New sorbents for hydrogen storage by hydrogen spillover – a review
Lifeng
Wang
and
Ralph T.
Yang
*
Department of Chemical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: yang@umich.edu; Fax: (+1 734) 764-7453
First published on 24th June 2008
Abstract
The utilization of hydrogen as an energy source or carrier for fuel-cell powered vehicles is limited by the lack of a safe and effective hydrogen storage system. Recent advances in the application of hydrogen spillover for hydrogen storage have provided a promising route for hydrogen storage. An overview of the progress made on hydrogen storage by spillover at ambient temperature on various adsorbents, including carbons, metal organic frameworks (MOFs) and other nanostructured materials, is given in this review. New techniques for facilitating hydrogen spillover that were developed in our laboratory are discussed, along with future directions.
 Lifeng Wang | Lifeng Wang received his BS degree in 2002 and PhD degree in 2007 in Chemistry from Jilin University, China. From 2007, he has been a postdoctoral fellow in the group of Professor Ralph T. Yang at the University of Michigan. |
 Ralph T. Yang | Ralph T. Yang is Dwight F. Benton Professor of Chemical Engineering at University of Michigan. He received his PhD from Yale University in 1971. He has published two books: “Gas Separation by Adsorption Processes” and “Adsorbents: Fundamentals and Applications”. He has received three national awards from AIChE, one ACS National Award, and he is a member of the U.S. National Academy of Engineering. |
1. Introduction
Hydrogen storage is the crucially missing link to a future “hydrogen economy.” Hydrogen can be stored in compressed tanks, in liquefied form, and in compressed tanks filled with sorbent materials. The most promising technique is the sorbent approach.1–3 The sorbent approach includes metal hydrides and adsorbents.4 For transportation applications, the US Department of Energy (DOE) has set 6.5 wt% and 62 kg H2 m−3 as the system capacity targets for on-board hydrogen storage in fuel cell applications in vehicles, at ambient temperature.3,5,6 The pressure is not specified, but 100 atm has been a nominal pressure for research. As a reference, for a compact passenger vehicle powered by fuel cell, 4 kg H2 is needed for a driving range of 400 km. To store hydrogen in gas tanks at the normal ambient temperature, 700 atm is needed for a compressed tank (of a reasonable size), which is unacceptably high. To store hydrogen in the liquid form requires large energy consumption for liquefaction at 20 K and it also suffers from the “boil-off” problem. Other examples for mobile applications are storage for portable electronics such as laptop computers and cell phones that are powered by fuel cells, as well as for non-automobile transportation applications such as motorcycles. The research targets in hydrogen storage capacities depend on the specific applications. The most challenging targets are for automobile application.Metal hydrides have been studied intensively for over four decades.7,8 Studies on adsorbents are more recent. Carbon materials, metal organic frameworks (MOFs) and other nanostructured and porous materials have received considerable interest due to their high surface areas.9–15 These materials have exhibited promising hydrogen storage capacities at 77 K. However, among the currently available candidate storage materials, none is capable of meeting the DOE criteria for personal transportation vehicles at moderate temperatures and pressures.16–18 The hydrogen adsorption capacities at the ambient temperature on all known sorbents are below 0.6–0.8 wt% at 298 K and 100 atm. This is true for all sorbent materials including the MOFs and templated carbons. In fact, the capacities for MOF-177 and MIL-101 are well below 0.7 wt% at 298 K and 100 atm, although these two materials are claimed to have the highest BET surface areas and storage capacities at 77 K.19–24
Based on experimental results and molecular orbital calculations, Yang et al. proposed to use the hydrogen “spillover” approach for hydrogen storage at ambient temperature.25,26Hydrogen spillover may be broadly defined as the transport (i.e., via surface diffusion) of dissociated hydrogen adsorbed or formed on a first surface onto another surface. The first surface is typically a metal (that dissociates H2) and the second surface is typically the support on which the metal is doped. Hydrogen spillover is a well documented phenomenon in the catalysis literature, and has been known in the catalysis community for over four decades, although it is still not well understood.27,28 Much evidence has been shown in the literature on its roles played in catalytic reactions. Very little has been studied on hydrogen storage by spillover at ambient temperature, although it is also known to occur at such temperatures. To exploit spillover for storage, among the key questions are whether spillover is reversible at ambient temperature and if the desorption rates at ambient temperature are fast enough for automotive applications.
By using the spillover approach, significant amounts of hydrogen storage capacities at 298 K have been obtained on a number of sorbents, including single-walled carbon nanotubes (SWNTs) and multi-walled carbon nanotubes (MWNTs)25,29,30 and graphite nanofibers,30,31 activated carbons,32,33 MOFs,34,35 covalent organic frameworks (COFs),23 zeolites,36 and mesoporous silica.37 It has been found in our studies that the hydrogen isotherms are reversible at ambient temperature and that the discharge rates are fast enough to meet the DOE targets on discharge rates. This review is aimed at summarizing the recent developments in hydrogen storage by spillover, as well as a fundamental understanding of the hydrogen spillover phenomena for hydrogen storage. The literatures on hydrogen storage in carbon, MOFs, metal hydride and other sorbents without using hydrogen spillover have been reviewed and discussed by many authors,3,4,7–15 which will not be focused on in this review.
2. Hydrogen storage by hydrogen spillover
2.1. Hydrogen storage on carbon by spillover
Since the first report on hydrogen storage in carbon nanotubes by Dillon et al.,38carbon materials have attracted considerable attention for hydrogen storage. High hydrogen adsorption capacities have been reported in various carbon nanostructures.39–42 However, the large amounts of hydrogen stored in carbon materials were disputed because of the difficulties in obtaining reproducible adsorption capacities from different laboratories.2 This discrepancy in the hydrogen storage abilities of carbon nanostructures is considered to be due to difficulties in accurate measurements, impurities in the carbon samples and in the hydrogen gas, and poor understanding of the hydrogen sorption mechanism.43–45 The controversy was partly or largely caused by the large experimental errors involved in the high-pressure measurements of hydrogen storage.4 This is true for both volumetric and gravimetric measurements. The volumetric technique is the most commonly used method for measuring high pressure isotherms.46 The experimental pitfalls in volumetric measurements for hydrogen storage have been discussed in detail by Tibbetts et al.47 and Darkrim et al.48 A more detailed analysis and discussion on all issues on the accuracies of volumetric measurements at high pressures have been given by us recently.49 A very small error in the experiment can cause large errors in the result. For example, in a typical setup, an artefact of 2.6 wt% adsorption can be caused by a 1 °C temperature rise. Calibration was sometimes not done properly. LaNi5 (a known metal hydride) was often used for calibration. The hydrogen uptake in LaNi5 (to form LaNi5H 6, or 1.37 wt% gain) occurs at 2 bar at 298 K, in one step. Thus, the high pressure range (2–100 atm), where the large errors occur, was not calibrated. Helium is typically used for calibration of dead spaces; ignoring adsorption by He also causes significant errors.49 We have established a calibration procedure using the commercially available superactivated carbon, AX-21 (with BET surface area ∼2600–2800 m2 g−1), as the calibration material,30,49 which yields 0.6 wt% storage at 100 atm and 298 K, with a slightly concave isotherm from 0–100 atm. The same result has been reported by several laboratories.50 When all errors were eliminated from the experimental measurements, the hydrogen storage capacities for all purified carbon nanotubes (both single-walled and multi-walled) were below 0.3 wt% at 298 K and 100 atm. More recently, it was claimed that an exceptionally high hydrogen storage capacity of 6.9 wt% at 77 K and 20 bars over all known sorbents was obtained on a microporous carbon (with a BET surface area of 3200 m2 g−1) that was synthesized by templating using zeolite beta.51 Our recent work showed that the hydrogen storage capacity in a microporous carbon with an ultra-high BET surface area of >3700 m2 g−1 was still less than 1 wt% at 298 K and 100 atm. On the basis of numerous theoretical studies and careful experimental validation, several reports have claimed that at ambient temperature the hydrogen storage capacities in pure carbon materials are all well below 1 wt%.52–54 However, recent results have shown that hydrogen storage in carbon materials can be significantly enhanced by spillover techniques.Mixing a catalytic material with an otherwise inert secondary material has been used to demonstrate hydrogen spillover by Srinivas and Rao.55 They studied the effect of H2 spillover on benzene hydrogenation over a Pt/C catalyst and a Pt/C catalyst diluted with carbon. From chemisorption results a direct observation of hydrogen spillover at room temperature was made.
