Fuel cell cathode catalyst layers from “green” catalyst inks†
Zhong
Xie
a,
Xinsheng
Zhao
a,
Makoto
Adachi
ab,
Zhiqing
Shi
a,
Tetsuya
Mashio
c,
Atsushi
Ohma
c,
Kazuhiko
Shinohara
c,
Steven
Holdcroft
*ab and
Titichai
Navessin
*a
aInstitute for Fuel Cell Innovation, National Research Council Canada, 4250 Wesbrook Mall, Vancouver, BC, Canada V6T 1 W5
bDepartment of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, BC, Canada V5A 1S6
cNissan Research Center, Nissan Motor Co. Ltd., 1, Natsushima, Yokosuka, Kanagawa 237-8523, Japan
First published on 7th July 2008
Abstract
Fuel cell cathode catalyst layers deposited from a water-based catalyst ink formulation, using high water content and minimum volatile organic compounds, are investigated. Cathodes fabricated from a dispersion medium containing 96 wt% water are compared with cathodes fabricated from conventional alcohol-based inks containing 1-propanol–water 3 : 1 (w/w). The morphology of the two catalyst layers are similar, as are electrochemically-active surface areas at relative humidities of 100, 70 and 30% RH. Oxygen reduction kinetics obtained under fully humidified H2/O2 conditions exhibit similar Tafel slopes, 67 ± 3 mV per dec. However, cathodes prepared from water-based inks exhibit a lower H2/air fuel cell performance under 100, 70 and 30% RH while its porosity, obtained using mercury porosimetry, is slightly higher. EIS measurements obtained under high current density indicate that the mass transport resistance of the water-based catalyst layer is lower, which is consistent with porosimetric data, and suggests that factors other than mass transport limit the performance of the water-based cathode. The protonic resistance of the catalyst layers was found to be 105 and 145 mΩ cm2 for the propanol- and water-based catalyst layers, respectively. The differences are more pronounced when RH is decreased from 100 to 30%. This trend is consistent with the observed decrease in fuel cell performance under conditions of lower RH, and indicates that the higher proton resistance of the water-based catalyst layer is the cause of its lower fuel cell performance.
1. Introduction
A high-performance membrane-electrode assembly (MEA) for proton-exchange membrane fuel cells (PEMFC) requires an effective three-phase boundary, high Pt utilization, good ion conduction, and facile reactant and water transport to and from active sites distributed throughout the catalyst layer. Due to improvements in both the development of component materials and the fabrication of MEAs, the power density of PEMFCs has substantially increased over the past decade. A vital component of the MEA is the catalyst layer, which is usually deposited from dispersions of catalyst and proton-conducting polymer in the form of an ink or slurry. A critical component of the ink is the dispersion medium, which governs physico-chemical properties such as aggregation, particle size and size distribution of the catalyst–ionomer agglomerates, the viscosity of the ink, the rate of evaporation, the morphology of the catalyst layer, and, ultimately, its performance. The selection of dispersion media, either in the form of pure solvent, solvent mixtures, with or without high boiling point additives, depends on the method of ink deposition, which subsequently is dictated by research or technological objectives. For example, screen-printing or roll-coating processes usually require a viscous ink formulation, containing a high solids content (>5 wt%) and high boiling point additives, such as ethylene glycol; whereas spray coating generally requires a low solids content (< 2 wt%) and alcohol/water-based solvents.Pioneering work on the influence of dispersion media on the catalyst layer structure, and its correlation with fuel cell performance was reported by Uchida et al.1,2 Here, the effect of dispersion media on the dispersability of ionomer in the ink was investigated. It was reported that perfluorosulfonated ionomer (PFSI) can be solubilized in organic solvents having a dielectric constant (ε) > 10; whereas colloidal solutions are obtained for solvents having a dielectric constant between 3 and 10. PFSI is reported to precipitate for solvents with ε < 3. Shin et al.3 reported that catalyst layers prepared via the colloidal method using n-butyl acetate (ε = 5.0) provided higher performing electrodes compared to those prepared by the solution method. It was suggested that the colloidal method yields a better continuity of the ionomer network and higher porosity of the catalyst layer. Kim et al.4 also concluded that the performance of gas-diffusion electrodes improved when less polar dispersion media, e.g., dipropyl ketone (ε = 12.6) or n-butyl acetate, was used; compared to methanol (ε = 33) and IPA (ε = 18). The improvement was ascribed to an increase in ionic conductivity of the catalyst layer and enlargement of the secondary pore structure. Chikasa et al.5 investigated the effect of glycerol on the micro/nano structure of catalyst layers fabricated using the decal transfer method. They found that decreasing the glycerol concentration increased the secondary pore volume, while proton conductivity decreased, resulting in an overall decrease in cell performance. Yang et al.6 investigated the effects of n-butyl acetate, iso-amyl alcohol, diethyl oxalate, ethylene glycol and ethylene glycol dimethyl ether as dispersion media on cell performance, discussing the results in terms of the distribution of Nafion ionomer on Pt particles and robustness of the catalyst structure.
