Synthesis of sodium amidoborane (NaNH2BH3) for hydrogen production
Zhitao
Xiong
a,
Guotao
Wu
c,
Yong Shen
Chua
b,
Jianjiang
Hu
a,
Teng
He
c,
Weiliang
Xu
b and
Ping
Chen
*abc
aDepartment of Physics, National University of Singapore, 10 Kent Ridge Crescent, Singapore 117542. E-mail: phychenp@nus.edu.sg; Fax: +65-67776126; Tel: +65-65165100
bDepartment of Chemistry, National University of Singapore, 10 Kent Ridge Crescent, Singapore 117542
cDalian Institute of Chemical Physics, Dalian, China 116023
First published on 23rd June 2008
Abstract
The prospect of building a future energy system on hydrogen has stimulated much research effort in developing hydrogen storage technologies. One of the potential materials newly developed is sodium amidoborane (NaNH2BH3) which evolves ∼7.5 wt% hydrogen at temperatures as low as 91 °C. In this paper, two methods of synthesizing pure NaNH2BH3 were reported. One method is by reacting NaH and ammonia borane in THF at low temperatures, and the other is by reacting NaNH2 and ammonia borane in THF at ambient temperature. Non-isothermal testing on the thermolysis of solid NaNH2BH3 showed that hydrogen evolution was composed of two exothermic steps. More than 1 equiv. H2 was evolved rapidly at temperatures below 87 °C. After evolving 2 equiv. H2, NaH was identified in solid products and coexisted with amorphous BN.
Introduction
One of the pending issues in the implementation of hydrogen fuel cell technology in the automobile industry is the lack of safe and highly efficient hydrogen storage materials.1,2 More research efforts in material development are now applied to chemicals comprising light elements and possessing high hydrogen content.3–8 Because of its high hydrogen capacity (19.6 wt%) as well as its stability in air and non-toxicity, ammonia borane (NH3BH3) was considered as one of the most promising candidates for future transportation applications.9,10 The thermal decomposition of NH3BH3 is composed of three steps evolving one equivalent H2 per step. In the first two steps, ∼2 equiv. H2 are evolved at temperatures of ca. 110 and ca. 150 °C,11,12 which are higher than the operation temperature of PEM (Proton Exchange Membrane) fuel cells. Another issue with NH3BH3 is that the hydrogen evolution is accompanied by the production of borazine, which is a volatile compound and will poison fuel cells. In an attempt to enhance the performance of thermolysis, NH3BH3 was deposited into mesoporous silica scaffolds and the onset temperature for hydrogen production was found to be reduced by approximately 15 °C.13 Furthermore, the formation of borazine was largely suppressed. Similar improvement was achieved by loading NH3BH3 on carbon cryogels.14 We have recently reported a derivative of ammonia borane, i.e., sodium amidoborane, NaNH2BH3, obtained from the solid-state interaction between NH3BH3 and sodium hydride (NaH). Sodium amidoborane evolves ∼7.4 wt% hydrogen at ca. 91 °C, considerably lower than that of neat NH3BH3.15 In the meantime, borazine is undetectable. Since NH3BH3 is plastic at room temperature, the mechano-synthetic route for NaNH2BH3 needs the help of inert additives, which enhance the efficiency of mechanical milling.15 However, the existence of these additives in the sample lowered the system gravimetric density of hydrogen. In this study, a wet-chemical route was developed to synthesize pure NaNH2BH3. NH3BH3 was first dissolved in THF and then reacted with NaH or sodium amide (NaNH2) at given temperatures. H2 or NH3 was generated during the reaction. Upon completion of the reaction, highly pure NaNH2BH3 can be obtained by removing THF. Subsequent hydrogen evolution from the NaNH2BH3 synthesized was investigated by Temperature Programmed Desorption (TPD), volumetric release and XRD; 7.5 wt% of hydrogen was found to be released from the sample. NaH was identified in the fully dehydrogenated solid residue.Experimental
Synthesis of NaNH2BH3