Lueking and Yang investigated hydrogen storage on various carbon materials (SWCNTs, MWCNTs, graphite nanofiber, activated carbon and carbon fibers) by secondary spillover.30 It was found that a supported catalyst as a hydrogen source enhanced the overall hydrogen uptake of the carbon materials: by simple mixing of carbon nanotubes with supported palladium, the hydrogen uptake on carbon nanotubes was increased by a factor of three. The adsorption of spiltover hydrogen on carbon was the predominant factor in the magnitude of the overall hydrogen uptake.30
Secondary spillover provides a method to clearly delineate the role of carbon surface functionalities; maintaining the primary metal–carbon material and varying the secondary carbon establishes a constant hydrogen source to the secondary material. This eliminates several variables inherent to primary spillover, including doping efficiencies, carbon–metal interface, and metal content.
Conner and Falconer proposed that secondary spillover requires intimate contact between the two unlike materials and there may be an energy barrier to transfer hydrogen from one material to another.28
Lueking and Yang studied the hydrogen storage on MWNTs with various degree of catalyst removal (Ni, Mg).25 In that study, they observed that removal of the catalyst decreased the uptake from 0.6 wt% to below detection limits. Normalization by metal content and temperature-programmed desorption studies suggested hydrogen dissociation and subsequent spillover to the MWNT. It was found that metal–support interactions were key to the spillover.
To improve the contact between the spillover source and the secondary receptor, a bridge-building technique was proposed by Yang et al.58 The bridge building technique involves mixing the catalyst (a small amount) and the sorbent with a small amount of carbon precursor such as glucose, followed by a temperature programmed heating protocol to carbonize the precursor. By using this simple technique, carbon “nanobridges” can be built between a spillover source particle and a secondary receptor particle.
The bridge-building techniques were first applied to two typical carbon materials of SWNTs and superactivated carbon (AX-21) due to their relative chemical stabilities and high surface areas. In this case, a supported catalyst (Pd/C) served as the source of hydrogen atoms via dissociation and primary spillover and AX-21 or SWNTs was the secondary spillover receptor. Fig. 1 depicts the spillover process in a supported catalyst system. The spillover source is shown in Fig. 1a, along with an adsorbed hydrogen molecule. Fig. 1b is a schematic of the low-capacity receptor, which could be bundles of SWNTs or AX-21. The size of the hydrogen molecule precludes it from accessing the interstices in the SWNT bundle or additional micropores of AX-21. The molecular size may also prevent transport of hydrogen through any defects in an individual SWNT surface; thus, the adsorption at endohedral sites is dependent upon diffusion to uncapped ends and further transport inside the tube to these sites. Examples of primary and secondary spillover are shown schematically in parts c and d, respectively, of Fig. 1. Dissociation is assumed to take place on the metal particle, and atomic hydrogen spills over to the support. The atoms are transported to the receptorvia diffusion across the bridge and can access additional sites on the receptor. It is noted that the creation of more intimate contacts between the metal particle and the support may also contribute to primary spillover enhancement, as depicted in Fig. 1e.
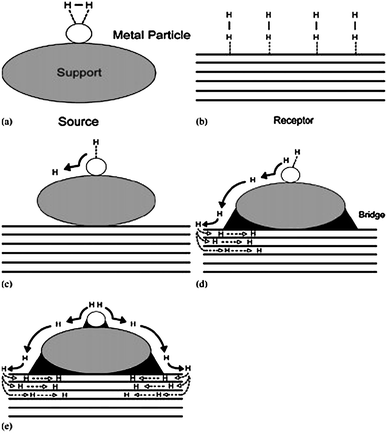 | ||
| Fig. 1 Hydrogen spillover in a supported catalyst system: (a) adsorption of hydrogen on a supported metal particle; (b) the low-capacity receptor; (c) primary spillover of atomic hydrogen to the support; (d) secondary spillover to the receptor enhanced by a physical bridge; (e) primary and secondary spillover enhancement by improved contacts and bridges. (Reprinted with permission from ref. 32. Copyright 2005 American Chemical Society). | ||
By using our bridge building technique, the hydrogen adsorption amounts were increased by a factor of 2.9 on the AX-21 receptor and 1.6 on the SWNT receptor at 298 K and 100 kPa. Similar results were also obtained at 10 MPa, indicating that the enhancement factor was a weak function of pressure. Moreover, reversibility was demonstrated through desorption and readsorption at 298 K.32
Another way to introduce metal onto the receptor material is by chemical-doping. Chemical doping has been developed extensively, particularly in catalysis. Compared with physical mixing, chemical doping produces identical samples and is more reproducible. Several studies showed that hydrogen uptake on nanostructured carbons (CNTs, active carbon, carbon nanofibers, etc.) was enhanced by doping with transition metals (Ni, Pd, Pt, etc.), and the maximum uptake could be achieved by optimizing adsorbents and treatment conditions.59–64
Back et al. investigated the hydrogen storage capacities of Pd-doped sepiolite-derived carbon nanofibers (SDCNs) at different Pd particle sizes and different Pd doping levels.59 It was found that the hydrogen storage capacity was dependent on the Pd particle size and doping amount as the Pd particle surface area and the carbon BET surface area changed with them. The maximum hydrogen uptake of about 0.59 wt% was obtained at 298 K and 90 bar for the 3 wt% Pd-doped SDCN due to the smaller Pd particle size and relatively high surface area of carbon.
Anson et al. prepared palladium-loaded single-walled carbon nanotubes and palladium-loaded MAXSORB activated carbon by reaction of the raw carbon support with Pd2(dba)3·CHCl3.60 A maximum hydrogen capacity of 0.7 wt% was obtained at 90 bar in a palladium-loaded MAXSORB sample, while the capacity for the raw MAXSORB at the same pressure was 0.42 wt%. They observed that the H/Pd atomic ratios in the palladium-loaded samples were always higher than in the bulk Pd. The spillover effect was considered as the cause for the high H/Pd atomic ratios.
Bettahar et al. investigated hydrogen storage on nickel catalysts supported on amorphous activated carbon.61 They found that the hydrogen storage capacity depended on the metal content and the temperature of pre-treatments.
Among the aforementioned catalysts, the reversible hydrogen capacities were all below 1.0 wt% at high pressure and room temperature. More recently, Kim et al. reported a higher hydrogen capacity of 2.8 wt% on Ni doped on multi-walled carbon nanotubes at a moderate temperature.64 But the hydrogen adsorbed on the material could only be released at temperatures higher than 340 K (340–520 K).
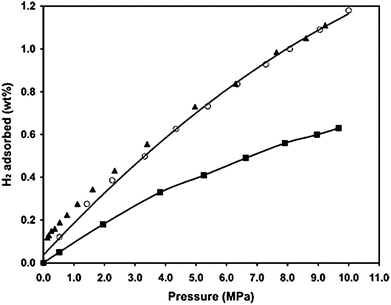 | ||
| Fig. 2 High-pressure hydrogen isotherms at 298 K for pure AX-21 (■), and the Pt/AX-21 sample: adsorption (○), and desorption (▲). (Reprinted with permission from ref. 33. Copyright 2007 American Chemical Society). | ||
Our recent studies indicated that uniform nanoparticles of Ru, Pt and Ni could be also effectively doped on porous carbon supports by using the ultrasonication assisted doping method.
As shown in Fig. 3, the equilibrium hydrogen adsorption isotherms on Norit activated carbon (NAC), 3 wt% Pt doped on Norit carbon prepared by hydrogen reduction, and 3 wt% Pt doped on Norit carbon prepared by plasma reduction are compared. The Norit carbon with a BET surface area of about 1000 m2 g−1 had a hydrogen storage capacity of 0.3 wt% at 298 K and 10 MPa. After doping the Norit carbon with 3 wt% Pt by hydrogen reduction, the hydrogen storage capacity was increased by about 50%, due to spillover. For the Pt-doped carbon treated with plasma, the hydrogen storage capacity was increased by approximately 270% from the value of the plain carbon. From TEM results and thermal stability tests, it was concluded that the Pt particles were highly dispersed and strongly anchored on the carbon supports. This large enhancement by plasma treatment was attributed to high dispersion of Pt particles and chemical bridges that were built between the Pt particles and the carbon surface, which enhanced the spillover.