As PFSI serves the role of physical binder in the catalyst layer, as well as proton conductor, the molecular interaction between the dispersion medium and PFSI is expected to play a key role in governing the microstructure and effective proton conductivity of catalyst layers. For more details on the state of understanding of the structure and morphology of Nafion, readers are referred to a comprehensive review by Mauritz and Moore.7 While literature reports on the morphology of the ionomer within the catalyst layer is sparse, there is a large body of literature concerning the conformation of Nafion in solution and the evolution of its morphology during their casting into films and membranes.8–10 A Nafion film prepared by casting a solution of Nafion from ethanol–water at room temperature, referred to as recast film, is reported to be brittle and can be re-dissolved in polar organic solvents. In contrast, as-received Nafion films are flexible, and insoluble in most polar solvents below 200 °C. The desirable properties of the as-received film can be reconstituted from solution-cast ionomer in the presence of a high boiling point solvent and using casting temperatures in excess of 160 °C, referred to as solution-processed films. As-received and solution-processed films possess similar degrees of crystallinity, whereas the recast film is amorphous. Earlier studies using small-angle neutron scattering (SANS)11 and small-angle X-ray scattering (SAXS)12 reveal that PFSI in water–alcohol is not completely dissolved as single chains but forms colloidal aggregates having an anisotropic structure with dimensions > 15 nm. The enhancement in crystallinity and physical properties of the solution-processed films is attributed to re-organization of the colloidal aggregates at elevated casting temperature to form a more entangled network. The brittleness and high solubility of the recast films led to the conclusion that the colloidal morphology of Nafion in solution remains intact in the recast state with little chain entanglement or coalescence between particles. Knowledge of the role of solvent and processing methods on PFSI morphology and film characteristics assists in the general understanding of the effect of dispersion media on the properties of a catalyst ink and on the ionomer network within the catalyst layer.
In the commercial production of catalyst layers, dispersion media used for preparing the ink typically comprises of water and organic solvents.13 Depending on the specific method of deposition and the resulting ink formulation required, the mass fraction of water in mixtures of dispersion media for catalyst inks ranges from ∼25 to 90 wt%. High water content (>90 wt%) ink formulae have been reported in patents.14,15 A survey of the literature indicates that dispersion media for catalyst inks are typically alcohol–water mixtures. In order to reduce the environmental impact of catalyst layer preparation, to comply with increasingly stringent governmental regulations, and to mitigate ink manufacturers from dealing with volatile toxic solvents and associated fire hazards, it would be prudent to reduce the use of volatile organic compounds during catalyst layer preparation. The printing industry, in response to increasing regulatory pressure, underwent a transition in the 1980's, from organic solvent-based inks to water-based inks.16 It is reasonable to assume that similar pressures will motivate MEA fabrication manufacturers to adopt “greener” chemical processes for catalyst layer deposition. The motivation for this work is therefore to understand the role of dispersion media in the catalyst ink and catalyst layer preparation in order to develop water-based ink formulae.
A question raised in the study of catalyst inks based on different dispersion media is what property of the ink does the dispersion medium control? How does this correlate to the architecture of the catalyst layer and how does the architecture influence the electrochemical kinetics of a fuel cell? In this study, cathode catalyst layers prepared from water-based catalyst inks are investigated. The dispersion medium comprises ∼97 wt% water, and is compared to an alcohol-based ink containing n-propanol–water (3 : 1 w/w ratio) – a mixture commonly employed in the industry and extensively reported in the literature.17–21 A systematic study of the cathode catalyst layers fabricated from the two ink formulae are reported, together with the results of a detailed electrochemical fuel cell analysis. The differences and similarities of the catalyst layer structures are identified and discussed.