NaH (95%) and NaNH2 (95%) were Sigma Aldrich products and used as received. NH3BH3 was synthesized on-site according to the literature.16 The resulting solid was dissolved in tetrahydrofuran (THF) and characterized by 11B NMR. A single –BH3 resonance was observed at δ −22.1, agreeing well with reference data.17 The crystal structure of the solid NH3BH3 was further confirmed by XRD analysis. The purity of synthesized NH3BH3 was estimated at 95%. The chemical reaction of NH3BH3 with NaH or NaNH2 in THF was conducted in a commercial Stirring Tank Reactor (STR). 0.008 mol NaH or NaNH2 and 0.008 mol NH3BH3 were added to 30 ml THF and stirred at constant temperature. As gas was evolved, the pressure variation with time inside the reactor was recorded automatically. At the end of the reaction, gaseous products were analyzed by a Mass Spectrometer (MS). The reaction solution was filtered and evaporated under reduced pressure at ambient temperature for crystallization.Characterization
Hydrogen evolution from the synthesized NaNH2BH3 was conducted on a custom-made Temperature Programmed Desorption (TPD) system. A Gas Chromatograph (GC)–Mass Spectrometer (MS) combined system was employed to trace the gaseous products. Detailed operation was described elsewhere.18 A 20 mg sample was ramp-heated in argon at 2 K min−1. Volumetric release for the quantitative measurement of hydrogen evolution was performed on a commercial gas reaction controller provided by Advanced Materials Corporation. Approximately 300 mg of sample was employed for each experiment and the heating rate was 1 K min−1. A Netzsch DSC 204 HP unit was applied to detect heat effects in the evolution process.11B {1H} magic-angle-spinning Nuclear Magnetic Resonance (NMR) experiments were carried out at room temperature on a Bruker Avance 400 NMR spectrometer operating at 9.7 T on 128.3 MHz 11B frequency. All chemical shifts are reported in ppm downfield.
Structural identifications were performed on a Bruker X-ray Diffractometer (XRD) equipped with an in situ cell. X-ray diffraction data were collected from 10 to 60° in 2θ with a scan step-width of 0.01° using Cu Kα radiation.
Results and discussion
Gas was evolved vigorously after dropping NaH into the NH3BH3–THF solution. As can be seen from Fig. 1, gas evolution was completed within 10 min at −3 °C. Mass spectrometer analysis showed that hydrogen was the only gaseous product. By applying the ideal equation of state, the amount of hydrogen evolved was ca. 1 equiv. H2 per [NaH–NH3BH3] mixture. NaH solid was completely consumed and a transparent solution was obtained at the end of the reaction. NMR characterization of the B species in the solution revealed that –BH3 with a chemical shift at −21.6 ppm was the dominant species. The resonance of –BH3 in NH3BH3 at −22.1 ppm weakened significantly (see Fig. 2). After removing THF at ambient temperature, a white powder with an orthorhombic structure (group Pbca) with lattice constants a = 7.4735 Å, b = 14.6452 Å, c = 5.6739 Å, was obtained, which is identical to the unit cell of NaNH2BH3 synthesized by mechano-chemical methods.15 Obviously, NaH and NH3BH3 converted to NaNH2BH3 and H2 in THF through reaction (1).| NaH (s) + NH3BH3–THF (l) → NaNH2BH3-THF (l) + H2 (g) | (1) |
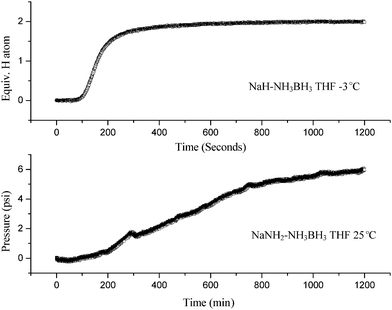 | ||
| Fig. 1 Time dependence of gas evolution from NaH–NH3BH3–THF and NaNH2–NH3BH3–THF solutions at temperatures of −3 °C and 25 °C, respectively. | ||
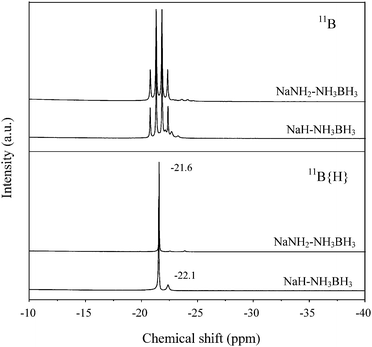 | ||
| Fig. 2 11B NMR spectra obtained at the end of gas evolution from NaH–NH3BH3–THF and NaNH2–NH3BH3–THF solutions. | ||
Comparatively, the chemical reaction between NaNH2 and NH3BH3 in THF was slow. Only 6 psi pressure inside the reactor was detected after the solution was stirred at 25 °C for 20 h (see Fig. 1). It was detected by MS that ammonia (NH3) was the dominant product. Observation of the transparent solution revealed the consumption of NaNH2. NH3BH3 was fully consumed as only the –BH3 resonance at −21.6 ppm was detected by NMR (see Fig. 2). Removal of the solvent through distillation led to the formation of NaNH2BH3 powder, which suggested the occurrence of reaction (2).