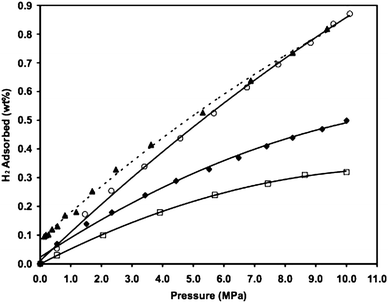 | ||
| Fig. 3 High-pressure hydrogen isotherms at 298 K for NAC (□), H2-reduced Pt/NAC-H (♦), and plasma-treated Pt/NAC-P sample: adsorption (○), and desorption (▲). (Reprinted with permission from ref. 73. Copyright 2007 American Chemical Society). | ||
In the preliminary study, glow discharge Ar plasma was used. The glow discharge was generated by applying 900 V to the planar electrodes. The temperature of the plasma was near the ambient temperature. Previous studies suggested that the ambient temperature during plasma treatment was the main reason for the high dispersion of metal particles. It is known that plasmas consist of ionized atoms and molecules as well as energized electrons. Other plasmas (microwave plasma, plasma spraying, radio frequency, etc.) and other gases (H2, O2, Air and CO2etc.) will give similar or even better results than the glow discharge Ar plasma. This would further reduce the costs of plasma treatment, and consequently the costs of sorbents for hydrogen storage.
2.2. Hydrogen storage on MOFs by spillover
Metal–organic frameworks (MOFs) are porous materials constructed by coordinate bonds between multidentate ligands and metal atoms or small metal-containing clusters. MOFs can be generally synthesized viaself-assembly from different organic linkers and metal nodules. Due to the variable building blocks, MOFs have very large surface areas, high porosities, uniform and adjustable pore sizes and well-defined hydrogen occupation sites. These features make MOFs promising candidates for hydrogen storage.13,14Yaghi et al. first reported a high hydrogen adsorption of 4.5 wt% on Zn4O(bdc)3 (bdc = 1,4-benzenedicarboxylate) at 77 K and less than 1 atm, which is known as MOF-5 or IRMOF-1.74 The reported hydrogen capacities were lower in their subsequent studies.75 The novelty of this work provided a promising candidate for hydrogen storage. A number of unique MOFs have been synthesized and evaluated for their hydrogen storage capacity.76–88 It has become clear that high surface area and pore volume are important factors for high hydrogen uptakes on MOFs at 77 K.77–80 The effects of surface area, pore volume and heat of adsorption on hydrogen uptake in MOFs were recently discussed by Snurr et al.81 Extensive work was directed toward the synthesis of MOFs with high surface areas and pore volumes. A series of isoreticular (meaning having the same underlying topology) metal organic frameworks, Zn4O(L)3, were constructed by changing the different linking zinc oxide clusters with linear carboxylates L, so as to get a high porosity.75–77 Recently, MOF-177 (Zn4O(BTB)2) was formed by linking the same clusters with a trigonal carboxylate. MOF-177 was claimed to have a high Langmuir surface area of 5640 m2 g−1, and the highest hydrogen storage of 7.5 wt% H2 at 77 K and 70 bar.21,22 The more meaningful surface area, BET surface area, for MOF-177 is around 3000 m2 g−1. More recently, Férey et al. reported a nanoporous chromium terephthalate-based material (MIL-101) with the highest Langmuir surface area (4500–5900 m2 g−1) among all MOFs. It was reported that the hydrogen storage capacities in this material at 8 MPa were 6.1 wt% at 77 K, and 0.43 wt% at 298 K.19,20 Although these MOFs have remarkable hydrogen capacities at 77 K, no significant hydrogen storage capacities were obtained with the MOFs at room temperature.23,24
2.2.1.1. Spillover by physical mixing. Significant hydrogen storage capacities at ambient temperature on nanostructured carbon materials have been achieved by using secondary spillover. It is expected that the hydrogen storage on MOFs at room temperature can also be much improved by hydrogen spillover techniques. The hydrogen spillover was first applied to MOF-5 (also known as IRMOF-1) and IRMOF-8, which were constructed by linking tetrahedral [Zn4O]6+ clusters with linear carboxylates.34
For spillover experiments, a commercial catalyst containing 5 wt% Pt supported on active carbon was used for dissociation of H2. Active carbon was the primary receptor for hydrogen spillover. The catalyst and the secondary spillover receptor (MOF-5 or IRMOF-8) (at a weight ratio of 1 : 9) were ground together to produce the physical mixture.
As shown in Fig. 4, pure MOF-5 had a hydrogen storage capacity of 0.4 wt% at 298 K and 10 MPa. Similar results were also observed by Panella and Hirscher82 and Rowsell et al.75 By thoroughly mixing MOF-5 with Pt/AC catalyst, the H2storage capacity of MOF-5 was enhanced to 1.5 wt% at 10 MPa. Considering the relatively low H2 uptake of Pt/AC (1.0 wt% at 10 MPa) and the small amount of Pt/AC in the mixture (10 wt%), the hydrogen uptake on MOF-5 was increased by a factor of 3.3. The enhanced storage capacity was clear evidence for secondary spillover of H atoms to MOF-5, as a secondary receptor. It is noted that there was no apparent saturation value for the physical mixture at 10 MPa. Reversibility was assessed by measuring the desorption branch down to 1 atm. The second adsorption branch was in complete agreement with the first adsorption branch.
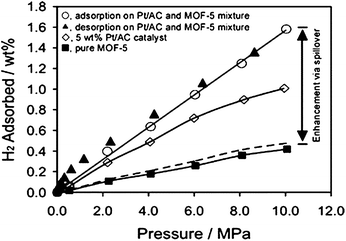 | ||
| Fig. 4 High-pressure hydrogen isotherms at 298 K for MOF-5. Dotted line is prediction based on the weighted average of the mixture. (Reprinted with permission from ref. 34. Copyright 2006 American Chemical Society). | ||
Similarly remarkable enhancement of H2storage capacity by spillover was also observed on IRMOF-8. The H2 uptake on pure IRMOF-8 was 0.5 wt% at 298 K and 10 MPa. By physically mixing IRMOF-8 with Pt/AC, the hydrogen uptake on IRMOF-8 was increased to 1.8 wt% under the same conditions. This uptake is equivalent to 8.3 H2 (or 16.6 H atoms) per formula unit, which is well below the saturation of IRMOF-8.74 Furthermore, the adsorption was also totally reversible and did not show any saturation up to 10 MPa, similar to results on MOF-5.
2.2.1.2. Spillover by bridge-building techniques. Secondary spillover requires intimate contacts between the two unlike materials because there exist tremendous physical/energy barriers for surface diffusion of hydrogen atoms from one material to another. It has been demonstrated that bridge-building technique could facilitate secondary spillover and hence increase the storage capacities in nanostructured carbon materials. Yang and Li further extended the bridge-building technique to MOFs.35
This work was first focused on IRMOF-1 and IRMOF-8, whose hydrogen uptakes had been proved to be enhanced via secondary spillover induced by physical mixing. In previous work, it was found that carbon bridges could be built between an activated carbon and a Pd/AC catalyst by carbonizing an added glucose precursor at 673 K.32,58 It is expected that carbon bridges can also be formed by carbonization of a hydrocarbon precursor that was previously introduced into a physical mixture of the MOF and catalyst. However, it is difficult to build carbon bridges in MOFs by using glucose precursor because MOFs are thermally unstable at temperatures >573 K.74 Yang et al. used sucrose instead of glucose as the carbon precursor. The ternary physical mixture of sucrose, Pt/AC catalyst, and MOF was first heated in He to 473 K (melting point of sucrose = 463 K) for 3 h to allow the sucrose to melt thoroughly and to fill the interstices (by capillarity) between the particles of the catalyst and the MOF (Fig. 5). Then the temperature was finally increased to 523 K to carbonize the sucrose. This temperature program was effective for building carbon bridges and avoiding the collapse of the MOF structure. Thermogravimetric analysis (TGA) confirmed the complete carbonization of sucrose by this heating protocol. X-Ray diffraction (XRD) and N2 adsorption results indicated that the structures of MOF materials were well retained during the bridge-building treatment.