2. Experimental
2.1 Preparation of catalyst inks and fabrication of membrane-electrode assemblies (MEA)
Both cathode and anode catalyst layers consisted of Pt/C catalyst (46.5 wt% Pt, TEC10E50E, Tanaka Kikinzoku Kogyo) (0.40 mg cm−2Pt) and 30 wt% Nafion ionomer (5 wt% solution, EW1000, Alfa Aeser). Catalyst inks were prepared using the same physical procedure but employing different dispersion media: 3 : 1 (w/w) nPA–H2O (Ink-p) and water (Ink-w), where nPA represents n-propanol. For ease of discussion, Ink-p and Ink-w represent propanol- and water-based inks. The ink compositions are summarized in Table 1. Cathode catalyst layers prepared from these inks are correspondingly termed CCL-p and CCL-w; and the corresponding membrane electrode assemblies are termed MEA-p and MEA–w, respectively.Nafion membranes (25 µm thick, NRE-211, DuPont) were used as-received. The anode catalyst layer for all MEAs was prepared from a catalyst ink based on 1 : 1 (w/w) MeOH–H2O; the cathode catalyst layers were prepared from inks based on different dispersion media. Catalyst-coated membranes (CCMs) having an electrode geometrical surface area of 25 cm2 were fabricated by direct, automated spray coating (EFD-Ultra TT Series). The variation of Pt loading was < 3% over the entire area of the CCM, i.e., < ± 0.01 mg Pt per cm2 for a 0.40 mg Pt per cm2 loading. The batch-to-batch reproducibility of fabricated CCMs was also verified to be > 97%, and the reproducibility of the measured electrochemical surface area (ESA) and fuel cell performance were also validated. Detailed procedures for the preparation of the catalyst inks, fabrication of CCMs, and evidence of reproducibility of CCMs are available in the ESI.†
2.2 Physical characterization of cathode catalyst layers (CCL)
SEM images of catalyst-coated membranes (CCM) were obtained using a scanning electron microscope (Hitachi S4700), by fracturing the CCMs after immersion in liquid nitrogen. The fractured samples were deposited with gold/palladium coating by sputtering.
Pore size and pore size distribution of catalyst layers were measured on half-CCMs using a Hg porosimeter (Micromeritics, Auto Pore IV9500,) using an applied pressure range of 177 to 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psi, which corresponds to pores 1 μm to 3 nm in diameter. Detailed procedures are available in the ESI.†
000 psi, which corresponds to pores 1 μm to 3 nm in diameter. Detailed procedures are available in the ESI.†
2.3 MEAs and single cell
CCMs, 25 cm2 in active geometric area, were sandwiched between two GDLs and assembled into a single cell possessing straight flow-through channels. The uniformity of cell compression was verified using pressure-sensitive film (Pressurex-Super Low, Sensor Products Inc.). The single cell was evaluated in a fuel cell test station (800 W, Hydrogenics). Humidification of gases was controlled by the temperature of the humidification tanks. The humidified gases were calibrated using the wet/dry bulb method. Gas flow rates were calibrated using a digital flow meter (Alicat Scientific). In order to verify the accuracy of the electrical output data, the cell current and potential were monitored using a 1 mΩ shunt and a high-resolution multimeter (Agilent 34401A). The high frequency resistance (HFR) of the cell was measured at 1 kHz using a digital resistance meter (Digital AC m-ohm tester 3566–01, Tsuruga Electric Corporation). Further details of the single cell are available in the ESI.†2.4 MEA testing protocol
MEAs were conditioned using a common procedure (details available in the ESI†). At the end of the conditioning period, the variation in the steady state potential was < ±1 mV.Electrochemical surface area (ESA) was measured by cyclic voltammetry (CV) using a potentiostat (1287A, Solartron Analytical). The anode and cathode were purged with humidified H2 (0.50 SLPM, 80 °C) and N2, (0.50 SLPM, 80 °C), respectively. Voltammograms were recorded using a 50 mV s−1 scan rate between 0.90 and 0.04 V vs. the anode, which served as the standard hydrogen electrode. The electrochemically active area was determined by integration of the voltammetric peak for hydrogen desorption and adsorption using 210 μC cm−2 as the conversion factor for charge to area.22
The H2 cross-over current density (iHX) was measured by potential step voltammetry under operating conditions similar to those described in the cyclic voltammetric measurements, except that H2 and N2 gases were continuously supplied to the cell during the measurement. The anode served as the reference standard hydrogen electrode. The cathode (working electrode) potential was stepped from 0.2 to 0.5 V in 0.1 V increments and 3 min equilibration times. The steady-state current density corresponds to the H2 cross-over current density (iHX).
The protonic conductivity in the catalyst layer was characterized using electrochemical impedance spectroscopy (EIS), conducted on MEAs using the same operating conditions as the potential step voltammetry measurements described above. In addition, the potentiostat was connected to a frequency response analyzer (1260A FRA, Solartron Analytical). The amplitude of sinusoidal current signal for AC impedance was set at 10 mV (root mean square) over a frequency range of 20 kHz to 0.1 Hz. To determine the ionic conductivity of the cathode catalyst layer, the cathode was purged with humidified N2, and the anode was fed with humidified H2 at a flow rate of 500 ml min−1. EIS spectra were measured under a DC bias potential of 0.45 V vs.SHE (the anode).
ORR kinetics were determined on single cells maintained at 80 °C and high flow rates of reactant gases (4 SLPM of H2 and 8 SLPM of O2). After holding the cell at OCP, the current load was increased to ∼1 A cm−2 and gradually decreased in intervals to 0.024 A cm−2. The cell potential was plotted against the log of current density, and the Tafel slope was extracted.