| NaNH2 (s) + NH3BH3–THF (l) → NaNH2BH3–THF (l) + NH3 (g) | (2) |
The weight difference between NaNH2 plus NH3BH3 (as starting chemicals) and the resulting solid after the removal of THF was found to be equal to the amount of NH3 evolution (ca. 1 equiv NH3), further validating reaction (2). The low NH3 pressure inside the reactor is due to the solubility of NH3 in THF. It is rather interesting to investigate the mechanism of reaction (2). It could be a direct substitution of NH3 in NH3BH3 by NaNH2 or by metathesis of Na in NaNH2 with H in NH3 in NH3BH3. It is also worthy of emphasis that the interaction between NaNH2 and NH3BH3 provides a facile pathway for the synthesis of high purity metal amidoboranes through the reaction of other amides and NH3BH3. As a matter of fact, LiNH2BH3 can be synthesized by reacting LiNH2 and NH3BH3 at room temperature.
Illustrated in Fig. 3 are TPD, DSC and volumetric release measurements on hydrogen evolution from solid-state NaNH2BH3. It can be seen that the TPD-H2 spectrum exhibits three main evolution steps. A burst was observed at 87 °C, and then broad peaks were found at ca. 174 °C and 325 °C, respectively. Borazine was undetectable, agreeing well with our previous investigations.15 DSC measurement revealed that the hydrogen evolution in the first two steps was exothermic. An endothermic signal at 57 °C is due to the melting of NaNH2BH3. In our previous study, NaNH2BH3 produced by a mechano-chemical process was of smaller particle size so that it exhibited better kinetics during dehydrogenation. Quantitative measurements on hydrogen evolution from NaNH2BH3 showed that the first and second steps evolved 4.4 wt% H2 (2.3 equiv. H) and 3.1 wt% H2 (1.7 equiv. H), respectively. Therefore, ca. 2 equiv. H2 were detached from one NaNH2BH3 molecule at temperatures below 200 °C. By holding the reaction temperature at ca. 91 °C for longer period of time, almost all H2 can be evolved. As hydrogen evolution in the first two steps is exothermic, the re-hydrogenation may not be thermodynamically favored. Our attempt to re-charge the dehydrogenated product at 120 bar hydrogen was not successful. The third evolution step evolved ca. 1 equiv. H. Due to its high operation temperature this step is not considered for practical usage.
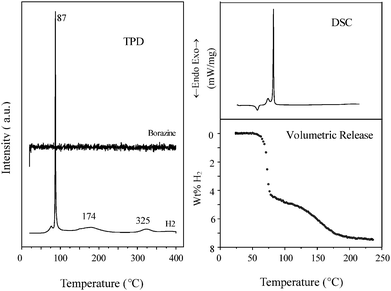 | ||
| Fig. 3 TPD, DSC and volumetric release measurements on synthesized NaNH2BH3 solid. | ||
To gain insight into the hydrogen evolution from NaNH2BH3, we collected the samples upon decomposition at 90 °C and 200 °C, respectively, for XRD characterization. As shown in Fig. 4, no phase can be identified from the solid residue collected after the first desorption step, indicating the amorphous nature of the dehydrogenation product. The sample collected at the end of the second desorption step presented a NaH phase, which reminds us that the third desorption step should be with the decomposition of NaH. In fact, our previous investigations showed that NaH decomposed to H2 at ca. 320 °C.19 Given that 1 equiv. H was evolved in the third desorption step, one unit of NaH should be in the residue after the second desorption step. Therefore, together with 2 equiv. H2 evolution in the first two steps, thermolysis of NaNH2BH3 at temperatures below 200 °C can be described by reaction (3).