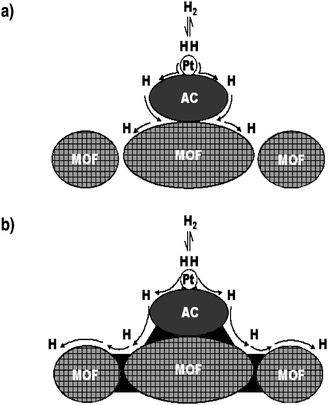 | ||
| Fig. 5 (a) Primary spillover of atomic hydrogen from Pt metal to the activated carbon support and secondary spillover to the MOF receptor that has limited contacts with the support. (b) Facilitated primary and secondary spillover by using carbon bridges (dark black zone). (Reprinted with permission from ref. 35. Copyright 2006 American Chemical Society). | ||
By building carbon bridges, the bridged sample exhibited a hydrogen uptake of 3 wt% at 10 MPa. It represented an enhancement factor of 2 compared with the physical mixture without bridges. This enhancement indicated the built bridges further facilitated secondary spillover of hydrogen atoms from the catalyst to the surface of the MOF material. Compared with pure IRMOF-1, the hydrogen adsorption amount of bridged IRMOF-1 has been enhanced by a factor of 7.5. These results indicated that the creation of carbon bridges was crucially important for achieving a high hydrogen adsorption capacity by spillover.
Similar enhancement of hydrogen storage by spillover was also observed on IRMOF-8. As shown in Fig. 6, by using the bridge-building technique, the H2 uptake was increased from 0.5 wt% on pure IRMOF-8 to 4 wt% at 10 MPa on bridged IRMOF-8. This uptake amount is equivalent to ∼34 H per formula unit (Zn4O(C12H6O4)3), which is much lower than the total number of atoms in IRMOF-8, assuming 1 H per surface atom. Furthermore, the ab initio molecular orbital calculation results indicated that it was favorable for one surface atom (such as oxygen) to adsorb two H atoms at the same time. Thus, the theoretical maximum storage would be at least 6.5 wt%. No apparent saturation value was approached for the bridged sample; as the isotherm was linear even at 10 MPa. The absence of a saturation value suggests that a further increase in capacity can be expected at pressures >10 MPa; for example, 6 wt% storage is expected at 15 MPa, a viable pressure for practical automotive applications.
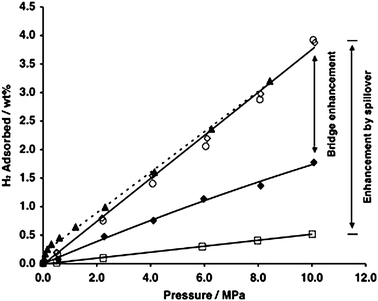 | ||
| Fig. 6 High-pressure hydrogen isotherms at 298 K for pure IRMOF-8 (□), Pt/AC and IRMOF-8 physical mixture (1 : 9 weight ratio; ♦) and for a bridged sample of Pt/AC-bridges-IRMOF-8: first adsorption (○), desorption (▲), and second adsorption (◇). (Reprinted with permission from ref. 35. Copyright 2006 American Chemical Society). | ||
| BET surface area/m2 g−1 | H2 at 77 K, 1 atm (wt%) | H2 at 298 K, 100 atm (wt%) | Bridged sample, H2 at 298 K, 100 atm (wt%) | ΔH (bridged)/kJ mol−1 | |
|---|---|---|---|---|---|
| IRMOF-8 | 548 | 1.4 | 0.4 | 2.2–4.0 | −21 |
| COF-1 | 628 | 1.1 | 0.3 | 0.7 | −7 |
| HKUST-1 | 1296 | 2.2 | 0.3 | 1.1 | −9 |
| MIL-101 | 2930 | 1.8 | 0.5 | 1.5 | −13 |
| MOF-177 | 3100 | 1.5 | 0.6 | 1.5 | −10 |
Recently, two kinds of new MOFs, MIL-10119 and HKUST-1,89 have received more attention due to their relative stabilities in air when compared with other MOFs. HKUST-1 and MIL-101 were synthesized in the presence of H2O, so their structures are stable upon water adsorption. HKUST-1 is a copper benzenetricarboxylate porous material reported by Williams et al.89 Recent research showed that it was a good adsorbent for H2 and NO storage.90 The high gas capacities in HKUST-1 were due to the presence of a metal site in the walls of the material that could interact strongly with gas molecules.91 MIL-101 was synthesized from benzene-1,4-dicarboxylate and trimetric chromium(III) octahedral clusters by Férey's group. It was reported to have the largest surface area (Langmuir surface area, 4500–5900 m2 g−1) among all MOFs.19 A high hydrogen storage capacity of 3.75 wt% was obtained on MIL-101 at 77 K and 2 MPa first, then it was reported that the hydrogen storage capacity in this material at 8 MPa was 6.1 wt% at 77 K, and 0.43 wt% at 298 K.20
Yang and Li investigated the hydrogen capacities of MIL-101 and HKUST (Fig. 7 and 8).23 The hydrogen uptakes on the pure MIL-101 and HKUST are 0.51 wt% and 0.35 wt%, respectively. By simply mixing with a small amount of Pt/AC catalyst (at 9 : 1 mass ratio), their hydrogen uptakes were enhanced to 0.9 wt% and 0.7 wt% up to 10 MPa. By building carbon bridges between the MOFs and the Pt/AC catalyst, the hydrogen capacities were further enhanced to 1.5 wt% on bridged-MIL-101 and 1.1 wt% on bridged-HKUST. There was no apparent saturation for the bridged samples and the adsorption in the bridged samples was fully reversible at 298 K, as found previously on the bridged IRMOFs.
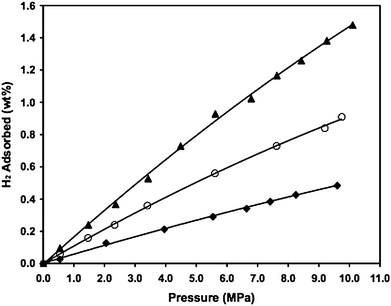 | ||
| Fig. 7 High-pressure hydrogen adsorption at 298 K for the MIL-101 samples. ♦: pure MIL-101; ○: Pt/AC and MIL-101 physical mixture; ▲: MIL-101–bridges–Pt/AC. (Reprinted with permission from ref. 23. Copyright 2008 Wiley). | ||
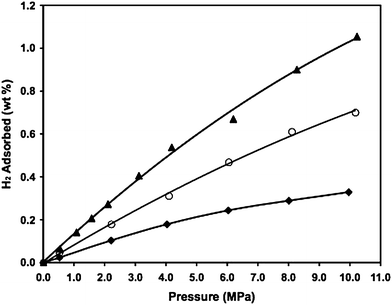 | ||
| Fig. 8 High-pressure hydrogen adsorption at 298 K for the HKUST-1 samples. ♦: pure HKUST-1; ○: Pt/AC and HKUST-1 physical mixture; ▲: HKUST-1–bridges–Pt/AC. (Reprinted with permission from ref. 23. Copyright 2008 Wiley). | ||
Recently, Liu et al. studied the hydrogen storage properties of doped (physical mixing with Pt/C) and bridged MIL-101 and MIL-53 samples under mild conditions.92 Both doped and bridged MIL-101 and MIL-53 samples showed enhanced hydrogen storage capacities compared with pristine samples. The gravimetric storage capacities of bridged MIL-101 and MIL-53 samples were 1.14 and 0.63 wt% at 293 K and 5 MPa.
2.3. Hydrogen storage on other adsorbents by spillover
Yang et al. investigated the hydrogen capacity of COF-1.23 The hydrogen uptake on pure COF-1 was 1.28 wt% at 77 K and 1 atm and 0.26 wt% at 298 K and 10 MPa. By using the bridge-building technique, the bridged COF-1 sample had a hydrogen storage capacity of 0.7 wt%, which was enhanced by a factor of 2.6. The relatively low hydrogen capacity was due to the lower surface area of COF-1.
The overall heats of adsorption (ΔH) shown in the table were obtained from the temperature dependence of the isotherms, i.e., by using the Clausius–Clapeyron equation. The isosteric heats of adsorption declined with loading; the values shown in the table were those after leveling off. The detailed relationship between the overall heat of adsorption thus obtained and the binding energy between the spiltover H and the MOF sites is not known, but they are obviously related; i.e., a larger heat of adsorption indicates a stronger binding energy. The ΔH values (for convenience, referring to the absolute values) are somewhat related to the contents of the metal clusters in the MOF. For example, COF contains no metal, and shows the lowest ΔH. IRMOF-8 and MOF-177 have the same metal clusters (Zn4O), and are directly comparable. The metal content in IRMOF-8 is higher than that in MOF-177, and the ΔH value is higher for IRMOF-8.
