | 0.45 (2.67), 60 bar | 4.1 | 44 | ||||
| 572 | 1014 | 0.28 | 1.6 (9.49), 10 bar | 0.2 (1.19), 67 bar | 45 | |||
| 2885 | 1.18 | 1.15 | 46 | |||||
| 3362 | 1.32 | 47 | ||||||
| 3800 | 4400 | 7.1 (42.1) 40 bar | 48 | |||||
| 10.0 (66), 100 bard | 48 | |||||||
| 2296 | 3840 | 4.7 (27.9), 50 bar | 0.28 (1.66), 65 bar | 37 | ||||
| 1.32 | 4.3 (25.5), 30 bar | 4.8 | 49 | |||||
| 4.5 | 9 | |||||||
| IRMOF-1 + Pd | 958 | 0.39 | 1.86 | 46 | ||||
| IRMOF-1 + Pt/Ac | 3.0, 100 bar | 50 | ||||||
| IRMOF-2, Zn4O(bbdc)3 | 1722 | 2544 | 0.88 | 1.21 | 40 | |||
| IRMOF-3, Zn4O(abdc)3 | 2446 | 3062 | 1.07 | 1.42 | 40 | |||
| IRMOF-6, Zn4O(cbbdc)3 | 2476 | 3263 | 1.14 | 1.48 | 4.9 (34.9), 32 bar | 40 | ||
| 2804 | 3300 | 4.8 (32), 45 bar | 39 | |||||
| IRMOF-8, Zn4O(ndc)3 | 1818 | 3.6 (20.9), 15 bar | 0.4 (2.32), 30 bar | 6.1 | 44 | |||
| 1466 | 1.5 | 47 | ||||||
| IRMOF-8 + Pt/Ac | 1.8, 100 bar | 51 | ||||||
| 4.0, 100 bar | 50 | |||||||
| IRMOF-9, Zn4O(bpdc)3 | 1904 | 2613 | 0.9 | 1.17 | 40 | |||
| IRMOF-11, Zn4O(hpdc)3 | 1984 | 2340 | 3.5 (27), 34 bar | 5.1–9.1 | 39 | |||
| 1911 | 1.62 | 47 | ||||||
| IRMOF-13, Zn4O(pydc)3 | 1551 | 2100 | 0.73 | 1.73 | 40 | |||
| IRMOF-18, Zn4O(tmbdc)3 | 1501 | 0.89 | 47 | |||||
| IRMOF-20, Zn4O(ttdc)3 | 3409 | 4346 | 1.53 | 1.35 | 6.7 (43.9), 70 bar | 40 | ||
| 4024 | 4590 | 6.7 (34), 80 bar | 39 | |||||
| JUC-48, Cd3(bpdc)3 | 880 | 0.19 | 2.8 (20.0), 40 bar | 1.1 (7.84), 100 bar | 52 | |||
| MAMS-1, Ni8(5-bbdc)6 | 0.6 | 53 | ||||||
| α-[Mg3(HCO2)6] | 150 | 0.043 | 0.6 | 54 | ||||
| Mg3(ndc)3 | 520 | 0.78 | 55 | |||||
| 190 | 0.46c | 7.0–9.5 | 56 | |||||
| MIL-53(Al), Al(OH)(bdc) | 1100 | 1590 | 0.59 | 2.1 | 3.8 (37.0), 16 bar | 57 | ||
| MIL-53(Cr), Cr(OH)(bdc) | 1100 | 1500 | 0.56 | 1.8 | 3.2 (33.2), 16 bar | 57 | ||
| MIL-53 + Pt/Ac | 0.63, 50 bar | 58 | ||||||
| MIL-96, Al3O(btc)3 | 1.91 (27.9), 3 bar | 59 | ||||||
| MIL-100(Cr), Cr3OF(btc)2 | 2700 | 1 | 3.3 (23.0), 25 bar | 0.15 (1.04), 73 bar | 5.6–6.3 | 60 | ||
| MIL-101(Cr), Cr3OF(bdc)3 | 5500 | 1.9 | 2.5 | 6.1 (26.1), 60 bar | 0.43 (1.84), 80 bar | 9.3–10.0 | 60 | |
| MIL-101 + Pt/Ac | 1.43, 100 bar | 42 | ||||||
| 1.14, 50 bar | 58 | |||||||
| MIL-102, Cr3OF(ntc)3/2 | 42.1 | 0.12 | 1.0 (16), 35 bar | 0.05 (0.8), 35 bar | 6 | 61 | ||
| Mn(HCO2)2 | 240 | 0.9 | 62 | |||||
| Mn(ndc) | 191 | 0.068 | 0.57 | 63 | ||||
| Mn3(bdt)8Cl2 | 530 | 0.82c | 6.0–8.8 | 19 | ||||
| Mn3(bdt)3 | 290 | 0.97c | 6.3–8.4 | 19 | ||||
| Mn3[(Mn4Cl)3(tpt-3tz)8]2 | 1580 | 1700 | 3.7(25.0), 25 bar | 0.5 (3.38), 65 bar | 7.6 | 27 | ||
| 4.5 (37), 80 bard | ||||||||
| Mn4Cl(btt)3/8 | 2100 | 2.2b | 5.1 (43), 50 bar | 10.1 | 64 | |||
| 6.9 (60), 90 bard | ||||||||
| Mn4Cl(btt)3/8 (Co) | 2096 | 2268 | 2.12b | 5.6–10.5 | 26 | |||
| Mn4Cl(btt)3/8 (Cu, Mn) | 1911 | 2072 | 2.00b | 5.6–9.9 | 26 | |||
| Mn4Cl(btt)3/8 (Fe) | 2033 | 2201 | 2.21b | 5.5–10.2 | 26 | |||
| Mn4Cl(btt)3/8 (Li) | 1904 | 2057 | 2.06b | 5.4–8.9 | 26 | |||
| Mn4Cl(btt)3/8 (Ni) | 2110 | 2282 | 2.29b | 5.2–9.1 | 26 | |||
| Zn0.7Mn3.3Cl(btt)3/8 (Zn) | 1927 | 2079 | 2.10b | 5.5–9.6 | 26 | |||
| MOF-177, Zn4O(btb)2 | 4750 | 5640 | 7.6 (32), 66 bar | 4.4 | 65 | |||
| 11.4 (49), 78 bard | ||||||||
| 4746 | 5640 | 7.5 (32), 70 bar | 39 | |||||
| 4526 | 1.25 | 47 | ||||||
| MOF-177 + Pt/Ac | 1.5, 100 bar | 5.8–11.3 | 66 | |||||
| MOF-505, Cu2(bptc) | 1670 | 1830 | 0.68 | 2.59 | 4.02 (37.3), 20 bar | 25 | ||
| 0.63 | 2.47 | 67 | ||||||
| MOF-74, Zn3(dhbdc)3 | 950 | 1070 | 2.3 (27), 26 bar | 39 | ||||
| 783 | 1132 | 0.39 | 1.77 | 8.3 | 40 | |||
| 870 | 2.8 (32.9) 30 bar | 68 | ||||||
| NaNi3(OH)(sip)2 | 743 | 0.94 | 9.4–10.4 | 69 | ||||
| Ni(4,4′-bipy)[Ni(CN)4] | 234 | 1.84 | 7.5 | 13 | ||||
| Ni(4,4′-bipy)[Pd(CN)4] | 220 | 7 | 13 | |||||
| Ni(cyclam)(bpydc) | 817 | 0.37 | 1.1 | 70 | ||||
| Ni(dpac)[Ni(CN)4] | 398 | 2.24 | 6 | 13 | ||||
| Ni(ox)(4,4′-bipy) | 0.16 | 12 | ||||||
| Ni(pyz)[Ni(CN)4] | 124 | 1.76 | 7.2 | 13 | ||||
| Ni2(4,4′-bipy)3(NO3)4 | 0.181 | 0.987 | 71 | |||||
| Ni2(4,4′-bipy)3(NO3)4 | 0.149 | 0.653 | 71 | |||||
| Ni2(dhbdc) | 1083 | 0.41 | 1.8 (21.5), 70 bar | 0.3 (3.58) 65 bar | 72 | |||
| Ni3(btc)2(3-pic)6 | 0.63 | 71 | ||||||
| Ni3(OH)(pbpc)3 | 1553 | 1.99 | 4.15 (43.9), 20 bar | 33 | ||||
| PCN-5, Ni3O(tatb)2 | 225 | 0.13 | 0.63 | 73 | ||||
| PCN-6, Cu3(tatb)2 | 3800 | 1.45 | 1.9 | 74 | ||||
| PCN-6′, Cu3(tatb)2 | 2700 | 1.045 | 1.35 | 74 | ||||
| PCN-9, Co4O(tatb)8/3 | 1355 | 0.51 | 1.53 | 10.1 | 75 | |||
| PCN-10, Cu2(abtc) | 1407 | 1779 | 0.67 | 2.34 | 4.33 (33.2), 20 bar | 4–7 | 76 | |
| PCN-11, Cu2(sbtc) | 1931 | 2442 | 0.91 | 2.55 | 5.05(37.8), 20 bar | 4–7 | 76 | |
| PCN-13, Zn4O(adc)3 | 150 | 0.1 | 0.41 | 77 | ||||
| PCN-17, Yb(tatb)8/3(SO4)2 | 820 | 0.34 | 0.94 | 78 | ||||
| Pd(2-pymo)2 | 600 | 1.29b | 17 | |||||
| rho-ZMOF, Cd(2-pmc)2 | 1168 | 0.474 | 1.16 | 8.7 | 79 | |||
| sod-ZMOF, In(4,6-pmdc)2 | 616 | 0.245 | 0.9 | 8.4 | 79 | |||
| Sc(bdc)3 | 721 | 0.332 | 1.5 | 80 | ||||
| Sm2Zn3(oxdc)6 | 718.8 | 0.31 | 1.19 (18.6), 34 bar | 0.54 (8.4), 35 bar | 81 | |||
| TUDMOF-1, Mo3(btc)2 | 1280 | 2010 | 0.67 | 1.75 | 82 | |||
| UMCM-150, Cu3(bhtc)2 | 2300 | 3100 | 1 | 2.1 | 5.7 (36), 45 bar | 7.3 | 83 | |
| Y2(pdc)3 | 676 | 32 | ||||||
| ZIF-11, Zn(phim)2 | 1676 | 0.582 | 1.37 | 84 | ||||
| ZIF-8, Zn(meim)2 | 1.3 | 3.3 (35.6), 30 bar | 0.13 (1.40), 30 bar | 4.5 | 49 | |||