| nNaNH2BH3 → nNaH + nBN + 2nH2 7.55 wt% H2 | (3) |
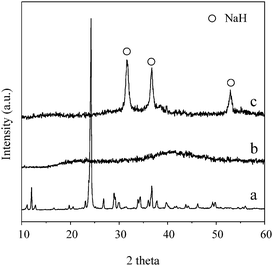 | ||
| Fig. 4 XRD patterns of a) synthesized NaNH2BH3 and its dehydrogenation product collected at b) 90 °C, c) 200 °C. | ||
BN should be in an amorphous form as its crystallization requires high temperatures.20
By summarizing reactions (1) and (3), one could have an overall reaction (4), which allows more than 10.9 wt% of hydrogen to be desorbed from NaH and NH3BH3.
| NaH + NH3BH3 → NaNH2BH3 + H2 → NaH + BN + 3 H2 10.9 wt% H2 | (4) |
Due to the exceptionally high hydrogen content and low reacting temperatures, the NaNH2BH3 and NaH–NH3BH3 systems are potential candidates for on-board hydrogen storage and, therefore, are worthy of further investigation.
Conclusion
Sodium amidoborane (NaNH2BH3) was synthesized through the reaction of NaH and NH3BH3 or NaNH2 and NH3BH3 in THF. Investigations on sodium amidoborane revealed that, at temperatures below 200 °C, its thermolysis was composed of two steps. More than 7.5 wt% of hydrogen can be evolved. NaH and BN are the solid products.Acknowledgements
The present work is financially supported by the Agency for Science, Technology & Research (A*STAR) in Singapore. Z. Xiong, G. Wu and P. Chen would like to thank the Dalian Institute of Chemical Physics (DICP) in China for startup funding support on laboratory building.References
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353 CrossRef
.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283 CrossRef CAS
- B. Bogdanovic and M. Schwickardi, J. Alloys Compd., 1997, 1, 253
- A. Zuttel, P. Wenger, S. Rentsch, P. Sudan, P. Mauron and C. Emmenegger, J. Power Sources, 2003, 118, 1 CrossRef CAS
- P. Chen, Z. T. Xiong, J. Z. Luo, J. Y. Lin and K. L. Tan, Nature, 2002, 420, 302 CrossRef CAS
- Z. T. Xiong, G. T. Wu, J. J. Hu and P. Chen, Adv. Mater., 2004, 16, 1522 CrossRef CAS
- Z. T. Xiong, G. T. Wu, J. J. Hu, Y. F. Liu, P. Chen, W. F. Luo and J. Wang, Adv. Funct. Mater., 2007, 17, 1137 CrossRef CAS
- F. E. Pinkerton, G. P. Meisner, M. S. Meyer, M. P. Balogh and M. D. Kundrat, J. Phys. Chem. B, 2004, 109, 6
- T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116 CrossRef CAS
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613 RSC
- V. Sit, R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1987, 113, 379 CrossRef CAS
- M. G. Hu, R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1978, 23, 249 CrossRef CAS
- A. C. Stowe, W. J. Shaw, J. C. Linehan, B. Schmid and T. Autrey, Phys. Chem. Chem. Phys., 2007, 1831 RSC
- A. Feaver, S. Sepehri, P. Shamberger, A. Stowe, T. Autrey and G. Cao, J. Phys. Chem. B, 2007, 111, 7469 CrossRef CAS
- Z. T. Xiong, C. K. Yong, G. T. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138 CrossRef CAS
- P. V. Ramachandran and P. D. Gagare, Inorg. Chem., 2007, 46, 7810 CrossRef CAS
- J. S. Wang and R. A. Geanangel, Inorg. Chim. Acta, 1988, 148, 185 CrossRef CAS
- Z. T. Xiong, P. Chen, G. T. Wu, J. Y. Lin and K. L. Tan, J. Mater. Chem., 2003, 13, 1 RSC
- Z. T. Xiong, G. T. Wu, J. J. Hu and P. Chen, Catal. Today, 2007, 120, 287 CrossRef CAS
- R. T. Paine and C. K. Narula, Chem. Rev., 1990, 90, 73 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2008 |