For pure MOFs, not only are surface area and pore volume important factors for hydrogen storage capacity, framework composition is another important factor. This is because interactions other than the van der Waals interactions exist between H2 and MOF. Electric charges (on metal oxide sites) play a significant role in these systems.88 The effects of different factors on hydrogen uptake in pure MOFs have been discussed by many authors.77–81
For the bridged samples, IRMOF-8 has the highest storage capacity because of the large ΔH value; whereas MIL-101 and MOF-177 also show high storage capacities, due to their large BET surface area. Thus, it is clear that both surface area and the ΔH values are important for storage by spillover.
Recent studies have shown that the amount of hydrogen adsorbed on zeolites depended on the framework structure, composition, and acid–base nature of the zeolites.96–107 The cations in the zeolite create strong electric fields (and field gradients) that will favor gas adsorption.108 It is expected that high H2storage capacities could be obtained on zeolites with high cation densities.
Yang et al. reported that low-silica (thus, high charge density) type X (LSX) zeolite (Si/Al = 1) had a hydrogen storage capacity of 0.6 wt% at 298 K and 10 MPa.36 By hydrogen spillover, bridged Li–LSX had a hydrogen storage capacity of 1.6 wt% at 298 K and 10 MPa, which was enhanced by a factor of 2.6 compared with that on pure Li–LSX. The storage capacity of bridged Li–LSX is among the highest for zeolite materials at ambient temperature. Li–LSX zeolite was selected as the hydrogen adsorbent because it had the largest number of charge-compensating cations per unit cell among all faujasites.
Ramachandran et al. evaluated the feasibility of Al–MCM-41 for hydrogen storage.37 The hydrogen storage properties of Al–MCM-41 with varying contents of aluminium and different metal oxide doped Al–MCM-41 were compared. Their gas chromatographic studies indicated that hydrogen uptake in Al–MCM-41 samples was strongly dependent on the density of Brønsted acid sites and the amount of metal oxide. Hydrogen spillover was mainly responsible for the enhanced hydrogen adsorption for the doped Al–MCM-41. The studies indicate the potential of mesoporous materials as good candidates to form and stabilize nanomaterials within their pores. Mesoporous materials could also be exploited for better adsorption properties by producing mono-, bi-, and tri-metallic alloys for hydrogen storage.
3. Mechanistic and kinetics studies of hydrogen storage by spillover
3.1. Evidence of hydrogen spillover
Although hydrogen storage by spillover is a rather new field, the phenomenon of “hydrogen spillover” has been known for a long time.111 Atomic hydrogen spillover was first observed during studies of ethylene hydrogenation via heterogeneous catalysis by Sinfelt and Lucchesi.112 Further evidence was observed by Khoobiar.113 They found the formation of the hydrogen bronze HxWO3 from WO3 at room temperature after WO3 was exposed to hydrogen in the presence of a platinum-containing catalyst. The experimental result could only be interpreted by the diffusion of atomic hydrogen from the platinum catalyst to WO3. During the last four decades, hydrogen spillover has been extensively studied particularly in the field of catalysis. The voluminous literature on atomic hydrogen spillover has been reviewed.27,28,114 Recently, detailed structural characterization of the hydrogen-adsorbed species has added insight into the mechanism of hydrogen adsorption. Previously, Schwarz investigated the hydrogen adsorption on an activated carbon mixed with transition metal from room temperature to 77 K, and observed that the spillover effects were strongest at 77 K and nearly nil at room temperature.115 This observation was not correct.116–122 Direct evidence for spillover of atomic hydrogen at room temperature has been reported. Inelastic neutron scattering studies have shown the atomic hydrogen spillover from Pt to carbon at room temperature.116,117 Infrared spectroscopic study provided evidence of spillover of atomic hydrogen from Au nanoparticles to TiO2 also at room temperature.118 Evidence has also been shown by deuterium isotope tracer studies at room temperature on Pt/TiO2.119,120 The temperature programmed desorption (TPD) technique was extended to study the spillover on Pt/Al2O3 by Chen and White.121 A number of theoretical studies have been done on the mechanism and energetics of spillover. The more recent DFT study by Chen et al. illustrated facile pathways for spillover from a Pt particle onto a graphene basal plane via “physisorbed H atoms.”1223.2. Kinetics of adsorption and desorption
The kinetics of adsorption and desorption via spillover storage have been studied for both metal-doped carbons and catalyst-bridged MOFs in our laboratory. Our recent kinetics studies on bridged MIL-101 are shown in Fig. 9, as a representative result.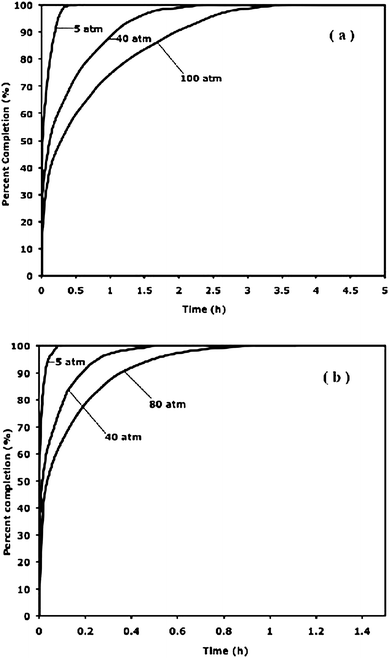 | ||
| Fig. 9 Rates of adsorption (a) and desorption (b) during stepped increases and decreases in pressure, at 298 K, for bridged MIL-101 sample. | ||
In the kinetics study, the pressure was increased or decreased in steps, all at 298 K. Thus, the pressure was increased from 0, to 5, 20, 40, 60, 80 and 100 atm (approximate values) during adsorption, and then the steps were reversed in desorption. At each end pressure, the percent completion with respect to the final (equilibrium) value was recorded. These are shown in Fig. 9. It is clear that the rates depended on the loading; the rates were slower at higher loading. This is another manifestation of spillover and that the surface diffusion step was the rate-limiting step, as the distance for surface diffusion increased with loading.
3.3. Mechanistic isotherm model for spillover storage
The mechanism of hydrogen spillover has also been investigated.123–134 It was found that hydrogen molecules were dissociated rapidly on metal sites and then diffused slowly away from them to the receptor. Surface diffusion of hydrogen atoms has been proposed to be the rate-determining step in hydrogen spillover.Hattori et al. investigated the kinetics of hydrogen adsorption on SO42−–ZrO2, and WO3–ZrO2 supported platinum catalysts in the temperature range 323–573 K.132,133 They observed very slow uptake rates of hydrogen on these two supported platinum samples.
Yang et al. studied the mechanism of hydrogen adsorption on bridged IRMOF-8 samples at room temperature. A two-dimension sectional plot of the spherical model was proposed for evaluating the diffusion time constants from the uptake rates, as shown in Fig. 10.134 In this model, H2 is dissociated on the surface of the Pt particle, where equilibrium is maintained. The atomic hydrogen undergoes surface diffusion onto the surface of the activated carbon particle, with radius R1, followed by diffusion onto the surface of the IRMOF-8 receptor. The average sphere of diffusion for the IRMOF-8 receptor is taken as R2, which depends on the connectivity through bridging as well as the ratio of Pt/AC catalyst over receptor. Based on this model, it was found that the diffusion time constant, D/R2, decreased sharply with surface concentration. This result clearly indicates that the diffusion distance, R2, was increasing with surface concentration. The concentration dependence for surface diffusion has been discussed in detail and explained satisfactorily by models by Higashi et al.135 and Yang et al.46,136,137 Surface diffusion is the result of a hopping process, for both physical adsorption and chemisorption of small molecules or atoms. Within the monolayer coverage, the surface diffusivity usually increases with surface concentration.
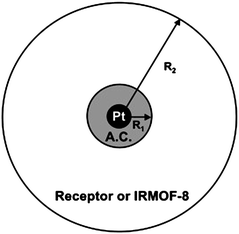 | ||
| Fig. 10 Two-dimension sectional plot of spherical model for hydrogen spillover on bridged IRMOF-8 sample, including Pt/AC catalyst (AC: activated carbon support) bridged with a receptor sorbent. (Reprinted with permission from ref. 134. Copyright 2007 American Chemical Society). | ||
The mechanistic model was further extended to explain the hydrogen adsorption on Pt/AX-21 systems.33 It was also observed that the diffusion time constant (D/R2) decreased with surface concentration. The proposed mechanistic model for hydrogen spillover could interpret the experimental results.