| 1630 | 1810 | 0.636 | 1.29 | 3.1 (33.4), 55 bar | 84 | |||
| Zn(adc)(4,4′-bpe)1/2 | 100 | 0.62 | 85 | |||||
| Zn(bdc)(4,4′-bipy)1/2 | 946 | 0.8 | 86 | |||||
| Zn(bdc)(dabco)1/2 | 0.73 | 2.1 | 5.0–5.3 | 87 | ||||
| 1794 | 0.65 | 2.1 | 18 | |||||
| Zn(2,6-ndc)(4,4′-bpe)1/2 | 303 | 0.2 | 0.80 | 2.0 (17.9), 40 bar | 0.3(2.7), 65 bar | 88 | ||
| Zn2(1,4-bdc)(tmbdc)(dabco) | 1100 | 1670 | 0.59 | 2.08 | 89 | |||
| Zn2(1,4-bdc)2(dabco) | 1450 | 2090 | 0.75 | 2.01 | 89 | |||
| Zn2(1,4-ndc)2(dabco) | 1000 | 1450 | 0.52 | 1.7 | 89 | |||
| Zn2(bdc)2(dabco) | 1603 | 2420 | 0.86 | 1.8 | 90 | |||
| Zn2(bpytc) | 312.7 | 0.146 | 1.08(16.7), 4 bar | 0.057(0.88), 4 bar | 5.12 | 91 | ||
| Zn2(detkb) | 252 | 0.2 | 24 | |||||
| Zn2(2,6-ndc)2(dipyni) | 802 | 0.30 | 0.93 | 5.6 | 92 | |||
| Zn2(2,6-ndc)2(dipyni) + Li+ | 756 | 0.34 | 1.63 | 6.1 | 92 | |||
| Zn2(tfbdc)2(dabco) | 1070 | 1610 | 0.57 | 1.78 | 89 | |||
| Zn2(tmbdc)2(4,4′-bipy) | 1120 | 1740 | 0.62 | 1.68 | 89 | |||
| Zn2(tmbdc)2(dabco) | 920 | 1400 | 0.5 | 1.85 | 89 | |||
| Zn3(bdc)3[Cu(pyen)] | 0.257 | 1.10 | 12.29 | 93 | ||||
| Zn3(bdt)3 | 640 | 1.46c | 6.8–8.7 | 19 | ||||
| Zn3(bpdc)3(4,4′-bipy) | 792 | 0.33 | 1.74 | 7.1 | 15 | |||
| Zn3(OH)(p-cdc)5/2 | 152 | 2.1 | 7 | 94 | ||||
| Zn3(tatb)2(HCOO) | 1100 | 1.3 | 95 | |||||
| Zn4(trz)4(1,4-ndc)2 | 362.1 | 0.84 | 96 | |||||
| Zn4(trz)4(2,6-ndc)2 | 584.1 | 1.11 | 96 | |||||
| Zn4O(D2-tcppda)3/2 | 2095 | 0.8 | 16 | |||||
| Zn4O(dbbd)3 | 396 | 0.13 | 0.98 (10), 48 bar | 97 | ||||
| Zn4O(debd)3 | 502 | 0.2 | 1.12 (10.3), 48 bar | 97 | ||||
| Zn4O(ntb)2 | 1121 | 0.51 | 1.9 | 98 | ||||
| Zn7O2(pda)5 | 0.17 | 1.01 (4.35), 71.4 bar | 99 | |||||
2.1. Characteristics of MOFs
MOFs are crystalline coordination polymers, containing organic ligands as the linkers and metal ions or clusters (secondary building units, SBUs) as the nodes (Fig. 1).100–102 The solvent or guest molecules, which occupy the pore spaces in MOFs can be removed upon solvent exchange and heating under vacuum to generate a stable porous structure. Compared to their porous counterparts, such as zeolite and activated carbon, MOFs have a much higher surface area (the record holder is MIL-101 with a Langmuir surface area of 5900 m2 g−1).103 The combination of different organic ligands and SBUs gives MOFs almost infinite geometrical and chemical variation. The availability of high-resolution crystal structures of MOFs allows direct observation and comparison of pore size and geometry, which is almost impossible in other porous materials.104 Potential reaction sites on the organic ligands in MOFs renders post-synthetic modification possible, leading to the introduction of additional active sites for stronger hydrogen binding.105 All these characteristics of MOFs make them one of the most promising sorbent materials for hydrogen storage. | ||
| Fig. 1 Illustration of MOF synthesis and chemical composition. | ||
2.2. Characterization of hydrogen uptake in MOFs
The two main methods for the measurement of hydrogen uptake of MOFs are gravimetry (gravimetric method) and volumetry (volumetric method). In the gravimetric method, the hydrogen uptake is directly measured by the mass change of the sorbent using a highly-sensitive balance. In the volumetric method, however, the hydrogen uptake is deduced from the decrease in hydrogen pressure in a fixed volume. These two methods have both advantages and disadvantages. For instance, results from the gravimetric method can be greatly affected by the impurities in the hydrogen source while the volumetric method suffers from accumulative errors.106 From a practical point of view, the volumetric method is more favorable in terms of ease of operation and availability of instrumentation.Although the DOE targets require hydrogen uptake measurements carried out at ambient temperature (from −40 to 85 °C) and applicable pressure (up to 100 atm), most reported hydrogen uptake measurements in MOFs are carried out at 77 K and 1 atm. This is largely because of the availability of that condition in commercially available gas adsorption equipment. Although far from DOE-target conditions, hydrogen uptake data obtained at low temperature and pressure are still useful in the initial exploration and comparison of hydrogen uptake capacities in different MOFs. As high pressure hydrogen uptake measurement equipment becomes more widely available, more and more data of high pressure hydrogen uptake in MOFs have been reported. At room temperature, the binding energy between hydrogen and the MOFs is comparable to thermal vibration energies, which leads to very poor hydrogen uptake (typically less than 1 wt%) and makes the comparison and discussion of hydrogen uptake capacity ambiguous and difficult. At the cryogenic condition, however, the relatively strong interaction between hydrogen and MOFs (as compared to thermal energies) gives rise to greatly enhanced hydrogen uptake. This enhancement is so pronounced that some saturation hydrogen uptake data obtained at 77 K have reached or even surpassed the DOE targets.39,48,64 Thus the comparison of saturation hydrogen uptake data at 77 K is more reliable and instructive in determining the MOFs' hydrogen uptake capacity.