Beginning of life (BOL) polarization curves of the MEAs were evaluated at 80 °C under different relative humidities (70 and 30% RH for both anode and cathode) and back pressure (ambient pressure and 200 kPa absolute). The cell was supplied with 4.0 SLPM of H2 and 8.0 SLPM of air to the anode and cathode, respectively. After the OCP (>0.95 V) was achieved and the variation was <5 mV over 10 min, i.e., stable OCP, the polarization curve was obtained galvanostatically. Further details of the MEA testing protocol are available in the ESI.†
3 Results and discussion
3.1 Microstructure of the catalyst layer
SEM image of the catalysts layer are shown in Fig. 1. The catalyst layers exhibit similar features of catalyst aggregates, which can be generalized as being spherical in nature, ranging from 50 to 100 nm in diameter. These aggregates appear to be randomly distributed. The pore space between aggregates corresponding to 10–100 nm is in agreement with the pore size distribution measured by Hg porosimetry (see below). Significantly larger void spaces of ∼200–500 nm in size are also observed, but the porosimetry results (see below) indicate that the cumulative volume of these large voids is negligible. Moreover, these pores are believed to be an artifact formed when the samples are fractured. The SEM images, while revealing essential features of aggregation of catalyst agglomeration and overall morphology, do not reveal significant differences in microstructure between catalyst layers fabricated from propanol- and water-based inks. | ||
| Fig. 1 SEM images of catalyst layers fabricated from water-based ink (CCL-w, left) and propanol–water 3 : 1 (w/w) (CCL-p, right). Typical pore diameters of 50 nm (solid line circles) and 100 nm (dash line circles) are indicated. | ||
3.2 Porosimetry of the catalyst layer
The pore diameter of the catalyst layers, which is calculated from eqn (1), see ESI†, is plotted against the differential intrusion volume in Fig. 2. This reveals the pore size distribution (PSD) of the catalyst layers. The cumulative intrusion curves, plotted on the secondary y-axis of Fig. 2, represent the integral volume of Hg penetrated into the pore diameters from 1 μm to 5 nm. At high intrusion pressure, the total volume of Hg penetrated into the pore of catalyst layer represents the total pore volume (Vpore). The porosity of the different catalyst layers was subsequently calculated using eqn (2), see ESI.† | ||
Fig. 2
Pore size distribution of the half-catalyst-coated membrane samples fabricated from, water-based ink (○, —) and propanol–water 3 : 1 (w/w) (X, ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ). ). | ||
In general, the pore size distribution of the CCLs range from 10 to 200 nm. These represent meso (2–50 nm diameter) and macro (> 50 nm) pores as defined by the International Unit of Pure and Applied Chemistry (IUPAC). The peak pore size for CCL-p is ∼40 nm, whereas for CCL-w it is ∼30 nm. CCL-w shows a broader pore distribution profile than CCL-p, the importance of which on fuel cell performance will be discussed later. Porosity increases from 38% for CCL-p to 51% for CCL-w – the same trend as the increasing cumulative mercury intrusion at high pressure (small pore diameter, 5–10 nm) as shown in Fig. 2.
3.3 Electrochemical fuel cell analysis
 | ||
| Fig. 3 Typical cyclic voltammograms of cathode catalyst layers in MEAs. MEA-w (—) and MEA-p (). Conditions: 80 °C, 100% RH. | ||
Calculated ESAs and double layer capacitances (Cdl) at three different relative humidities, are listed in Table 2. The cathode catalyst layers exhibit the same distinguishing features of hydrogen adsorption/desorption, between 0.05 and 0.40 V, and oxide layer formation and oxide reduction between 0.50 and 0.90 V. These signature peaks are typical for Pt/C electrodes. The ESA values obtained from desorption peaks at 100% RH are 58.6 and 58.0 m2 g−1 for CCL-p and CCL-w, respectively. Similarly, ESA values are 46.2, and 47.0 m2 g−1 at RH 70%, and 40.4 and 39.7 at RH 30%. For a given set of adsorption and desorption derived data, obtained at a given RH, the ESAs for the MEAs lie within experimental error. These ESA values are averaged to be 58.3 ± 0.3 (for RH 100%), 46.6 ± 0.4 (RH 70%), and 40.0 ± 0.4 (RH 30%) m2 g−1. A similar trend is observed for ESA values obtained from the adsorption peaks. These ESA values are averaged to be 60.4 ± 3.2 (RH 100%), 49.6 ± 3.1 (RH 70%) and 44.5 ± 0.8 (RH 30%) m2 g−1, respectively.
The double layer capacitance (Cdl) for the MEAs at RH 100% are 45.8 and 44.8 mF cm−2 for CCL-p and CCL-w, respectively. Similarly, at RH 70%, the values of Cdl are 44.0 and 44.4 mF cm−2; and at RH 30%, they are 33.4 and 32.8 mF cm−2, respectively. For a given RH, the Cdl for the different catalyst layers are within 3% of each other. These Cdl values are averaged to be 45.3 ± 0.5 (RH 100%), 44.2 ± 0.2 (RH 70%) and 33.1 ± 0.3 (RH 30%) mF cm−2, respectively.
It is concluded that there is no significant deviation in the ESA, nor Cdl, between the different CCLs, which indicates that the available area of Pt in the cathode catalyst layer is the same for the two types of catalyst layers when operated under fuel cell conditions. However, both ESA and Cdl consistently decrease for both types of CCL when RH is reduced. This is expected because under lower RH, the water content within the CCL is reduced, together with the contact area between the ionomer and Pt.
| Limiting current density/mA cm−2 | ||
|---|---|---|
| E (W.E.)/Vvs.SHE | MEA-w | MEA-p |
| 0.2 | 1.01 | 1.6 |
| 0.3 | 1.06 | 1.88 |
| 0.4 | 1.28 | 2.14 |
| 0.5 | 1.34 | 2.43 |
| Linear fitting | ||
| Slope (mS cm2) | 1.21 | 2.75 |
| y-Intercept (mA cm−2) | 0.75 | 1.05 |
| Short resistance (Ω cm2) | 826 | 364 |
 | ||
| Fig. 4 Potential step voltammograms (a) for determination of hydrogen cross-over current density from the plot of limiting current density vs.electrode potential (b). MEA-w (—, ○) and MEA-p (, X). | ||
The values of iHX are 1.05 and 0.75 mA cm−2 for MEA-p and MEA-w, respectively. The corresponding values of electronic leakage resistances are 364 and 826 Ω cm2. When iHX is < 2.0 mA cm−2, the loss of efficiency caused by H2 cross-over is considered negligible, i.e., for a fuel cell operating under practical conditions (0.8–0.5 V, 0.06–1.10 A cm−2, see polarization curves below), iHX accounts for < 3% of current density (∼3% when operating at 60 mA cm−2, and ∼ 0.2% when operating at 1.0 A cm−2). Similarly, the values of electronic leakage resistance > 300 Ω cm2 would account for less than 3% of the Faradaic resistance, i.e., the Faradaic resistance in an operating cell is ∼10−1 to 101 Ω cm2. Therefore, the values of H2 cross-over and electronic leakage for the MEAs are considered negligible.
| Dispersion media | MEA-w/mΩ cm2 | MEA-p/mΩ cm2 |
|---|---|---|
| Resistance | ||
| RH 100% | 149 | 105 |
| RH 70% | 498 | 250 |
| RH 30% | 1023 | 763 |
 | ||
| Fig. 5 Nyquist plots of MEA-w (○) and MEA-p (X) operated under H2/N2 at 80 °C and RH 100% (a), 70% (b) and 30% (c). The experimental results (symbols) are fitted to the equivalent circuit (d). The fitted data are shown as solid lines. | ||
 | ||
| Fig. 6 Tafel plot of H2/O2 polarization at 80 °C, 100% RH cathode and anode. MEA-w (○) and MEA-p (X). | ||
 | ||
| Fig. 7 H2/air polarization curves at 80 °C and 70% RH (anode/cathode) (a) and iR compensated polarization curves (b). MEA-w (○) and MEA-p (X). | ||
In order to determine the effect of the cell resistance on the polarization curve, iRHFR – compensated curves for MEAs operated under 70% RH and ambient pressure are plotted in Fig. 7b. The compensated polarization curves correspond to the performance free from Ohmic resistances, i.e., the curves are limited only by electrochemical kinetics, mass transport resistances, and proton transport resistances in the catalyst layers. As expected, the iRHFR-compensated polarization curves exhibit higher electrode potentials for a given current density than uncompensated curves, but the difference in performance between the two MEAs remains significant. Since it was shown in the previous section that the MEAs possess similar ORR kinetics, the difference in the polarization curves are caused by either differences in mass transport resistance or proton resistance in the catalyst layers, or both.
When the RH of supplied gases was reduced to 30%, the performances of the MEAs significantly decreased and the difference between the MEAs was even more pronounced. The same trend of decreasing performance was observed: at 1 A cm−2 the voltages measured were 0.31 V for MEA-p and 0.18 V for MEA-w. This trend is consistent with the decrease in proton resistance in the cathode catalyst layer (Rp) from CCL-p to CCL-w (Fig. 8). The cathode catalyst layers fabricated from the two different dispersion media exhibit different degrees of tolerance towards drier operating conditions. This is discussed in the next section.

Operating MEAs at lower relative humidity enhances the rates of gas transport because the partial pressures of H2 and air are higher, as are the rates of permeation of gases due to the reduced likelihood of catalyst layer flooding, but the resistance to the proton transport in the ionomer (in the bulk membrane and CL) are significantly increased. Because the bulk membrane employed in the two types of MEA is the same, it is assumed that the membrane resistance under low RH is similar for both types of MEA. This leads to the conclusion that the difference in performance in Fig. 9 may be dominated by differences in Rp. To gain further insight into the predominant factors that cause the differences in performance, polarization curves were obtained at 200 KPa anode and cathode back pressure, and at 70% RH, such that the mass transport resistances are significantly reduced. The values of Rp for the two MEAs are sufficiently different so as to be observed (Fig. 10a) Under these conditions, the same trend of decreasing performance of the MEAs was observed: at 1.0 A cm−2: measured voltages are 0.65 V for MEA-p and 0.62 V for MEA-w. When compared to polarization curves obtained at ambient pressure, Fig. 7, the difference between the MEAs is much less pronounced. This is an indication that factors other than the kinetics and the mass transport resistances are affecting the performance, i.e., either membrane or ionomer resistances. iRHFR-compensated polarization curves (Fig. 10b) in which the contribution from the bulk membrane resistance is removed, exhibit small but still significantly different performances. At 1.0 A cm−2, the performance was 0.72 V for MEA-p and 0.69 V for MEA-w. The difference in performance is further confirmation that the dispersion media affects Rp and, consequently affects fuel cell performance.
 | ||
| Fig. 9 H2/air polarization curves at 80 °C and 30% RH (anode/cathode). MEA-w (○) and MEA-p (X). | ||
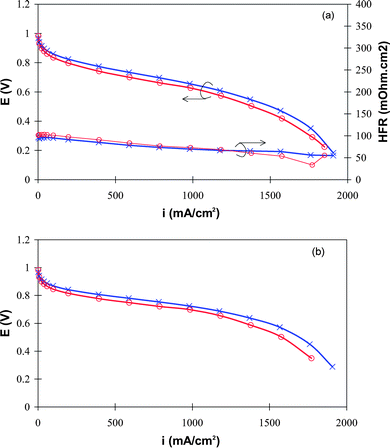 | ||
| Fig. 10 H2/air polarization curves at 80 °C, 70% RH (anode/cathode) and 200 KPa back pressures (a) and iR compensated polarization curves (b). MEA-w (○) and MEA-p (X). | ||
In order to further verify that, at 200 KPa back pressure and 70% RH, mass transport is not the rate-limiting process, in situelectrochemical impedance spectroscopy (EIS) was performed at 1.0 and 1.2 A cm−2, as shown in Fig. 11. For clarity of comparison, the high frequency intercepts, corresponding to the uncompensated resistance are off-set to zero. The diameter of the semi-circle or the intercept on the real impedance axis corresponds to the sum of the charge transfer resistance (Rct) and mass transport resistance (Rmt). Rct, which is due to ORR kinetics, is similar for the two MEAs, as proved previously (see Fig. 6), therefore, the differences in the impedance spectra are attributed to Rmt. An increase in mass transport resistance from MEA-w to MEA-p is concluded. However, this trend is opposite to the observed trend of increasing fuel cell performance, i.e., MEA-w, which exhibits the lower fuel cell performance, has the smaller resistance to mass transport, and indicates that differences in mass transport of oxygen in the cathode catalyst layers does not account for the differences in performance between MEAs when operated under 200 KPa back pressure. It is noteworthy that the trend of increasing resistance to mass transport, Rmt, from MEA-w to MEA-p correlates with the porosimetric data: CL-w possesses a much higher porosity (52%) than CL-p (38%).
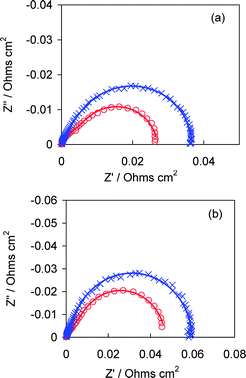 | ||
| Fig. 11 In situ electrochemical impedance spectra obtained at 70% RH while the cell is operated at (a) 1.0 and (b) 1.2 A cm−2. MEA-w (○) and MEA-p (X). | ||
In summary, the results from porosimetric and EIS measurements suggest that the two MEAs exhibit different porosity and potentially different gas mass transport resistances. However, when the MEAs were operated under high stoichiometric flow rates and elevated back pressures (200 KPa), their performances are not dominated by either the porosity of CCLs nor gas mass transport resistance. In fact, the poorer performing MEA (MEA-w) possessed a lower resistance to mass transport. Rather, EIS measurements reveal differences in ionic resistance of the catalyst layer (Rp). The observation of increasing Rp, from MEA-p to MEA-w, directly correlates to the trend of decreasing performance particularly when operated under low RH (30%). This leads to the conclusion that the dispersion media, in addition to affecting mass transport properties, significantly affects ionomer resistances in the cathode catalyst layer, and ultimately exerts a large impact on fuel cell performance. The fundamental causes of these experimental observations are further discussed below.
3.4 Discussion
The primary role of the dispersion media in the catalyst ink is to enable an intimate mixing of Pt/C and ionomer in order to allow creation of a three-phase interface (gas, electrolyte, electrode). A complex interaction exists between the dispersion media and the solid components (Pt, C and ionomer). It is believed that the overall morphology of the catalyst layer is primarily dictated by the microstructure of carbon and ionomer composite because, for the catalyst layer specification employed, the volume fractions for carbon (0.29 vol. fraction) and ionomer (0.23) are much larger than that of Pt (0.02). Given that the cathode catalyst layer fabrication processes are the same, the difference in porosity observed in Fig. 2 arise from the differences in the physico-chemical properties of the inks (viscosity, evaporation rate and dispersability), which gives rise to different microstructures during the spray deposition. This appears to be primarily a nanoscopic effect rather than a micro-scale effect since high magnification SEM images of the CCLs are similar. Another important aspect of this discussion is the correlation between the ionic resistance of the catalyst layer (Rp) with catalyst ink formulation. The effects of dispersion medium on the structural conformation of Nafion in solution have been extensively reported in the literature.11,12,25–32 According to measurements of the cohesive energy density of Nafion,26Nafion is characterized as possessing two Hildeband solubility parameters (δ): 9.7 (cal cm−3)1/2 for the perfluorocarbon backbone (δ1); and 17.3 (cal cm−3)1/2 for the sulfonated vinyl ether side chain (δ2). For comparison, the solubility parameters for n-propanol and water are 11.9 and 23.4 (cal cm−3)1/2, respectively. Using light scattering measurements, various authors27,29,32,33 report that two modes of particle size distributions are observed for Nafion dispersed in water–alcohol mixtures possessing δ >> δ1; a fast moving mode which corresponds to single polymer chains (hydrodynamic radii ∼102 nm), and a slow moving mode corresponding to aggregates of chains (radii >103 nm). The extent of aggregation, which may be loosely quantified by the ratio of the aggregates to single polymer chains, decreases when δ of the dispersion media is close to δ1. Dispersion media which are more compatible with the fluorous Nafion backbone such as N,N′-dimethylformamide (DMF, δ = 12.2 (cal cm−3)1/2) and N,N′-dimethylacetamide (DMAc, δ = 10.8 (cal cm−3)1/2) significantly reduce chain aggregation. On the other hand, dispersion media with low δ values such as n-hexane (7.2 (cal cm−3)1/2) are not compatible with δ2 and consequently Nafion does not disperse but forms precipitates. A trend of decreasing aggregation with decreasing δ of dispersion media from methanol–water, to 2-propanol–water and to DMA was reported.34 Lin et al.34 illustrated that the morphology of membranes is strongly influenced by the chain conformation of Nafion in solution. A high degree of chain aggregation in water–alcohol mixtures leads to the large scale hydrophobic and hydrophilic phase separation – an important feature to achieve high proton conductivity.Based on the trend observed in the literature, a qualitative correlation to our observed results reveals that for the water-based ink (CCL-w), δ for water is much larger than δ1 and δ2. Among the two ink formulae, the water-based ink is the least compatible with Nafion. It is hypothesized that Nafion in the ink is not as effectively dispersed, which presumably influences the morphology and connectivity of ionomers in the catalyst layer to the point that the rod-like protonic channels required for high proton conductivity35,36 is impeded, and hence Rp is lower for the CCL-w. In contrast, for the n-propanol-rich medium used to fabricate CCL-p, δ for n-propanol is closer in value to δ1 while the presence of water (22 wt%) assists in the association with the ionic groups. Presumably, this ink formulation disperses Nafion more effectively.
4. Conclusions
Comparing cathode catalyst layers prepared from the water-based ink with catalyst layers prepared from propanol-based inks the following observations are made of CCL-w. They exhibit (i) similar electrochemically active Pt surface area to CCL-p, (ii) similar ORR kinetics, (ii) lower proton conductivity, (iii) higher porosity, and (iv) lower resistance to mass transport. Employing a fuel cell testing protocol based on a judicious choice of electroanalytical methods, the performance-limiting factor of the water-based CCL is identified to be its proton conductivity. The dispersion medium is thus considered to exert a strong influence on the ionomer microstructure/network affecting the proton conductivity in the catalyst layer, and on the physico-chemical properties of the ink. Further study of the correlation of dispersion medium on the aggregation and morphology within these systems would appear warranted.Acknowledgements
The authors thank Dr Norbert Wagner of DLR, Germany for useful discussions and technical comments on EIS analysis.References
- M. Uchida, Y. Aoyama, N. Eda and A. Ohta, J. Electrochem. Soc., 1995, 142, 4143 CAS.
- M. Uchida, Y. Aoyama, N. Eda and A. Ohta, J. Electrochem. Soc., 1995, 142, 463 CAS.
- S.-J. Shin, J.-K. Lee, H.-Y. Ha, S.-A. Hong, H.-S. Chun and I.-H. Oh, J. Power Sources, 2002, 106, 146 CAS.
- J. H. Kim, H. Y. Ha, I. H. Oh, S. A. Hong and H. I. Lee, J. Power Sources, 2004, 135, 29 CrossRef CAS.
- M. Chisaka and H. Daiguji, Electrochim. Acta, 2005, 51, 4828.
- T. H. Yang, Y. G. Yoon, G. G. Park, W. Y. Lee and C. S. Kim, J. Power Sources, 2004, 127, 230 CrossRef CAS.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535 CrossRef CAS.
- R. B. Moore and C. R. Martin, Anal. Chem., 1986, 58, 2569 CrossRef CAS.
- R. B. Moore and C. R. Martin, Macromolecules, 1988, 21, 1334 CrossRef CAS.
- R. B. Moore and C. R. Martin, Macromolecules, 1989, 22, 3594 CrossRef CAS.
- P. Aldebert, B. Dreyfus and M. Pineri, Macromolecules, 1986, 19, 2651 CrossRef CAS.
- B. Loppinet, G. Gebel and C. E. Williams, J. Phys. Chem. B, 1997, 101, 1884 CrossRef CAS.
-
R. D. Mussell, US Pat., 5869416, 1999.
-
J. Denton, J. M. Gascoyne, D. Thompsett, US Pat., 5716437, 1998.
-
D. Heer, M. Pieter, D. Brujin, F. Albert, US Pat., 7186665, 2001.
- P. Laden, Chemistry and Technology of Water Based Inks, Blackie Academic & Professional, London, 1997 Search PubMed.
- X. Cheng, B. Yi, M. Han, J. Zhang, Y. Qiao and J. Yu, J. Power Sources, 1999, 79, 75 CrossRef.
- J. M. Song, S. Y. Cha and W. M. Lee, J. Power Sources, 2001, 94, 78 CrossRef CAS.
- S. Kontou, V. Stergiopoulos, S. Song and P. Tsiakaras, J. Power Sources, 2007, 171, 1 CrossRef CAS.
- V. A. Paganin, E. A. Ticianelli and E. R. Gonzalez, J. Appl. Electrochem., 1996, 26, 297 CAS.
- H. S. Park, Y. H. Cho, Y. H. Cho, C. R. Jung, J. H. Jang and Y. E. Sung, Electrochim. Acta, 2007, 53, 763 CrossRef CAS.
- K. Kinoshita and P. Stonehart, in Modern Aspects of Electrochemistry, ed. J. O'M. Bokris and B. E. Conway, Plenum Press, New York, 1977, vol. 12, pp. 183–266 Search PubMed.
- J. Jiang and A. Kucernak, J. Electroanal. Chem., 2004, 567, 123 CrossRef CAS.
- M. C. Lefebvre, R. B. Martin and P. G. Pickup, Electrochem. Solid–State Lett., 1999, 2, 259 CrossRef CAS.
- E. Szajdzinska-Pietek, S. Schlick and A. Plonka, Langmuir, 1994, 10, 2188 CrossRef CAS.
- R. S. Yeo, Polymer, 1980, 21, 432 CrossRef CAS.
- S. J. Lee, T. L. Yu, H. L. Lin, W. H. Liu and C. L. Lai, Polymer, 2004, 45, 2853 CrossRef CAS.
- P. A. Cirkel, T. Okada and S. Kinugasa, Macromolecules, 1999, 32, 531 CrossRef CAS.
- S. Jiang, K. Q. Xia and G. Xu, Macromolecules, 2001, 34, 7783 CrossRef CAS.
- S. J. Lee, T. L. Yu, H. L. Lin, W. H. Liu and C. L. Lai, Polymer, 2004, 45, 2853 CrossRef CAS.
- R. D. Lousenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 43, 421.
- W. H. Liu, T. Y. Yu, T. L. Yu and W. H. Liu, E-Polymers, 2007, 109, 1 Search PubMed.
- S. Wang, G. Sun, Z. Wu and Q. Xin, J. Power Sources, 2007, 165, 128 CrossRef CAS.
- H. L. Lin, T. L. Yu, C. H. Huang and T. L. Lin, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3044 CrossRef CAS.
- O. Diat and G. Gebel, Nat. Mater., 2008, 7, 13 CrossRef CAS.
- K. Schmidt-Rohr and Q. Chen, Nat. Mater., 2008, 7, 75 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental information on the preparation of catalyst inks, fabrication of MEAs, physical characterization of CCLs, single cell construction, MEA testing protocols, and reproducibility of MEAs. See DOI: 10.1039/b808613n |
| This journal is © The Royal Society of Chemistry 2008 |