Based on this mechanistic model, an equilibrium isotherm for the spillover system was derived by Yang and co-workers, and the isotherm was given as in eqn (1):134
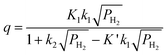 | (1) |
Here, q is the equilibrium amount adsorbed at hydrogen pressure PH2, and the constants were defined in ref. 134 and can, in principle, be determined independently.134 This equation can explain the nearly linear behavior of the isotherms that have been observed for all spillover sorbent systems investigated so far.
More recently, Lachawiec and Yang used a deuterium isotope sequential dosing and TPD technique to study the mechanism and kinetics of spillover of atomic hydrogen on Pt/AX-21 and IRMOF-8 at room temperature.138 The sorbent sample was dosed sequentially with H2 and D2 and the sample was then quickly “frozen.” Temperature programmed desorption followed. TPD product distributions and HD formation showed that desorption of the isotopes followed the reverse sequence of that of dosing, indicating reverse spillover during desorption. The HD was clearly formed by scrambling of the surface spiltover H and D atoms.
4. Summary and outlook
Significant hydrogen storage capacity has been obtained at ambient temperature by spillover. Further enhanced hydrogen capacity and understanding of hydrogen spillover are still needed for meeting the DOE targets. Several aspects should be further developed to improve the hydrogen capacity by spillover.4.1. Heats of adsorption
Hydrogen storage by spillover is an atomic hydrogen adsorption process. The surface adsorption sites of the adsorbent determine the storage capacity by spillover. It is understood that an adsorbent with more and stronger surface adsorption sites for hydrogen atoms should have higher hydrogen capacity by spillover. Our recent work on a series of MOFs showed that the hydrogen uptakes were mainly related with the heats of adsorption of hydrogen on these adsorbents. Higher hydrogen uptakes were obtained on MOFs with higher heats of adsorption (stronger surface adsorption sites) even some MOFs had relatively lower surface areas. For example, IRMOF-8 has the highest storage capacity among the MOFs studied in our work. It has a high heat of adsorption (20 kJ mol−1) and a relative low BET surface area (about 600 m2 g−1). The heat of adsorption for hydrogen on the adsorbent is the key factor in the hydrogen adsorption by spillover.4.2. Pore volume and surface area
It was suggested that the hydrogen uptake on an adsorbent at 77 K and high pressures was related to its pore volume. This is easily understood because at 77 K and high pressures, the hydrogen storage in an adsorbent is mainly by condensation of H2 molecules into its porosity, while adsorption by spillover is by the interaction of hydrogen atoms with the surface sites of the adsorbent. Therefore, the surface area and not the pore volume of the adsorbent should be an important factor in hydrogen adsorption by spillover. An adsorbent with high surface area would provide more adsorption sites for hydrogen adsorption via spillover. For example, Pt was doped on carbons with different surface areas. The highest hydrogen uptake was obtained on Pt/templated carbon with the highest surface area among the carbons studied.4.3. Contacts between the source and the receptor
Contacts between the source and the receptor are very important for hydrogen spillover. Enhanced storage capacity has been achieved by several methods such as bridge building, plasma assisted doping and ultrasound assisted building. However, the contact effect is still least understood in spillover. It is suggested that the enhanced contacts between the source and receptor would facilitate the spillover and hence increase the storage capacity. Significant hydrogen uptakes could be expected by enhancing contact strength between the source and receptor, creating more contact sites and option of the ratio of source and receptor.Acknowledgements
The authors acknowledge the funding provided by the US Department of Energy's Office of Energy Efficiency and Renewable Energy within the Hydrogen Sorption Center of Excellence (HS CoE), and funding from NSF.References
- A. C. Dillon and M. J. Heben, Appl. Phys. A, 2001, 72, 133 CrossRef CAS.
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353 CrossRef.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246 CrossRef CAS.
- R. T. Yang, Adsorbents: Fundamentals and Applications, Wiley, New York, 2003 Search PubMed.
- Department of Energy (DOE), A Multiyear Plan for the Hydrogen R&D Program, US Department of Energy (Office of Power Delivery, Office of Power Technologies, Energy Efficiency and Renewable Energy), August, 1999 Search PubMed.
- S. Hynek, W. Fuller and J. Bentley, Int. J. Hydrogen Energy, 1997, 22, 601 CrossRef CAS.
- G. Sandrock, S. Suda and L. Schlapbach, Hydrogen in Intermetallic Compounds II, Topics in Applied Physics, Springer-Verlag, 1992, vol. 67(5), pp. 197 Search PubMed; G. Sandrock and Y. Yurum, Hydrogen Energy Systems: Production and Utilization of Hydrogen and Future Aspects, NATO ASI Series, Kluwer Academic Publishers, 1994, pp. 253 Search PubMed.
- S.-i. Orimo, Y. Nakamori, J. R. Eliseo, A. Zuttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111 CrossRef CAS.
- R. Q. Snurr, J. T. Hupp and S. T. Nguyen, AIChE J., 2004, 50, 1090 CrossRef CAS.
- M. Dinca, A. Dailly, Y. Lin, C. M. Brown, D. A. Newmann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876 CrossRef CAS.
- A. M. Seayad and D. M. Antonelli, Adv. Mater., 2004, 16, 765 CrossRef CAS.
- F. Lamari Darkrim, P. Malbrunot and G. P. Tartaglia, Int. J. Hydrogen Energy, 2002, 27, 193 CrossRef CAS.
- J. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS.
- D. J. Collins and H.-C. Zhou, J. Mater. Chem., 2007, 30, 3154 RSC.
- R. Ströbel, J. Garche, P. T. Moseley, L. Jörisen and G. Wolf, J. Power Sources, 2006, 159, 781 CrossRef; P. Kowalczyk, R. Holyst, M. Terrones and H. Terrones, Phys. Chem. Chem. Phys., 2007, 9, 1786 RSC.
- S. Suda and G. Sandrock, Z. Phys. Chem., 1994, 183, 149 CAS.
- A. W. C. van den Berg and C. O. Areán, Chem. Commun., 2008, 668 RSC.
- F. E. Pinkerton, B. G. Wicke, C. H. Olk, G. G. Tibbetts, G. P. Meisner, M. S. Meyer and J. F. Herbst, J. Phys. Chem. B, 2000, 104, 9460 CrossRef CAS.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS.
- M. Latroche, S. Surblé, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J.-H. Lee, J.-S. Chang, S. H. Jhung and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 8227 CrossRef CAS.
- H. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523 CrossRef CAS.
- H. Furukawa, M. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197 RSC.
- Y. Li and R. T. Yang, AIChE J., 2008, 54, 269 CrossRef CAS.
- Y. Li and R. T. Yang, Langmuir, 2007, 23, 12937 CrossRef CAS.
- A. D. Lueking and R. T. Yang, J. Catal., 2002, 206, 165 CrossRef CAS.
- F. H. Yang and R. T. Yang, Carbon, 2002, 40, 437 CrossRef CAS.
- A. J. Robell, E. V. Ballou and M. Boudart, J. Phys. Chem., 1964, 68, 2748 CAS.
- W. C. Conner, Jr. and J. L. Falconer, Chem. Rev., 1995, 95, 759 CrossRef CAS.
- F. H. Yang, A. J. Lachawiec and R. T. Yang, J. Phys. Chem. B, 2006, 110, 6236 CrossRef CAS.
- A. D. Lueking and R. T. Yang, Appl. Catal., A, 2004, 265, 259 CrossRef CAS.
- A. D. Lueking, R. T. Yang, N. M. Rodriguez and R. T. K. Baker, Langmuir, 2004, 20, 714 CrossRef CAS.
- A. J. Lachawiec, G. Qi and R. T. Yang, Langmuir, 2005, 21, 11418 CrossRef CAS.
- Y. Li and R. T. Yang, J. Phys. Chem. C, 2007, 111, 11086 CrossRef CAS.
- Y. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 726 CrossRef CAS.
- Y. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 8136 CrossRef CAS.
- Y. Li and R. T. Yang, J. Phys. Chem. B, 2006, 110, 17175 CrossRef CAS.
- S. Ramachandran, J.-H. Ha and D. K. Kim, Catal. Commun., 2007, 8, 1934 CrossRef CAS.