By applying the Clausius–Clapeyron equation to two sets of hydrogen adsorption data collected at different temperatures (typically 77 K and 87 K), the isosteric heat of adsorption (∆Hads) can be deduced; this is an important criterion in judging how strongly hydrogen binds the MOFs.40,64 In order to guarantee the validity of these ∆Hads values, data sets should be collected at more than two different temperatures.
2.3. Criteria for hydrogen storage capacity
There are two criteria for the sorbent's hydrogen storage capacity: excess adsorption and absolute adsorption. Simply speaking, excess adsorption is a measurement of the gas molecules stuck to the surface of the sorbent, which is given directly from experimental measurements; while absolute adsorption includes both excess adsorption and gas molecules occupying the voids inside the sorbent.65 From a practical viewpoint, absolute adsorption is more relevant for hydrogen storage applications. However, due to the difficulty in obtaining absolute adsorption data, which is estimated theoretically, most hydrogen uptake data reported are based on excess adsorption. Furukawa et al. proposed a simple equation for the conversion of these two values:| Nabs = Nex + ρbulkVpore |
A large isosteric heat of adsorption would lead to a steep rise in the adsorption isotherm within the low pressure range (less than 3 atm), which at first was deemed to be helpful for hydrogen storage. However, in the DOE targets, the minimum delivery pressure is 3–4 atm for fuel-cells and 35 atm for internal combustion engines, which means that any hydrogen that is stored at a pressure less than 3 atm would not be fully utilized.4 Furukawa et al. proposed the deliverable capacity as another criterion for judging material's hydrogen storage uptake, based on the absolute adsorbed amount of hydrogen in the sorbent materials from 1.5 to 100 bar.65 The same idea has been addressed by other research groups.87,107
In order to directly judge the sorbent materials' gas uptake capacities, Zhou et al. introduced the “effective adsorption” concept, which compares the amount of gas held in a container with and without the sorbent materials.49 According to their high pressure hydrogen uptake measurements at room temperature, the effective hydrogen adsorption in MOF-5 and ZIF-8 are nearly zero and negative respectively, indicating no positive contribution to hydrogen storage compared with a high pressure tank. However, the high pressure hydrogen uptake measurements at 77 K conducted by Mueller et al. show that the container filled with MOF-5 takes up higher amounts of hydrogen than the empty container.108 Among the MOFs they tested, HKUST-1, a MOF composed of copper ions and 1,3,5-benzenetricarboxylate,109 reaches an effective adsorption of +44%, with a volumetric hydrogen uptake capacity of 18.5 g L−1. They also pointed out that because of the many volume-limited fuel-cell applications and the low density of MOFs, the volumetric hydrogen uptake capacity should also be addressed along with the gravimetric one.
3. Factors influencing hydrogen uptake
One of the characteristics that differentiate MOFs from other hydrogen storage materials is the richness of factors that influence their hydrogen uptake capacity. These factors will be summarized and discussed in the following.3.1. Surface area and pore volume
The first notable characteristic of porous materials is surface area. In the case of MOFs, since there is a positive relationship between surface area and pore volume (Fig. 2), these two criteria will be discussed together here. | ||
| Fig. 2 Correlation between surface area (red: Langmuir; blue: BET) and total pore volume. | ||
There is a well-established positive relationship between the surface area and the hydrogen uptake in carbon-based sorbents.110,111 Note that the linker portion of most MOFs is composed of aromatic ring motifs, which give rigidity to the framework. Such chemical composition is very similar to that of carbon materials, which are largely composed of sp2-hybridized carbon atoms. A positive, roughly linear relationship between specific surface area and hydrogen uptake in MOFs can be observed by plotting the surface areas versus the 77 K saturation hydrogen uptake data (Fig. 3).39,104 The slope of the linear relationship is 1.45 × 10−3 wt% (m2 g−1)−1 for the Langmuir surface area and 1.92 × 10−3 wt% (m2 g−1)−1 for the BET surface area, which is comparable to the theoretical value for carbon (2.28 × 10−3 wt% (m2 g−1)−1).112 Although it has been suggested that adsorption in MOFs occurs through a pore-filling mechanism rather than layer formation, grand canonical Monte Carlo (GCMC) simulations performed on a series of MOFs have verified the validity of the BET theory in determining the surface areas of MOFs.113
 | ||
| Fig. 3 Correlation between surface area and saturation hydrogen uptake at 77 K (red: MOFs, Langmuir method; blue: MOFs, BET method; black: sp2 carbon, theoretical). | ||
Among MOFs, MOF-177 ranks highest for gravimetric hydrogen uptake, with a value of 7.6 wt% at 77 K and 70 bar.65 It also has a very large surface area of around 4500 m2 g−1. In the case of MIL-101, although it possesses the highest surface area (Langmuir surface area is ∼5900 m2 g−1),103 the high pressure hydrogen uptake at 77 K is not the highest.60 However, according to the author, the sample has not been fully activated due to the small opening within the cage structure.
It can be safely concluded that in most cases, the saturation hydrogen uptake in MOFs at 77 K is mainly determined by the surface area and the pore volume regardless of the chemical composition.114 Theoretically, a minimum surface area of ∼1400 m2 g−1 is needed for a material to reach an excess adsorption of 6 wt% (or ∼2100 m2 g−1 for 9 wt%).68
3.2. Pore size and geometry
The hydrogen uptake capacity of MOFs is not determined solely by the surface area. In the case of MIL-101, for example, the very large empty space generated by large cages contributes very little to hydrogen storage.60 Small pores, which have stronger interaction with hydrogen due to the overlap of the potential fields from both sides of the pore walls, should have higher affinity towards hydrogen and thus higher hydrogen uptake capacity. One can deduce this conclusion from the hydrogen-binding energies in pure carbon materials, which range from 4 to 15 kJ mol−1.115 The lower value is common in flat carbon structures such as graphite, while the higher one is typical of internal and interstitial sites in carbon nanostructures, an indication that confined geometry can greatly enhance hydrogen-binding energy. Note that the kinetic diameter of hydrogen is 2.9 Å. Calculations based on carbon materials showed that a slit pore with a width of 6 Å has the highest hydrogen uptake at very low pressures because it exhibits the strongest interaction potential. A larger width of 9 Å was recommended for maximum hydrogen uptake capacity at high pressure.116In the case of HKUST-1, both neutron powder diffraction and inelastic neutron scattering (INS) data support that hydrogen molecules are adsorbed into the smaller cage before the larger one, indicating a stronger interaction between hydrogen and the smaller pore.117,118 This conclusion is confirmed indirectly by desorption studies of hydrogen in various MOFs (HKUST-1, MIL-53, MOF-5, and IRMOF-8), where the hydrogen desorbed first from the larger pore, then the smaller pore with increasing temperature.119
One way to reduce pore size is by introducing bulky groups in the ligands. Farha et al. used a bulky carborane ligand to construct a MOF with 2.1 wt% of hydrogen uptake at 77 K and 1 atm.94 Pan et al. constructed a microporous MOF with a ligand decorated with the bulky trifluoromethyl group.22 Due to the curved internal surfaces and the reduced pore size, the room temperature hydrogen uptake capacity of this MOF is close to 1 wt% at 48 atm, which is comparable to the best-performing carbon nanotubes they examined. The same strategy was adopted by Yang et al., who generated a MOF with volumetric hydrogen uptake capacity of 41 g L−1 at 77 K and 64 bar, close to the DOE 2010 volumetric target of 45 g L−1.36 However, one should bear in mind that the improved gravimetric hydrogen uptake by this strategy is partially counteracted by the increased framework density given by the decorating groups.40
3.3. Catenation
One direct way to reduce the pore size is catenation, in which two or more identical frameworks interpenetrate with each other to generate reduced pore size.120Because catenation will decrease the free volume, whether catenation is helpful for hydrogen uptake is determined by the compromise between the increased hydrogen density within the pores and the decreased free volume from catenation.121 The GCMC simulation on IRMOF-9 and IRMOF-10 suggested that catenation is not a promising option to increase the hydrogen uptake at high pressure due to the reduced pore volume.121 At low pressure and 77 K, however, the experimental study of catenated IRMOF-11 showed higher hydrogen uptake compared to non-catenated IRMOFs with the same topology, which has been supported by GCMC simulation.40,122
Using oxalic acid as a template molecule, Ma et al. have generated a catenated MOF, PCN-6, and its non-catenated counterpart, PCN-6′, making the study of catenation's effect on hydrogen uptake in MOFs as an independent criterion possible.74 In this case, the low pressure and 77 K gas sorption results showed that catenation leads to a 41% increase in Langmuir surface area and 133% of enhancement in volumetric hydrogen uptake (29% in gravimetric). The high pressure and 77 K hydrogen uptake measurements done by Dincă et al. on one pair of ligand-directed catenated and non-catenated MOFs indicated 41% increase in surface area and 32% increase in gravimetric hydrogen uptake by catenation.27
Up to now, catenation has shown some improvement in hydrogen uptake in some MOF systems at 77 K. The study of catenation's effect on MOFs' hydrogen uptake capacity at ambient temperature is being undertaken by our group and will be published soon.
3.4. Ligand structure and functionalization
The influence of ligand structure on hydrogen uptake capacity of MOFs can be traced back to the first reported hydrogen uptake in MOFs, in which the authors proposed that using larger aromatic ligands would increase the hydrogen uptake capacity.9 This idea is supported by other theoretical studies.123,124 In some of these proposed MOFs, the organic linkers dominate the hydrogen adsorption while the metal clusters play only a lesser role,123 in contrast to the initial INS study.9 The same phenomenon is observed in zeolitic imidazolate frameworks, a kind of MOF using imidazolate as the organic ligands. The neutron powder diffraction data on ZIF-8 indicated that the imidazolate organic linker was primarily responsible for hydrogen adsorption, suggesting a larger effect of linker modification on the hydrogen storage capacity.125In contrast to using a larger aromatic ring, the VASP ab initio computer calculations indicated that there is little effect on the hydrogen uptake capacity if the aromatic ring is substituted with halogen, and the added mass would be detrimental to the hydrogen uptake capacity.126 This is confirmed experimentally. A systematic study by Chun et al. on the influence in hydrogen uptake capacity brought by the modulation of the organic ligands resulted in no direct relationship between the hydrogen uptake capacity and the chemical composition of the organic ligands.89 On the contrary, the authors suggested that the shape and size of channels instead of the ligands' chemical nature should be responsible for the hydrogen uptake trend in their study. The same conclusion is drawn by Rowsell et al., in which the low pressure hydrogen uptake measurements were done on a series of IRMOFs.40
3.5. Unsaturated metal sites
In some MOFs, the solvent or guest molecules coordinatively bound to the metal node can be removed without the collapse of the framework by heating under vacuum. These MOFs, bearing coordinatively unsaturated metal sites, show higher hydrogen-binding energy. One example is the dicopper paddlewheel SBU, in which the removal of the axial ligands has been confirmed by single crystal X-ray diffraction.127 The stronger interaction between hydrogen and the copper open sites has been demonstrated by IR spectroscopy.128 Additionally, neutron powder diffraction revealed that the most favorable hydrogen binding sites are the unsaturated axial sites of the dicopper SBUs.117 This is also consistent with an INS study.118The unsaturated metal sites are not exclusive to copper-containing MOFs. Inspired by the entatic state in biological systems, Ma et al. constructed a MOF, PCN-9, in which cobalt atoms are five-coordinate with square pyramidal geometry, leading to a hydrogen adsorption heat of 10.1 kJ mol−1.75 The neutron powder diffraction study done on MOF-74 revealed the strong interaction between hydrogen and the exposed Zn2+ ions and indicated a strong correlation between the existence of unsaturated metal sites and the high hydrogen surface packing density.68 Dincă et al. constructed a MOF with both exposed Mn2+ coordination sites and free Mn2+ within the channel.64 Neutron powder diffraction data showed direct hydrogen binding at the unsaturated Mn2+ within the framework, with the maximum isosteric heat of adsorption 10.1 kJ mol−1. The absolute hydrogen uptake is 6.9 wt% at 77 K and 90 bar with the density of the stored hydrogen 85% of that of liquid hydrogen. By replacing the coordinated Mn2+ with Cu2+, a more robust MOF is generated, which can be fully desolvated to expose a larger number of open metal sites.28 A slightly decreased heat of adsorption of the generated copper MOF as compared to its manganese counterpart was explained by Jahn–Teller distortion of the coordination environment of the Cu2+ ions. Another explanation using spin state has been given by a computational study, which demonstrated that binding energy can be tuned in a range of about 10 to 50 kJ mol−1 using different transition metal ions.129 Another theoretical study pointed out that the interaction is not of the expected Kubas-type but only comes from classical Coulomb interaction.130 Using the same MOF, Dincă et al. replaced the free Mn2+ cation with other cations to generate a series of isostructural MOFs.26 There is an adsorption heat difference of 2 kJ mol−1 between the weakest and strongest hydrogen-binding MOFs; among them the Co2+-exchanged MOF exhibits the highest heat of adsorption, 10.5 kJ mol−1.
A combined DFT and GCMC simulation study on MOF-505 showed that open metal sites have favorable impact on hydrogen adsorption in MOFs at low pressures, and the hydrogen molecule is inclined to expose the negative lobe of its quadrupole to the exposed copper atoms, which act as Lewis acids.131 According to another simulation study, if the open metal sites could be incorporated on the organic linkers, the metal–hydrogen dissociation energy could go as high as 84 kJ mol−1, a potential route to achieve reversible sorption at ambient conditions.132
Although there are still some arguments about whether the unsaturated metal sites are the main reason for the increased interaction between hydrogen and the framework,63 the combination of unsaturated metal sites with the appropriate pore size and geometry discussed above gives rise to some MOFs with strong hydrogen binding energies. In the case of Mg3(O2C–C10H6–CO2)3, where the Mg2+ centers are unsaturated and the pore dimensions are constricted, the hydrogen isosteric heat of adsorption reaches 9.5 kJ mol−1.56 Chen et al. immobilized unsaturated metal sites within ultramicropores to generate a mixed zinc/copper MOF with an isosteric heat of hydrogen adsorption of up to 12.29 kJ mol−1, the highest reported so far.93 With the increasing amount of hydrogen adsorbed, the heat decreases and reaches a plateau where all the open metal sites become saturated by the hydrogen. Even more interestingly, because the pore size is so small, the quantum effects in the sorption of H2 and D2 are observable, demonstrating potential applications for isotope separation.
3.6. Chemical doping and spillover
Another way to increase the binding energy between hydrogen and MOF is through chemical doping. Li+ has been calculated to show a strong affinity for hydrogen (∼24 kJ mol−1),133 and ab initio calculations for Li-decorated MOF-5 indicated that a hydrogen uptake of 2.9 wt% at 200 K and 2.0 wt% at 300 K is achievable.134 A combination of quantum and classical calculations done on MOFs modified by lithium alkoxide groups gave the hydrogen uptake capacity of 10 wt% at 77 K and 4.5 wt% at room temperature, both at 100 bar.135 Other calculations indicated that the 2010 DOE targets of 6.0 wt% could be reached on Li-MOF-C30 at −30 °C and 100 bar.136 By direct reduction of the organic ligand with lithium metal, Mulfort et al. experimentally introduced lithium cations into MOFs.92 Compared to the pristine MOF, the Li-doped MOF demonstrates a 75% increase in gravimetric hydrogen uptake, up to 1.63 wt% at 77 K and 1 atm. Furthermore, the isosteric heat of adsorption is greater in the Li-doped MOF over the entire loading range.In addition to cations, anions can also be helpful in hydrogen adsorption. For example, charge-separated ammonium fluorides are calculated to have enhanced binding energy towards molecular hydrogen.137
Because it is well demonstrated that the hydrogen molecule can be dissociated into monoatomic hydrogen by certain heavy transition metals (e.g. Pt), making use of this “dissociation/spillover” in a MOF-based hydrogen-storage system leads to hydrogen uptake enhancement, increasing adsorbed hydrogen by a factor of 3.3 for MOF-5 and 3.1 for IRMOF-8.51 In the latter exploration, by using a carbon bridge to facilitate the secondary spillover, the enhancement factor for IRMOF-8 has been increased to 8, resulting in a hydrogen uptake of 4 wt% at room temperature and 10 MPa, the highest among all the MOFs, with the entire process completely reversible.50 The spillover effect has been reproduced by Liu et al.58 Their results show the storage capacity of 1.14 and 0.63 wt% for MIL-101 and MIL-53 at 5.0 MPa and 293 K, which is greatly increased from that of pristine samples (0.37 wt% and none). In another approach, in which palladium was doped into MOF-5 via solution infiltration, the hydrogen adsorption capacity is increased by 62% to 1.86 wt% at 77 K and 1 atm.46 However, according to the authors, the increase at low pressures does not necessarily imply a higher capacity at high pressures.
3.7. Sample preparation
Sample preparation has been recognized in hydrogen uptake studies in MOFs as a key to obtaining repeatable and reliable data. For example, differences in the reported hydrogen uptake capacity in HKUST-1 are attributed to sample purity and activation.38 During the degassing process, around 10 mPa is suggested as a satisfactory residual pressure.106 The degassing temperature and time is also important. Navarro et al. demonstrated that activation at 105 °C gave almost negligible hydrogen sorption in their sample, while 120 °C activation resulted in much better hydrogen uptake.17 Generally speaking, the higher the temperature, the shorter the degassing time required. Within the temperature range in which the framework remains intact, the plot between the measured surface areas versus the degassing time gives the optimum degassing time where the plot reaches a plateau. In practice, a rough estimation about whether the degassing is complete can be made if the pressure keeps a stable value close to zero during degassing. Powder X-ray diffraction should be used to monitor the quality of the crystal before each hydrogen uptake measurement.Besides the sample quality and activation conditions, sample size can also potentially affect the accuracy of the measurement.106 Too small a sample size would lead to larger uptake while too large a sample size may need more time for the sorption to reach equilibrium. For the volumetric method, an appropriate sample size is typically about 100 mg.
In our own study, we have observed that a sample with small particle size has a larger hydrogen uptake than one with larger particle size. The possibility of the surface area difference caused by the particle size has been ruled out because the external surface area increase due to smaller particle size is neglectable compared to the much larger internal surface area. One possible explanation would be that in larger particles, the longer diffusion path limits access into the interior of the particle, either for the guest molecules coming out from the frameworks during activation or the hydrogen molecules going into the frameworks during adsorption. Besides, the chemical difference between the terminal and the inner parts of the particle should also be considered. It is possible that more unsaturated metal sites would be exposed at the surface of smaller particles than that of the larger one, leading to increased hydrogen uptake.
Solvent exchange is also a crucial step in sample activation. By replacing the high-boiling-point and strongly-bound solvent or guest molecules (e.g. amide) with low-boiling-point and weakly-bound molecules (e.g. dichloromethane, chloroform, and methanol), the void inside the MOFs could be evacuated under moderate condition without framework collapse.
Sample preparation is also important for material stability. For example, MOF-5, containing the Zn4O motif, is proven to be unstable upon contact with moisture.42,45,48,66,138 Kaye et al. modified the previously reported method to obtain a sample of MOF-5 with the highest surface area among the reported data, in which the exposure to water and air was minimized.48 Their sample adsorbed 7.1 excess wt% hydrogen, and the absolute hydrogen uptake climbed to 11.5 wt% at 170 bar, with a volumetric storage density of 77 g L−1, which is greater than the density of liquid hydrogen (70.8 g L−1).
4. Future developments
4.1. Balancing between surface area and pore size
GCMC simulations done on a series of isoreticular MOFs indicated three adsorption regimes.121,139 At low pressure (less than 1 atm), the hydrogen uptake correlates with the heat of adsorption, while at high pressure the surface area and free volume become more important. Lin et al. constructed a series of MOFs with the same linkage but different length of ligands, leading to MOFs with the same topology but different pore sizes and surface areas. Hydrogen uptake measurements carried out at 77 K and under either 1 or 20 bar indicate that the MOF with the smallest pore size shows the strongest hydrogen-binding energy, while the highest gravimetric hydrogen uptake is achieved in the sample with the largest surface area.25 A similar conclusion was drawn by Culp et al.13 It follows that the ideal MOFs for hydrogen storage purpose should have high surface area for higher capacity and appropriate pore size for strong binding with hydrogen.114 However, there is no simple relationship between these two factors. In the above two cases, these two factors are linked, with the smaller surface area MOF having smaller pores and the larger surface area MOF having larger pores. More work should be done to optimize the trade-off between surface area and pore size.4.2. Enhancing interaction between hydrogen and framework
Up to now, the bottleneck for a MOF-based hydrogen storage system is the weak interaction between hydrogen and the framework. Fig. 4 summarizes the current status of MOFs' hydrogen storage capacity at 77 K. Some of them have reached or even surpassed the DOE targets. However, the hydrogen uptake at ambient condition is almost negligible. How strong an interaction between hydrogen and MOFs is needed to reach the DOE targets? The calculation done by Bhatia et al. indicated that an adsorption enthalpy of −15.1 kJ mol−1 is needed for optimum delivery of hydrogen between pressures of 1.5 and 30 bar at 298 K.140 More importantly, this value of enthalpy should be kept consistently high throughout the hydrogen coverage range.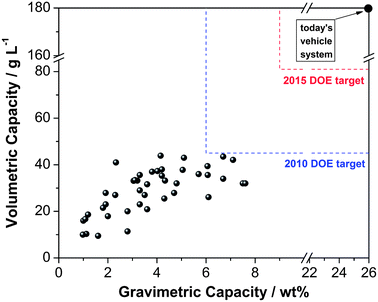
The nature of the primary interaction between hydrogen and MOFs is also unclear. Theoretically, the interaction forces between molecular hydrogen with any system include weak van der Waals forces, electrostatic interactions, orbital interactions, and non-classical sigma bonding (metal–dihydrogen complexes or the “Kubas complex”).133 In the case of MOFs, INS data indicated that there are two hydrogen-binding sites in MOF-5, with the stronger binding site associated with the metal–oxide cluster and the weaker one with the organic linker.9 These conclusions have been supported by both neutron powder diffraction and molecular dynamics simulation.141,142 The IR spectroscopic study conducted by Bordiga et al. demonstrates that the interaction between hydrogen and MOF-5 is largely due to van der Waals interactions with the internal wall structure and to weak electrostatic forces associated with the metal–oxide cluster.143 The isosteric heat of adsorption for hydrogen uptake in most MOFs lies in the range of 3.5 to 6.5 kJ mol−1, and these values tend to decrease with increasing amount of hydrogen due to the formation of a hydrogen monolayer on available surfaces.1
Clearly, the inherently weak interaction between hydrogen and MOFs would not meet the interaction requirement discussed above. Although the introduction of unsaturated metal sites into MOFs is an effective way to increase the hydrogen binding energy, the enhancement of hydrogen uptake in this way is limited due to the short-range nature of this interaction. In the case of HKUST-1, the enhancement is only around 1 wt% if each copper open site can bind one hydrogen molecule, and the enhanced uptake is limited to a very narrow pressure range (below 0.3 bar),128 leading to poor delivery capacity.65 Spillover might be a plausible method to strengthen the hydrogen binding were it not for the unpredictable hydrogen uptake enhancement factor. In contrast with the initial impressive report (enhancement factor of 8 for IRMOF-8),50 other studies only show a moderate effect (enhancement factor of 2.08–3.2).42,58,66 In addition, the usage of expensive and environmentally harmful heavy transition metals would greatly limit large scale application of this method.
Introducing charges into the MOFs would be a good method to increase the interaction. Since there is neither charge nor dipole moment in the dihydrogen molecule, the highest-energy interactions between a point charge and hydrogen are through the quadrupole moment, which is ∼3.5 kJ mol−1 at 3 Å separation, and via charge-induced dipole interaction, with an energy ∼6.8 kJ mol−1 at 3 Å separation.133 Calculations done by Garberoglio et al. find that electrostatic charges on MOFs would substantially increase the hydrogen uptake at 77 K and low pressure, but the effect on high pressure uptake is only marginal, with minimal effect on room temperature hydrogen uptake.144 Eddaoudi et al. have prepared ionic MOFs that show high hydrogen uptake and isosteric heat of adsorption, which, according to their explanation, is due to the narrow pore and highly localized charge density.43,79 The corresponding calculation study confirmed the speculation that polarization interactions are significantly enhanced by the presence of a charged framework with confined pores, which makes these MOFs excellent hydrogen storage candidates.145 The same conclusion has been drawn based on theoretical study of other charged carbon materials. GCMC simulation on charged single-walled carbon nanotubes demonstrates that a hydrogen uptake increase of ∼10%–20% for 298 K and 15%–30% for 77 K is achievable in realistically charged (0.1 e per carbon atom) nanotubes compared to uncharged ones.146 In order to achieve the DOE targets, however, the charges on the nanotubes need to be unrealistically large, which is both theoretically and experimentally impossible. A more optimistic conclusion is given by the first-principles calculations on charged fullerenes, in which the binding strength for hydrogen could be enhanced to a desirable range for potential near ambient applications with a maximum storage capacity of up to ∼8.0 wt%.147
4.3. Optimizing sorption isotherms
One of the special properties of MOFs that distinguishes them from the other porous materials is their framework-flexibility, which stems from ligand rotation, weak interactions (hydrogen bond, π–π stacking), etc.148 Zhao et al. designed a nanoporous MOF which shows hysteresis in hydrogen adsorption/desorption, a behavior that could be used to adsorb hydrogen at high pressures but store at lower pressures.71 The same hysteresis behavior has been observed in MOFs with strong hydrogen binding parts and some interpenetrated dynamic MOFs.69,73,85 Recently, Choi et al. demonstrated a broadly hysteretic hydrogen sorption in a microporous MOF, resulting a hysteresis loop with a width of 13 bar.10 Due to the weak interaction between hydrogen and the MOF, they attributed this hysteresis to the phase transitions in the MOF material.For hydrogen storage applications, however, the opposite trend in hysteresis would be more useful, leading to improved usable storage capacity due to a clean release (Fig. 5). There is no such sorption isotherm existing among the currently known isotherm types. In order to reach this optimized sorption isotherm, other methods besides pressure can be used to trigger the gas delivery, such as raising the temperature or exposure to UV light.107

5. Conclusions
The main hurdle to increase the hydrogen uptake in MOFs is the weak interaction between dihydrogen and the framework; this remains largely a challenge despite the recent surge in such studies.The DOE targets for on-board hydrogen storage pose a formidable challenge to those who are interested in solving such a fundamental but rewarding problem. The on-board hydrogen storage goal can only be achieved if theorists and experimentalists work together to find revolutionary systems based on basic studies including those reviewed above.
Acknowledgements
This work was supported by the U.S. Department of Energy (DE-FC36-07GO17033), the U.S. Defense Logistics Agency (N00164-07-P-1300), and the U.S. National Science Foundation (CHE-0449634). We thank David J. Collins and Mary E. O'Donnell for their helpful discussions.References
- A. W. C. van den Berg and C. O. Areán, Chem. Commun., 2008, 6, 668 RSC
.
- L. Schlapbach, MRS Bull., 2002, 27, 675 CAS
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef
- DOE Office of Energy Efficiency and Renewable Energy Hydrogen, Fuel Cells & Infrastructure Technologies Program Multi-Year Research, Development and Demonstration Plan, available at: http://www.eere.energy.gov/hydrogenandfuelcells/mypp.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283 CrossRef CAS
- B. Bogdanović and G. Sandrock, MRS Bull., 2002, 27, 712 CAS
- C. P. Baldé, B. P. C. Hereijgers, J. H. Bitter and K. P. de Jong, J. Am. Chem. Soc., 2008, 130, 6761 CrossRef CAS
- S. I. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111 CrossRef CAS
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS
- H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848 CrossRef CAS
- B. L. Chen, S. Q. Ma, E. J. Hurtado, E. B. Lobkovsky, C. D. Liang, H. G. Zhu and S. Dai, Inorg. Chem., 2007, 46, 8705 CrossRef CAS
- J. Y. Lee, J. Li and J. Jagiello, J. Solid State Chem., 2005, 178, 2527 CrossRef CAS
- J. T. Culp, S. Natesakhawat, M. R. Smith, E. Bittner, C. Matranga and B. Bockrath, J. Phys. Chem. C, 2008, 112, 7079 CrossRef CAS
- H. Chun, H. Jung, G. Koo, H. Jeong and D.-K. Kim, Inorg. Chem., 2008, 47, 5355 CrossRef CAS
- J. Y. Lee, L. Pan, S. P. Kelly, J. Jagiello, T. J. Emge and J. Li, Adv. Mater., 2005, 17, 2703 CrossRef CAS
- D. F. Sun, D. J. Collins, Y. X. Ke, J. L. Zuo and H. C. Zhou, Chem.–Eur. J., 2006, 12, 3768 CrossRef CAS
- J. A. R. Navarro, E. Barea, J. M. Salas, N. Masciocchi, S. Galli, A. Sironi, C. O. Ania and J. B. Parra, Inorg. Chem., 2006, 45, 2397 CrossRef CAS
- J. Y. Lee, D. H. Olson, L. Pan, T. J. Emge and J. Li, Adv. Funct. Mater., 2007, 17, 1255 CrossRef CAS
- M. Dincă, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904 CrossRef CAS
- W. B. Yang, X. Lin, J. H. Jia, A. J. Blake, C. Wilson, P. Hubberstey, N. R. Champness and M. Schröder, Chem. Commun., 2008, 3, 359 RSC
- B. L. Chen, S. Q. Ma, F. Zapata, F. R. Fronczek, E. B. Lobkovsky and H. C. Zhou, Inorg. Chem., 2007, 46, 1233 CrossRef CAS
- L. Pan, M. B. Sander, X. Y. Huang, J. Li, M. Smith, E. Bittner, B. Bockrath and J. K. Johnson, J. Am. Chem. Soc., 2004, 126, 1308 CrossRef CAS
- D. F. Sun, Y. X. Ke, T. M. Mattox, B. A. Ooro and H. C. Zhou, Chem. Commun., 2005, 43, 5447 Search PubMed
- L. Q. Ma, J. Y. Lee, J. Li and W. B. Lin, Inorg. Chem., 2008, 47, 3955 CrossRef CAS
- X. Lin, J. H. Jia, X. B. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358 CrossRef
- M. Dincă and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172 CrossRef CAS
- M. Dincă, A. Dailly, C. Tsay and J. R. Long, Inorg. Chem., 2008, 47, 11 CrossRef CAS
- M. Dincă, W. S. Han, Y. Liu, A. Dailly, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2007, 46, 1419 CrossRef CAS
- F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 1833 CrossRef CAS
- S. M. Humphrey, J. S. Chang, S. H. Jhung, J. W. Yoon and P. T. Wood, Angew. Chem., Int. Ed., 2007, 46, 272 CrossRef CAS
- X. D. Guo, G. S. Zhu, Z. Y. Li, F. X. Sun, Z. H. Yang and S. L. Qiu, Chem. Commun., 2006, 30, 3172 Search PubMed
- J. H. Jia, X. Lin, A. J. Blake, N. R. Champness, P. Hubberstey, L. M. Shao, G. Walker, C. Wilson and M. Schröder, Inorg. Chem., 2006, 45, 8838 CrossRef CAS
- J. H. Jia, X. Lin, C. Wilson, A. J. Blake, N. R. Champness, P. Hubberstey, G. Walker, E. J. Cussen and M. Schröder, Chem. Commun., 2007, 8, 840 Search PubMed
- J. H. Yoon, S. B. Choi, Y. J. Oh, M. J. Seo, Y. H. Jhon, T. B. Lee, D. Kim, S. H. Choi and J. Kim, Catal. Today, 2007, 120, 324 CrossRef CAS
- S. B. Choi, M. J. Seo, M. Cho, Y. Kim, M. K. Jin, D. Y. Jung, J. S. Choi, W. S. Ahn, J. L. C. Rowsell and J. Kim, Cryst. Growth Des., 2007, 7, 2290 CrossRef CAS
- C. Yang, X. P. Wang and M. A. Omary, J. Am. Chem. Soc., 2007, 129, 15454 CrossRef CAS
- B. Panella, M. Hirscher, H. Pütter and U. Müller, Adv. Funct. Mater., 2006, 16, 520 CrossRef CAS
- B. Xiao, P. S. Wheatley, X. B. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203 CrossRef CAS
- A. G. Wong-Foy, A. J. Matzger and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 3494 CrossRef CAS
- J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304 CrossRef CAS
- P. Krawiec, M. Kramer, M. Sabo, R. Kunschke, H. Fröde and S. Kaskel, Adv. Eng. Mater., 2006, 8, 293 CrossRef
- Y. W. Li and R. T. Yang, AICHE J., 2008, 54, 269 CrossRef CAS
- Y. L. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278 CrossRef CAS
- A. Dailly, J. J. Vajo and C. C. Ahn, J. Phys. Chem. B, 2006, 110, 1099 CrossRef CAS
- B. Panella and M. Hirscher, Adv. Mater., 2005, 17, 538 CrossRef
- M. Sabo, A. Henschel, H. Fröde, E. Klemm and S. Kaskel, J. Mater. Chem., 2007, 17, 3827 RSC
- J. L. C. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666 CrossRef CAS
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176 CrossRef CAS
- W. Zhou, H. Wu, M. R. Hartman and T. Yildirim, J. Phys. Chem. C, 2007, 111, 16131 CrossRef CAS
- Y. W. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 8136 CrossRef CAS
- Y. W. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 726 CrossRef CAS
- Q. R. Fang, G. S. Zhu, Z. Jin, Y. Y. Ji, J. W. Ye, M. Xue, H. Yang, Y. Wang and S. L. Qiu, Angew. Chem., Int. Ed., 2007, 46, 6638 CrossRef CAS
- S. Q. Ma, D. F. Sun, X. S. Wang and H. C. Zhou, Angew. Chem., Int. Ed., 2007, 46, 2458 CrossRef
- J. A. Rood, B. C. Noll and K. W. Henderson, Inorg. Chem., 2006, 45, 5521 CrossRef CAS
- I. Senkovska and S. Kaskel, Eur. J. Inorg. Chem., 2006, 4564 CrossRef CAS
- M. Dincă and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376 CrossRef CAS
- G. Férey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guégan, Chem. Commun., 2003, 24, 2976 RSC
- Y. Y. Liu, J. L. Zeng, J. Zhang, F. Xu and L. X. Sun, Int. J. Hydrogen Energy, 2007, 32, 4005 CrossRef CAS
- T. Loiseau, L. Lecroq, C. Volkringer, J. Marrot, G. Férey, M. Haouas, F. Taulelle, S. Bourrelly, P. L. Llewellyn and M. Latroche, J. Am. Chem. Soc., 2006, 128, 10223 CrossRef CAS
- M. Latroche, S. Surblé, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J. H. Lee, J. S. Chang, S. H. Jhung and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 8227 CrossRef CAS
- S. Surblé, F. Millange, C. Serre, T. Düren, M. Latroche, S. Bourrelly, P. L. Llewellyn and G. Férey, J. Am. Chem. Soc., 2006, 128, 14889 CrossRef CAS
- D. N. Dybtsev, H. Chun, S. H. Yoon, D. Kim and K. Kim, J. Am. Chem. Soc., 2004, 126, 32 CrossRef CAS
- H. R. Moon, N. Kobayashi and M. P. Suh, Inorg. Chem., 2006, 45, 8672 CrossRef CAS
- M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876 CrossRef CAS
- H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197 RSC
- Y. W. Li and R. T. Yang, Langmuir, 2007, 23, 12937 CrossRef CAS
- B. L. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745 CrossRef CAS
- Y. Liu, H. Kabbour, C. M. Brown, D. A. Neumann and C. C. Ahn, Langmuir, 2008, 24, 4772 CrossRef CAS
- P. M. Forster, J. Eckert, B. D. Heiken, J. B. Parise, J. W. Yoon, S. H. Jhung, J. S. Chang and A. K. Cheetham, J. Am. Chem. Soc., 2006, 128, 16846 CrossRef CAS
- E. Y. Lee and M. P. Suh, Angew. Chem., Int. Ed., 2004, 43, 2798 CrossRef CAS
- X. B. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012 CrossRef CAS
- P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvåg, Chem. Commun., 2006, 9, 959 Search PubMed
- S. Q. Ma, X. S. Wang, E. S. Manis, C. D. Collier and H. C. Zhou, Inorg. Chem., 2007, 46, 3432 CrossRef CAS
- S. Q. Ma, D. F. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H. C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858 CrossRef CAS
- S. Q. Ma and H. C. Zhou, J. Am. Chem. Soc., 2006, 128, 11734 CrossRef CAS
- X. S. Wang, S. Q. Ma, K. Rauch, J. M. Simmons, D. Q. Yuan, X. P. Wang, T. Yildirim, W. C. Cole, J. J. López, A. de Meijere and H. C. Zhou, Chem. Mater., 2008, 20, 3145 CrossRef CAS
- S. Q. Ma, X. S. Wang, C. D. Collier, E. S. Manis and H. C. Zhou, Inorg. Chem., 2007, 46, 8499 CrossRef CAS
- S. Q. Ma, X. S. Wang, D. Q. Yuan and H. C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4130 CrossRef CAS
- D. F. Sava, V. C. Kravtsov, F. Nouar, L. Wojtas, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 3768 CrossRef CAS
- J. Perles, M. Iglesias, M. A. Martín-Luengo, M. Á. Monge, C. Ruiz-Valero and N. Snejko, Chem. Mater., 2005, 17, 5837 CrossRef CAS
- Y. Wang, P. Cheng, J. Chen, D. Z. Liao and S. P. Yan, Inorg. Chem., 2007, 46, 4530 CrossRef CAS
- M. Kramer, U. Schwarz and S. Kaskel, J. Mater. Chem., 2006, 16, 2245 RSC
- A. G. Wong-Foy, O. Lebel and A. J. Matzger, J. Am. Chem. Soc., 2007, 129, 15740 CrossRef CAS
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS
- B. L. Chen, S. Q. Ma, E. J. Hurtado, E. B. Lobkovsky and H. C. Zhou, Inorg. Chem., 2007, 46, 8490 CrossRef CAS
- B. L. Chen, C. D. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390 CrossRef CAS
- J. C. Liu, J. Y. Lee, L. Pan, R. T. Obermyer, S. Simizu, B. Zande, J. Li, S. G. Sankar and J. K. Johnson, J. Phys. Chem. C, 2008, 112, 2911 CrossRef CAS
- B. L. Chen, S. Q. Ma, F. Zapata, E. B. Lobkovsky and J. Yang, Inorg. Chem., 2006, 45, 5718 CrossRef CAS
- H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521 CrossRef CAS
- H. Chun and J. Moon, Inorg. Chem., 2007, 46, 4371 CrossRef CAS
- X. Lin, A. J. Blake, C. Wilson, X. Z. Sun, N. R. Champness, M. W. George, P. Hubberstey, R. Mokaya and M. Schröder, J. Am. Chem. Soc., 2006, 128, 10745 CrossRef CAS
- K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 9604 CrossRef CAS
- B. L. Chen, X. B. Zhao, A. Putkham, K. Hong, E. B. Lobkovsky, E. J. Hurtado, A. J. Fletcher and K. M. Thomas, J. Am. Chem. Soc., 2008, 130, 6411 CrossRef CAS
- O. K. Farha, A. M. Spokoyny, K. L. Mulfort, M. F. Hawthorne, C. A. Mirkin and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 12680 CrossRef CAS
- D. F. Sun, Y. X. Ke, D. J. Collins, G. A. Lorigan and H. C. Zhou, Inorg. Chem., 2007, 46, 2725 CrossRef CAS
- H. Park, J. F. Britten, U. Mueller, J. Y. Lee, J. Li and J. B. Parise, Chem. Mater., 2007, 19, 1302 CrossRef CAS
- B. Kesanli, Y. Cui, M. R. Smith, E. W. Bittner, B. C. Bockrath and W. B. Lin, Angew. Chem., Int. Ed., 2005, 44, 72 CrossRef CAS
- E. Y. Lee, S. Y. Jang and M. P. Suh, J. Am. Chem. Soc., 2005, 127, 6374 CrossRef CAS
- Q. R. Fang, G. S. Zhu, M. Xue, Q. L. Zhang, J. Y. Sun, X. D. Guo, S. L. Qiu, S. T. Xu, P. Wang, D. J. Wang and Y. Wei, Chem.–Eur. J., 2006, 12, 3754 CrossRef CAS
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS
- G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS
- D. J. Collins and H. C. Zhou, J. Mater. Chem., 2007, 17, 3154 RSC
- Z. Q. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368 CrossRef CAS
- D. P. Broom, Int. J. Hydrogen Energy, 2007, 32, 4871 CrossRef CAS
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966 CrossRef
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastré, J. Mater. Chem., 2006, 16, 626 RSC
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS
- M. G. Nijkamp, J. E. M. J. Raaymakers, A. J. van Dillen and K. P. de Jong, Appl. Phys. A: Mater. Sci. Process., 2001, 72, 619 CrossRef CAS
- B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 43, 2209 CrossRef CAS
- A. Züttel, P. Sudan, P. Mauron and P. Wenger, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 941 CrossRef
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552 CrossRef CAS
- M. Hirscher and B. Panella, Scr. Mater., 2007, 56, 809 CrossRef CAS
- P. Bénard and R. Chahine, Scr. Mater., 2007, 56, 803 CrossRef CAS
- Q. Y. Wang and J. K. Johnson, J. Chem. Phys., 1999, 110, 577 CrossRef CAS
- V. K. Peterson, Y. Liu, C. M. Brown and C. J. Kepert, J. Am. Chem. Soc., 2006, 128, 15578 CrossRef CAS
- Y. Liu, C. M. Brown, D. A. Neumann, V. K. Peterson and C. J. Kepert, J. Alloys Compd., 2007, 446, 385 CrossRef
- B. Panella, K. Hönes, U. Müller, N. Trukhan, M. Schubert, H. Pütter and M. Hirscher, Angew. Chem., Int. Ed., 2008, 47, 2138 CrossRef CAS
- J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS
- H. Frost and R. Q. Snurr, J. Phys. Chem. C, 2007, 111, 18794 CrossRef CAS
- D. H. Jung, D. Kim, T. B. Lee, S. B. Choi, J. H. Yoon, J. Kim, K. Choi and S. H. Choi, J. Phys. Chem. B, 2006, 110, 22987 CrossRef CAS
- S. S. Han, W. Q. Deng and W. A. Goddard, Angew. Chem., Int. Ed., 2007, 46, 6289 CrossRef CAS
- T. Sagara, J. Ortony and E. Ganz, J. Chem. Phys., 2005, 123, 214707 CrossRef
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2007, 129, 5314 CrossRef CAS
- F. M. Mulder, T. J. Dingemans, M. Wagemaker and G. J. Kearley, Chem. Phys., 2005, 317, 113 CrossRef CAS
- B. L. Chen, M. Eddaoudi, T. M. Reineke, J. W. Kampf, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 11559 CrossRef CAS
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337 CrossRef CAS
- Y. Y. Sun, Y. H. Kim and S. B. Zhang, J. Am. Chem. Soc., 2007, 129, 12606 CrossRef CAS
- W. Zhou and T. Yildirim, J. Phys. Chem. C, 2008, 112, 8132 CrossRef CAS
- Q. Y. Yang and C. L. Zhong, J. Phys. Chem. B, 2006, 110, 655 CrossRef CAS
- R. C. Lochan, R. Z. Khaliullin and M. Head-Gordon, Inorg. Chem., 2008, 47, 4032 CrossRef CAS
- R. C. Lochan and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 1357 RSC
- A. Blomqvist, C. M. Araújo, P. Srepusharawoot and R. Ahuja, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20173 CrossRef CAS
- E. Klontzas, A. Mavrandonakis, E. Tylianakis and G. E. Froudakis, Nano Lett., 2008, 8, 1572 CrossRef
- S. S. Han and W. A. Goddard, J. Am. Chem. Soc., 2007, 129, 8422 CrossRef CAS
- A. Trewin, G. R. Darling and A. I. Cooper, New J. Chem., 2008, 32, 17 RSC
- E. Poirier, R. Chahine, P. Bénard, L. Lafi, G. Dorval-Douville and P. A. Chandonia, Langmuir, 2006, 22, 8784 CrossRef CAS
- H. Frost, T. Düren and R. Q. Snurr, J. Phys. Chem. B, 2006, 110, 9565 CrossRef CAS
- S. K. Bhatia and A. L. Myers, Langmuir, 2006, 22, 1688 CrossRef CAS
- T. Yildirim and M. R. Hartman, Phys. Rev. Lett., 2005, 95, 215504 CrossRef CAS
- T. Mueller and G. Ceder, J. Phys. Chem. B, 2005, 109, 17974 CrossRef CAS
- S. Bordiga, J. G. Vitillo, G. Ricchiardi, L. Regli, D. Cocina, A. Zecchina, B. Arstad, M. Bjørgen, J. Hafizovic and K. P. Lillerud, J. Phys. Chem. B, 2005, 109, 18237 CrossRef CAS
- G. Garberoglio, A. I. Skoulidas and J. K. Johnson, J. Phys. Chem. B, 2005, 109, 13094 CrossRef CAS
- J. L. Belof, A. C. Stern, M. Eddaoudi and B. Space, J. Am. Chem. Soc., 2007, 129, 15202 CrossRef CAS
- V. V. Simonyan, P. Diep and J. K. Johnson, J. Chem. Phys., 1999, 111, 9778 CrossRef CAS
- M. Yoon, S. Y. Yang, E. G. Wang and Z. Y. Zhang, Nano Lett., 2007, 7, 2578 CrossRef CAS
- K. Uemura, R. Matsuda and S. Kitagawa, J. Solid State Chem., 2005, 178, 2420 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Abbreviation, full name and chemical structure of the ligands listed in Table 1. See DOI: 10.1039/b808322n |
| This journal is © The Royal Society of Chemistry 2008 |