- A. C. Dillon, K. M. Johns, T. A. Bekkedahl, C. H. Klang, D. S. Bethune and M. J. Heben, Nature, 1997, 386, 377 CrossRef CAS.
- C. Liu, Y. Y. Fan, M. Liu, H. T. Cong, H. M. Cheng and M. S. Dresselhaus, Science, 1999, 286, 1127 CrossRef CAS; P. Chen, X. Wu, J. Lin and K. L. Tan, Science, 1999, 285, 91 CrossRef CAS.
- A. Chambers, C. Park, R. T. K. Baker and N. M. Rodriguez, J. Phys. Chem. B, 1998, 102, 4253 CrossRef CAS.
- Y. Ye, C. C. Ahn, C. Witham, B. Fultz, J. Liu, A. G. Rinzler, D. Colbert, K. A. Smith and R. E. Smalley, Appl. Phys. Lett., 1999, 74, 2307 CrossRef CAS.
- R. Zacharia, S.-u. Rather, S. W. Hwang and K. S. Nahm, Chem. Phys. Lett., 2007, 434, 286 CrossRef CAS; C.-H. Chen and C.-C. Huang, Microporous Mesoporous Mater., 2008, 109, 549 CrossRef CAS; B.-J. Kim, Y.-S. Lee and S.-J. Park, J. Colloid Interface Sci., 2008, 318, 530 CrossRef CAS.
- A. Zuttel, Mater. Today, 2003, 6, 24 CrossRef.
- R. T. Yang, Carbon, 2000, 38, 623 CrossRef CAS.
- M. Becher, M. Haluska, M. Hirscher, A. Quintel, V. Skakalova, U. Dettlaff-Weglikovska, X. Chen, M. Hulman, Y. Choi, S. Roth, V. Meregalli, M. Parrinello, R. Strobel, L. Jorissen, M. M. Kappes, J. Fink, A. Zuttel, I. Stepanek and P. Bernier, C. R. Phys., 2003, 4, 1055 Search PubMed.
- R. T. Yang, Gas Separation by Adsorption Processes, Butterworth, Boston and London, 1987. Paperback edition, Imperial College Press, London, 1997 Search PubMed.
- G. G. Tibbetts, G. P. Meisner and C. H. Olk, Carbon, 2001, 39, 2291 CrossRef CAS.
- F. Darkrim, P. Malbrunot and G. P. Tartaglia, Int. J. Hydrogen Energy, 2002, 27, 193 CrossRef CAS.
- A. J. Lachawiec, T. R. DiRamondo and R. T. Yang, Rev. Sci. Instrum., 2008, 79, 063906 CrossRef.
- E. Poirier, R. Chahine and T. K. Bose, Int. J. Hydrogen Energy, 2001, 26, 831 CrossRef CAS; M. K. Haas, J. M. Zielinski, G. Dantsin, C. G. Coe, G. P. Pez and A. C. Cooper, J. Mater. Res., 2005, 20, 3214 CrossRef CAS; L. Zhou, Y. Sun and Y. Zhou, Chem. Eng. Commun., 2006, 193, 564 CrossRef CAS.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673 CrossRef CAS.
- B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 43, 2209 CrossRef CAS; R. Dash, J. Chmiola, G. Yushin, Y. Gogotsi, G. Landisio, J. Singer, J. Fischer and S. Kucheyev, Carbon, 2006, 44, 2489 CrossRef CAS.
- M. Shiraishi, T. Takenobu, H. Kataura and M. Ata, Appl. Phys. A, 2004, 78, 947 CrossRef CAS; E. Poirier, R. Chahine, P. Bénard, D. Cossement, L. Lafi, E. Mélançon, T. K. Bose and S. Désilets, Appl. Phys. A, 2004, 78, 961 CrossRef CAS.
- B. Panella, M. Hirscher and B. Ludescher, Microporous Mesoporous Mater., 2007, 103, 230 CrossRef CAS.
- S. T. Srinivas and P. K. Rao, J. Catal., 1994, 148, 470 CrossRef CAS.
- M. Boudart, M. A. Vannice and J. E. Benson, Z. Phys. Chem. Neue Folge, 1969, 64, 171 CAS; R. B. Levy and M. Boudart, J. Catal., 1974, 32, 304 CrossRef CAS.
- W. C. Neikam and M. A. Vannice, J. Catal., 1972, 27, 207 CrossRef CAS.
- R. T. Yang, Y. Li and A. J. Lachawiec, Chemical Bridges for Enhancing Hydrogen Storage by Spillover and Methods Forming the Same, US patent application, Serial No. 11/442.898, 2006 Search PubMed.
- C. Back, G. Sandi, J. Prakash and J. Hranisavljevic, J. Phys. Chem. B, 2006, 110, 16225 CrossRef CAS.
- A. Anson, E. Lafuente, E. Urriolabeitia, R. Navarro, A. M. Benito, W. K. Maser and M. T. Martinez, J. Phys. Chem. B, 2006, 110, 6643 CrossRef CAS.
- M. Zielinski, R. Wojcieszak, S. Monteverdi, M. Mercy and M. M. Bettahar, Catal. Commun., 2005, 6, 777 CrossRef CAS.
- R. Zacharia, K. Y. Kim, A. K. M. Fazle Kibria and K. S. Nahm, Chem. Phys. Lett., 2005, 412, 369 CrossRef CAS; S.-u. Rather, R. Zacharia, S. W. Hwang, M.-u.-d. Naik and K. S. Nahm, Chem. Phys. Lett., 2007, 441, 261 CrossRef CAS.
- D. Lupu, A. R. Biris, I. Misan, A. Jianu, G. Holzhuter and E. Burkel, Int. J. Hydrogen Energy, 2004, 29, 97 CrossRef CAS; R. Campesi, F. Cuevas, R. Gadiou, E. Leroy, M. Hirscher, C. Vix-Guterl and M. Latroche, Carbon, 2008, 46, 206 CrossRef CAS.
- H. S. Kim, H. Lee, K. S. Han, J. H. Kim, M. S. Song, M. S. Park, J. Y. Lee and J. K. Kang, J. Phys. Chem. B, 2005, 109, 8983 CrossRef.
- X. Hao, L. Quach, J. Korah, W. A. Spieker and J. R. Regalbuto, J. Mol. Catal. A, 2004, 219, 97 CrossRef CAS.
- R. T. Yang, Y. Li and A. J. Lachawiec, Enhancing Hydrogen Spillover and Storage, US patent application filed, Serial No. 11/820,954, 2007 Search PubMed.
- B. S. Schueller and R. T. Yang, Ind. Eng. Chem. Res., 2001, 40, 4912 CrossRef CAS.
- C. J. Liu, G. P. Vissokov and B. W. L. Jang, Catal. Today, 2002, 72, 173 CrossRef CAS.
- J. C. Legrand, A. M. Diamy, G. Riahi, Z. Randriamanantenasoa, M. Polisset-Thfoin and J. Fraissard, Catal. Today, 2004, 89, 177 CrossRef CAS.
- I. G. Koo, M. S. Lee, J. H. Shim, J. H. Ahn and W. M. Lee, J. Mater. Chem., 2005, 15, 4125 RSC.
- X. L. Zhu, P. P. Huo, Y. P. Zhang and C. J. Liu, Ind. Eng. Chem. Res., 2006, 45, 8604 CrossRef CAS.
- J. J. Zhou, Y. P. Zhang and C. J. Liu, Langmuir, 2006, 22, 11388 CrossRef CAS.
- Y. Li, R. T. Yang, C.-J. Liu and Z. Wang, Ind. Eng. Chem. Res., 2007, 46, 8277 CrossRef CAS.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keefee and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS.
- J. L. C. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666 CrossRef CAS; J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS.
- A. G. Wong-Foy, A. J. Matzger and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 3494 CrossRef CAS.
- K. M. Thomas, Catal. Today, 2007, 120, 389 CrossRef CAS.
- X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358 CrossRef.
- J. Y. Lee, L. Pan, S. P. Kelly, J. Jagiello, T. J. Emge and J. Li, Adv. Mater., 2005, 17, 2703 CrossRef CAS.
- H. Frost, T. Düren and R. Q. Snurr, J. Phys. Chem. B, 2006, 110, 9565 CrossRef CAS; H. Frost and R. Q. Snurr, J. Phys. Chem. C, 2007, 111, 18794 CrossRef CAS.
- B. Panella and M. Hirscher, Adv. Mater., 2005, 17, 538 CrossRef.
- L. Pan, M. B. Sander, X. Y. Huang, J. Li, M. Smith, E. Bittner, B. Bockrath and J. K. Johnson, J. Am. Chem. Soc., 2004, 126, 1308 CrossRef CAS.
- M. Dinca and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376 CrossRef CAS; M. Dinca, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904 CrossRef CAS; S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176 CrossRef CAS.
- X. B. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012 CrossRef CAS.
- D. N. Dybtsev, H. Chun, S. H. Yoon, D. Kim and K. Kim, J. Am. Chem. Soc., 2004, 126, 32 CrossRef CAS; M. P. Suh, J. W. Ko and H. J. Choi, J. Am. Chem. Soc., 2002, 124, 10976 CrossRef CAS.
- B. Kesanli, Y. Cui, M. R. Smith, E. W. Bittner, B. C. Bockrath and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 72 CrossRef CAS; Y. Kubota, M. Takata, R. Matsuda, R. Kitaura, S. Kitagawa, K. Kato, M. Sakata and T. C. Kobayashi, Angew. Chem., Int. Ed., 2005, 44, 920 CrossRef CAS.
- D. Sun, S. Ma, Y. Ke, D. J. Collins and H.-C. Zhou, J. Am. Chem. Soc., 2006, 128, 3896 CrossRef CAS; S. Ma and H.-C. Zhou, J. Am. Chem. Soc., 2006, 128, 11734 CrossRef CAS; S. Ma, D. Sun, M. W. Ambrogio, J. A. Fillinger, S. Parkin and H.-C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858 CrossRef CAS.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS.
- B. Xiao, P. S. Wheatley, X. B. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203 CrossRef CAS.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337 CrossRef CAS.
- Y.-Y. Liu, J.-L. Zeng, J. Zhang, F. Xu and L.-X. Sun, Int. J. Hydrogen Energy, 2007, 32, 4005 CrossRef CAS.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS.
- M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keefe and O. M. Yaghi, Science, 2007, 316, 268 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS.
- L. Regli, A. Zecchina, J. G. Vitillo, D. Cocina, G. Spoto, C. Lamberti, K. P. Lillerud, U. Olsbye and S. Bordiga, Phys. Chem. Chem. Phys., 2005, 7, 3197 RSC.
- F. Stphanie-Victoire, A. M. Goulay and E. C. de Lara, Langmuir, 1998, 14, 7255 CrossRef CAS.
- V. B. Kazansky, J. Mol. Catal. A: Chem., 1999, 141, 83 CrossRef CAS.
- H. W. Langmi, D. Book, A. Walton, S. R. Johnson, M. M. Al-Mamouri, J. D. Speight, P. P. Edwards, I. R. Harris and P. A. Anderson, J. Alloys Compd., 2005, 404–406, 637 CrossRef CAS.
- V. B. Kazansky, V. Y. Borovkov, A. Serich and H. G. Karge, Microporous Mesoporous Mater., 1998, 22, 251 CrossRef CAS.
- M. A. Makarova, V. L. Zholobenko, K. M. Alghefaili, N. E. Thompson, J. Dewing and J. Dwyer, J. Chem. Soc., Faraday Trans., 1994, 90, 1047 RSC.
- M. G. Nijkamp, J. E. M. J. Raaymakers, A. J. van Dillen and K. P. de Jong, Appl. Phys. A, 2001, 72, 619 CrossRef CAS.
- A. Zecchina, S. Bordiga, J. G. Vitillo, G. Ricchiardi, C. Lamberti, G. Spoto, M. Bjrgen and K. P. Lillerud, J. Am. Chem. Soc., 2005, 127, 6361 CrossRef CAS.
- H. W. Langmi, A. Walton, M. M. Al-Mamouri, S. R. Johnson, D. Book, J. D. Speight, P. P. Edwards, I. Gameson, P. A. Anderson and I. R. Harris, J. Alloys Compd., 2003, 356–357, 710 CrossRef CAS.
- S. B. Kayiran and F. L. Darkrim, Surf. Interface Anal., 2002, 34, 100 CrossRef CAS.
- J. Weitkamp, M. Fritz and S. Ernst, Int. J. Hydrogen Energy, 1995, 20, 967 CrossRef CAS.
- J. G. Vitillo, G. Ricchiardi, G. Spoto and A. Zecchina, Phys. Chem. Chem. Phys., 2005, 7, 3948 RSC.
- C. O. Aren, O. V. Manoilova, B. Bonelli, M. R. Delgado, G. T. Palomino and E. Garrone, Chem. Phys. Lett., 2003, 370, 631 CrossRef.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS.
- H. Taylor, Annu. Rev. Phys. Chem., 1961, 12, 127 CrossRef CAS.
- J. H. Sinfelt and P. J. Lucchesi, J. Am. Chem. Soc., 1963, 85, 3365 CrossRef CAS.
- S. Khoobiar, J. Phys. Chem., 1964, 68, 411 CrossRef.
- G. M. Pajonk, Appl. Catal., A, 2000, 202, 157 CrossRef CAS.
- J. A. Schwarz, US Pat., 4 716 736, 1988.
- P. C. H. Mitchell, A. J. Ramirez-Cuesta, S. F. Parker, J. Tomkinson and D. Thompsett, J. Phys. Chem. B, 2003, 107, 6838 CrossRef CAS.
- P. C. H. Mitchell, A. J. Ramirez-Cuesta, S. F. Parker and J. Tomkinson, J. Mol. Struct., 2003, 651–653, 781 CrossRef CAS.
- D. A. Panayotov and J. T. Yates Jr., J. Phys. Chem. C, 2007, 111, 2959 CrossRef CAS.
- D. D. Beck and J. M. White, J. Phys. Chem., 1984, 88, 2764 CrossRef CAS.
- D. D. Beck, A. O. Bawagan and J. M. White, J. Phys. Chem., 1984, 88, 2771 CrossRef CAS.
- H.-W. Chen and J. M. White, J. Mol. Catal., 1986, 25, 355.
- L. Chen, A. C. Cooper, G. P. Pez and H. S. Cheng, J. Phys. Chem. C, 2007, 111, 18995 CrossRef CAS.
- U. Roland, T. Braunschweig and F. Roessner, J. Mol. Catal. A: Chem., 1997, 127, 61 CrossRef CAS.
- M. Jaroniec and R. Mady, Physical Adsorption on Heterogeneous Solids, Elsevier, Amsterdam, 1988 Search PubMed.
- W. Rudzinski and D. H. Everett, Adsorption of Gases on Heterogeneous Surfaces, Academic Press, San Diego, 1992 Search PubMed.
- A. J. Robell, E. V. Ballou and M. Boudart, J. Phys. Chem., 1964, 68, 2748 CAS.
- R. Kramer and M. Andre, J. Catal., 1979, 58, 287 CrossRef CAS.
- A. Borgschulte, R. J. Westerwaal, J. H. Rector, H. Schreuders, B. Dam and R. Griessen, J. Catal., 2006, 239, 263 CrossRef CAS.
- M. Boudart, A. W. Aldag and M. A. Vannice, J. Catal., 1970, 18, 46 CrossRef CAS.
- M. Arai, M. Fukushima and Y. Nishiyama, Appl. Surf. Sci., 1996, 99, 145 CrossRef CAS.
- K. J. Sladek, E. R. Gilliland and R. F. Baddour, Ind. Eng. Chem. Fundam., 1974, 13, 100 CrossRef CAS.
- S. Triwahyono, T. Yamada and H. Hattori, Appl. Catal., A, 2003, 250, 65 CrossRef CAS.
- N. Satoh, J. Hayashi and H. Hattori, Appl. Catal., A, 2000, 202, 207 CrossRef CAS.
- Y. Li, F. H. Yang and R. T. Yang, J. Phys. Chem. C, 2007, 111, 3405 CrossRef CAS.
- K. Higashi, H. Ito and J. Oishi, J. At. Energy Soc. Jpn., 1963, 5, 846 Search PubMed.
- R. T. Yang, J. B. Fenn and G. L. Haller, AIChE J., 1973, 19, 1052 CrossRef CAS.
- K. Kapoor and R. T. Yang, AIChE J., 1989, 35, 1735 CrossRef.
- A. J. Lachawiec and R. T. Yang, Langmuir, 2008, 24, 6159 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |