Anomalous reactivity of radical cations produced by photosensitized oxidation of 4-methoxybenzyl alcohol derivatives: role of the sensitizer†
Tiziana
Del Giacco
*,
Annalisa
Faltoni
and
Fausto
Elisei
Dipartimento di Chimica and Centro di Eccellenza Materiali Innovativi Nanostrutturati (CEMIN), Università di Perugia, Via Elce di Sotto 8, 06123, Perugia, Italy. E-mail: dgiacco@unipg.it.; Fax: +39 075 5855560; Tel: +039 075 5855559
First published on 16th October 2007
Abstract
Steady-state and nanosecond laser flash photolysis measurements of 4-methoxybenzyl alcohol (1a), 4-methoxy-α-methylbenzyl alcohol (1b), 4,4′-dimethoxydiphenylmethanol (1c) and 4-methoxy-α,α′-dimethylbenzyl alcohol (1d) were carried out in air-equilibrated CH2Cl2 and CH3CN solutions, in the presence of 9,10-dicyanoanthracene (DCA) and N-methylquinolinium tetrafluoroborate (NMQ+BF4−) as sensitizers. In particular, steady-state irradiation with DCA produced carbonyl compounds and, with NMQ+BF4−, carbonyl compounds, ethers (substrates 1a–c) and styrene (substrate 1d) while time-resolved investigations gave evidence of charged species produced upon irradiation. The effect of solvent polarity on the reactivity was investigated; in the case of DCA, the reactivity increased with the solvent polarity, while the opposite was obtained when NMQ+BF4− was used. Quantum mechanical calculations at semiempirical (INDO/1-CI) and DFT (B3LYP/6-311G(d)) levels were used to support transient assignments and to obtain the charge and spin density distributions, respectively. The different photooxidation mechanisms operative with the neutral and charged sensitizer were rationalized in terms of the reactivity of free and complexed radical cations, respectively.
Introduction
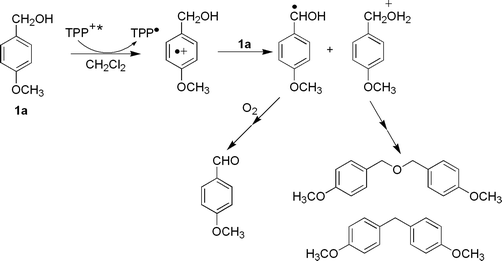
Studies on the electron transfer oxidation of benzyl alcohol derivatives [X-C6H4CH(OH)R] via radical cation intermediate have been extensively reported. This reaction has been investigated upon UV irradiation in homogeneous and heterogeneous media1–2 and by electrochemical3 and thermal4 conditions, in organic and aqueous solvents. In all cases, oxidation produces exclusively benzaldehydes (with R = H) or the corresponding ketone (for alcohols with R = CH3 and 4-YC6H4). The reaction mechanism proceeds through the deprotonation of the radical cation, formed in the first electron transfer step, to give an α-hydroxy X-benzyl radical. This intermediate is then converted into the carbonyl product through two different pathways, depending on the experimental conditions. In aerated solutions, the radical reacts quickly with oxygen to give a peroxyl radical that evolves to the final product, or it is oxidized in the reaction medium, producing the corresponding cation, which finally leads to the product (Scheme 1, path a and b, respectively). Only the latter path is operative in deaerated conditions. The mechanism is the same regardless of the way in which the radical cation is produced, however in aqueous solutions, it is only operative at pH ≤ 4.5,6 The mechanism has been supported by intermediate characterization and kinetics studies performed by laser flash photolysis, pulse radiolysis, and steady-state experiments.Different results were obtained when the radical cations of benzyl alcohols were generated by photoinduced electron transfer sensitized by 2,4,6-triphenylpyrylium tetrafluoroborate (TPP+BF4−). A first report showed the different kinds of products observed in the reaction of 4-X-benzyl alcohols both in CH2Cl2 and CH3CN, depending on the nature of the X group.7 As expected with X = H and Cl, the product obtained was the corresponding benzaldehyde, while with X = OMe (1a), bis(4-methoxybenzyl)ether, 4,4′-dimethoxydiphenylmethane and [4-methoxy-3-(4-methoxybenzyl)phenyl](4-methoxyphenyl)methane were produced. The absence of oxidation products, even in the presence of oxygen, has been explained in terms of the acid properties of the TPP+ excited state, which is able to catalyze the intermediate 4-methoxybenzyl cation involved in the product formation (Scheme 2).
 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
The driving force of this process seems to be the higher stability of the 4-methoxybenzyl cation compared with that of the benzyl cations formed from electron-poor benzyl alcohols.
The hypothesis of a Lewis acid action of the excited TPP+ on 4-methoxybenzyl alcohol seems to be in disagreement with the kinetics and product studies of the TPP+BF4− photosensitized oxidation of ring-methoxylated benzyl alcohols in air-saturated CH2Cl2.8 In particular, for the reaction of 4-methoxybenzyl alcohol, the oxidation product (4-methoxybenzaldehyde) was detected in addition to bis(4-methoxybenzyl)ether and 4,4′-dimethoxy-diphenylmethane. The product distribution was strongly dependent on the experimental conditions (irradiation time, presence of bases). The results of the product analysis, intermediate investigation by laser flash photolysis and DFT calculations of the relative stability of ring-methoxylated benzyl cations have been used to formulate the mechanism shown in Scheme 3, where the substrate reacts with the singlet excited state of TPP+ to give the couple radical cation/TPP˙. In CH2Cl2, the neutral substrate is thought to be the only base present in the system that is able to induce α-C–H deprotonation of the radical cation, to form an α-hydroxy-4-methoxbenzyl radical and a cation. The radical reacts with O2 to give 4-methoxybenzaldehyde, while the cation can form bis(4-methoxybenzyl)ether and 4,4′-dimethoxydiphenylmethane (Scheme 3).
 | ||
| Scheme 3 | ||
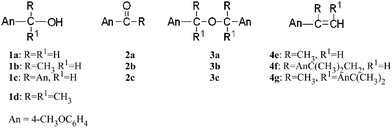
This research deals with the oxidation of 4-methoxybenzyl alcohol derivatives (see Scheme 4, where the molecular structures are reported together with those of the corresponding products) photosensitized by two well known electron acceptors, N-methylquinolinium tetrafluoroborate (NMQ+BF4−) and 9,10-dicyanoanthracene (DCA), that are able to form the radical cations of the substrates. The study was carried out by steady state, intermediate investigations (with nanosecond laser flash photolysis) and quantum mechanical calculations. The comparison of the results obtained with the two sensitizers in terms of product formation and intermediates involved in the mechanism suggests that the different behavior is due to specific electrostatic interactions between the BF4− anion and the radical cation.
 | ||
| Scheme 4 | ||
Results
Fluorescence quenching
All the alcohols examined (1a–d) efficiently quenched the fluorescence emission of NMQ+ in accordance with the Stern–Volmer eqn (1) where I0 and I are the emission intensities in the absence and in the presence of increasing concentrations of alcohol ([Q]), respectively. The quenching rate constants (kq) calculated from the slopes (Stern–Volmer constants KSV) of the linear plots (see ESI, Fig. S1)† divided by 3.3 ns (the lifetime of 1NMQ+* measured in air-equilibrated CH2Cl2) are reported in Table 1. The high kq values, close to the diffusional limit, indicate that the process is diffusion-controlled. | (1) |
| Alcohol | k q/109 M−1 s−1 | |
|---|---|---|
| NMQ+ | DCA | |
| 1a | 12 | 6.4 |
| 1b | 13 | 4.1 |
| 1c | 13 | 9.6 |
| 1d | 30 | 7 |
Steady-state photolysis
Steady-state experiments sensitized by DCA and NMQ+ were performed in air-equilibrated CH2Cl2 and CH3CN, as reported in the Experimental section. No products were detected in the absence of irradiation or sensitizer. After workup, the products were identified by 1H-NMR, 13C-NMR and GC-MS by comparison with authentic specimens. Quantitative analysis was performed by GC and 1H-NMR by using the internal standard method. The material balance was very satisfactory (>90%) in all experiments. The regeneration of the sensitizers was checked by UV-visible analysis. All the product structures are reported in Scheme 4.Photolysis of alcohols 1a, 1b and 1c (2.0 × 10−2 M) in the presence of catalytic amounts of DCA (3.0 × 10−4 M) by irradiation at 410 ± 20 nm in air-equilibrated condition gave the corresponding oxidation product, that is, 4-methoxybenzaldehyde (2a), 4-methoxyacetophenone (2b) and 4,4′-dimethoxybenzophenone (2c), respectively. As shown in Table 2, the product yields were remarkable in CH3CN, but very low in CH2Cl2 (1a was unreactive), despite longer irradiation times (up to four times longer) in this latter solvent. Only traces of 2b (2%) were detected in the case of alcohol 1d in CH3CN, while no products were detected in CH2Cl2.
Irradiation at 310 ± 20 nm of an air-equilibrated solution containing NMQ+BF4− (1.0 × 10−3 M), 1a, 1b and 1c (2.0 × 10−2 M) produced, together with the expected carbonyl compounds (2a, 2b and 2c, respectively), as well as the corresponding symmetrical ethers: bis(4-methoxybenzyl) ether (3a), bis[1-(4-methoxyphenyl)ethyl] ether (3b) and bis[4,4′-dimethoxydiphenylmethyl] ether (3c), respectively. High product yields were generally obtained with 1a–c, as shown in Table 3, especially in CH2Cl2, where comparable amounts of products were obtained with an irradiation time about three times shorter than in CH3CN. Experiments carried out under these experimental conditions at different irradiation times showed that the ether formed was not stable. In fact, separate experiments evidenced that, when the ethers were photolyzed under the same experimental conditions as those used for alcohols, they reacted to give the initial alcohol and the carbonyl product.
| Yield/% | |||||
|---|---|---|---|---|---|
| Alcohol | Solvent | Unreacted alcohol | AnCOR | (AnCRR1)2O | AnC(CH3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
| a [NMQ+BF4−] = 1.0 × 10−3 M; [alcohol] = 2.0 × 10−2 M. b Irradiation time = 30 min. c Irradiation time = 105 min. d A mixture of substituted p-methoxystyrenes was also recovered with 1d, 18% in CH2Cl2 and only traces in CH3CN (see Results section). | |||||
| 1a | CH2Cl2b | 66 | 18 | 14 | |
| CH3CNc | 73 | 25 | |||
| 1b | CH2Cl2b | 21 | 11 | 65 | |
| CH3CNc | 30 | 30 | 30 | ||
| 1c | CH2Cl2b | 41 | 12 | 40 | |
| CH3CNc | 37 | 43 | 13 | ||
| 1d | CH2Cl2b | 52 | 14d | ||
| CH3CNc | 69 | 28d | |||
The reaction of alcohol 1d with excited NMQ+BF4− produced 4-methoxyphenyl-α-methylstyrene (4e) as primary product (Table 3), together with an isomer mixture characterized by 2,4-bis(4-methoxyphenyl)-4-methyl-1-pentene (4f) and 2,4-bis(4-methoxyphenyl)-4-methyl-2-pentene (4g), in high yield in CH2Cl2, but only in traces in CH3CN. Experiments performed at different irradiation times indicated that these isomers were formed by the reaction of 4e; in fact, with short irradiation times, 4e was the only product detected, while with prolonged irradiation times, the formation of 4e slowed down, thus favoring the increase of the isomer mixture. As observed for the other alcohols, the total yield of 4e was higher in CH2Cl2 (32% after 30 min) than in CH3CN (28% after 105 min).
The photoproduct quantum yields were determined for the reactions of alcohols 1b, 1c and 1d with NMQ+BF4− irradiated around 313 nm in air-equilibrated CH2Cl2. From the data reported in Table 4, it can be observed that the ketone quantum yields were very high and independent of the irradiation time, while the ether quantum yields were not constant over time and reached values higher than unity, especially in the case of 1c. The result with 1d is much clearer, in fact the quantum yield for the formation of 4e was 0.64 and independent of the irradiation time.
Laser flash photolysis experiments
In the cases of the alcohol/NMQ+BF4− systems, only the laser flash photolysis results in CH2Cl2 are reported because much smaller transient signals were obtained in CH3CN; this is in line with the efficiencies observed in the steady state experiments. All experiments showed an electron transfer from the alcohol to the NMQ+BF4− excited state, confirmed by the detection of the radical cation–radical couple, as shown below. In order to increase the yield of separate radical cations, 1 M toluene was used as a co-sensitizer. Under these conditions, the sequence of reactions (2) and (3) takes place (A = alcohol, T = toluene). | (2) |
 | (3) |
 | ||
| Fig. 1 Time-resolved absorption spectra of the NMQ+BF4− (2.6 × 10−3 M)/toluene (1 M)/1b (1.0 × 10−2 M) system in air-equilibrated CH2Cl2 recorded 0.08 (△), 1.2 (▲) and 6.2 (○) μs after the laser pulse. λexc = 355 nm. Inset: decay kinetics recorded at 430 (A) and 540 (B) nm. | ||
The time-resolved absorption spectra of 1c in O2-saturated solution appear to be much more complex, due to the presence of three bands centered at 350, 420 and 550 nm just after the laser pulse (Fig. 2). The absorption at 550 nm was again ascribed to NMQ˙, while the one at 420 nm can be reasonably assigned to 1c++˙, the partner of NMQ˙ in the electron transfer reaction. Actually, the absorption spectrum of 1c++˙ generated by pulse radiolysis in aqueous solution6 shows three absorption bands at 290, 440 and 980 nm. The last one is typical of these systems and is ascribed to an intramolecular charge resonance interaction. In our case, the maximum at 290 nm is out of the detectable wavelength region, while the NIR absorption band was completely missing (Fig. 2). This spectral behavior is explained in the Discussion section. Moreover, the absorption at 350 nm is made up of two components, a short-lived one, probably due to the ketyl radical An2COH˙, that reacts quickly with molecular oxygen,6 and a longer-lived one coupled with the absorption at 420 nm. The kinetics analysis gave a lifetime of 6.6 μs for the long component, which is very close to the 6.8 μs value obtained at 420 nm. The rate constant calculated by first order fitting at 420 nm is 1.5 × 105 s−1 (Table 5). The time-evolution of the absorption spectra shows that the decay at 550 nm is well fitted by a first-order law (k = 7.7 × 106 s−1); it is faster than that obtained for 1a and 1b (k = 3.1 × 106 s−1) because of the higher O2 concentration. A narrow absorption band detected at 510 nm that builds up in the same amount time (τ = 7.0 μs) as the absorptions at 350 and 420 nm was assigned to the cation An2CH+ on the basis of literature data.13 This assignment is in agreement with the absence of O2 effects on the decay kinetics recorded at 510 nm (lifetimes of 49 and 50 μs were recorded in air-equilibrated and N2-saturated solutions, respectively).
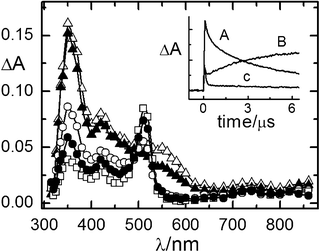 | ||
| Fig. 2 Time-resolved absorption spectra of the NMQ+BF4− (2.0 × 10−4 M)/toluene (1 M)/1c (9.0 × 10−3 M) system in O2-saturated CH2Cl2 recorded 0.08 (△), 0.13 (▲), 1.5 (○), 3.5 (●) and 6.4 (□) μs after the laser pulse. λecc = 308 nm. Inset: decay kinetics recorded at 350 (A), 510 (B) and 550 (C) nm. | ||
The time-resolved absorption spectra of 1d in air-equilibrated CH2Cl2 recorded by flash photolysis show the formation of both NMQ˙ (λmax = 540 nm)12 and 1d++˙ (λmax = 440 nm)5 within the laser pulse (Fig. 3). Their absorption is further replaced by the build up of the intense absorption signal at 370 nm ascribed to the cation AnC+(CH3)2 by comparison with its already reported spectrum.14 The nature of this transient is confirmed by the fact that molecular oxygen had little influence on its lifetime; in fact, τ values of 34 and 40 μs were measured in air-equilibrated and N2-saturated solution, respectively. In addition, it reacts quickly with water by a second-order kinetics (k2 = 5.0 × 106 M−1 s−1). By analyzing the kinetics, it is clear that NMQ˙ decays with a first-order kinetics (k = 3.1 × 106 M−1 s−1); the same rate constant value was found for 1a and 1b (this was expected because the experiments were performed with the same amount of O2). For 1d++˙ the change of absorbance at 440 nm follows a second order kinetics (k2/ε = 8.9 × 106 s−1 cm, Table 5), which is different from the cases of 1a++˙, 1b++˙ and 1c++˙ where a first order decay was found. Furthermore, it is reasonable to suppose that 1d++˙ is the precursor of the cation (λmax = 370 nm), even if the build up kinetics was prevented by its fast reaction.
 | ||
| Fig. 3 Time-resolved absorption spectra of the NMQ+BF4− (2.5 × 10−3 M)/toluene (1 M)/1d (1.0 × 10−2 M) system in air-equilibrated CH2Cl2 recorded 0.08 (△), 0.90 (▲), 3.5 (○) and 6.4 (●) μs after the laser pulse. λexc = 355 nm. Inset: decay kinetics recorded at 370 (A), 440 (B) and 540 (C) nm. | ||
Transient quantum yields were measured by flash photolysis experiments performed on 1c and 1d, according to the procedure reported in the Experimental section. In the case of 1c, the quantum yield of NMQ˙ is 0.60, with a value of 3800 M−1 cm−1 for the extinction coefficient in CH2Cl2 at 550 nm, measured with respect to the biphenyl radical cation (ε = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at λmax = 670 nm)15 in an independent experiment. In contrast, the An2CH+ quantum yield, obtained by using a value of 100000 M−1 cm−1 for the extinction coefficient of the cation,13 is quite low (0.034). Finally, concerning the experiments with 1d, a quantum yield of 0.69 was obtained for NMQ˙; this was close to the value of 0.62 obtained for the cation AnC+(CH3)2 by using a value of 11000 M−1 cm−1 for the extinction coefficient of cumyl cation.16
000 M−1 cm−1 at λmax = 670 nm)15 in an independent experiment. In contrast, the An2CH+ quantum yield, obtained by using a value of 100000 M−1 cm−1 for the extinction coefficient of the cation,13 is quite low (0.034). Finally, concerning the experiments with 1d, a quantum yield of 0.69 was obtained for NMQ˙; this was close to the value of 0.62 obtained for the cation AnC+(CH3)2 by using a value of 11000 M−1 cm−1 for the extinction coefficient of cumyl cation.16
The results of the steady-state photooxidation of alcohols 1a–d with DCA are in line with those reported for the well known oxidationvia photoinduced electron transfer of benzyl alcohol derivatives.1 In particular, substrates 1a–c, whose radical cations are able to deprotonate, form the respective carbonyl compounds, while compound 1d, whose radical cation cannot deprotonate, is practically unreactive. In order to gain a greater insight into the photooxidation mechanism, a detailed time-resolved investigation of substrate 1c was performed. Compounds 1a and 1b were not studied in detail because the cations generated by their radical cations absorb below 300 nm17 and could not be detected with our experimental setup.
The flash photolysis experiments were carried our in air-equilibrated CH3CN in the presence of 0.2 M biphenyl (BP) as a co-sensitizer; under these conditions the formation efficiencies of radical ions (eqns (4) and (5)) are higher.15
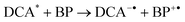 | (4) |
 | (5) |
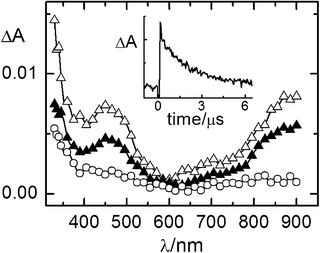 | ||
| Fig. 4 Time-resolved absorption spectra of the DCA (2.0 × 10−4 M)/biphenyl (0.2 M)/1c (1.0 × 10−2 M) system in air-equilibrated CH3CN recorded 0.32 (△), 1.9 (▲), 6.3 (○) μs after the laser pulse. λexc = 355 nm. Inset: decay kinetics recorded at 450 nm. | ||
Quantum mechanical calculations were performed at a semi-empirical level to investigate the effect of the interactions between 1c++˙ and BF4− on the UV-Vis absorption spectrum. The absorption spectra of the radical cation 1c++˙ and the complex (1c++˙/BF4−) calculated by INDO/1-CI, after geometrical optimization by PM3, are shown in Fig. 5. It was found that the most stable structure was due to a hydrogen bond between a fluorine atom of BF4− and the hydroxyl group of 1c++˙, where the main positive charge is predicted to be localized. These calculations predict that the 1c++˙/BF4− interaction shifts the absorption band from 400 to about 350 nm, in agreement with the absorption spectra obtained by laser flash photolysis of the DCA/1c and NMQ+BF4−/1c systems.
 | ||
| Fig. 5 UV-Vis absorption spectra of the free (a) and complexed with BF4− (b) radical cation 1c++˙ calculated by the INDO/1-CI semiempirical method after optimization with the PM3 Hamiltonian together with the respective oscillator strengths (bars). The singly excited configurations were built by using the 15 highest occupied MOs and the 15 lowest virtual MOs. | ||
In order to obtain more detailed information on the effect of BF4− on the reactivity of the radical cations here investigated, calculations at a DFT level were carried out on 1d++˙ (Fig. 6) by using the B3LYP/6-311G(d) model both in the absence and presence of the BF4− anion. The same theoretical model was also used to calculate the spin density and partial-charge distributions on the free and complexed radical cation 1d++˙; the results are summarized in Fig. 7.
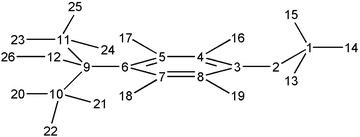 | ||
| Fig. 6 Numbering of 1d++˙ atoms. | ||
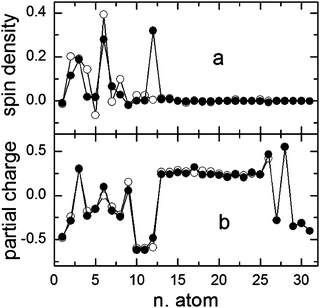 | ||
| Fig. 7 Spin-density (a) and partial-charge (b) distributions of the radical cation 1d++˙, free (open symbols) and associated with BF4− (full symbols) calculated by B3LYP/6-311G(d) after optimization with the same method. | ||
While the effect of the association between 1d++˙ and BF4− (also in this case the more stable complex was predicted to have a hydrogen bond between the hydroxyl hydrogen and a fluorine atom) on the charge distribution is practically negligible (Fig. 7b), the effect on the spin density is much more evident. In fact, a significant increase in spin density is predicted on the oxygen atom (number 12 of Fig. 6) which reaches a value of about 0.3 (compared with the value of about 1 calculated by the same theoretical model for the spin density on the oxygen atom of OH˙). Thus, a large part of the spin density of the complex is located on the C−O bond which therefore becomes more reactive than that in the radical cation (see Discussion).
Discussion
Reaction with DCA
The occurrence of an electron transfer as the primary step was confirmed by laser flash photolysis experiments that clearly showed the formation of the alcohol radical cations (Fig. 4). The high quenching rate constant values (Table 1) determined with DCA are close to the diffusion limit in CH2Cl2 (1.5 ×10 10 M−1 s−1)18 which is in line with an efficient electron transfer process. This is confirmed by the negative free energy change (ca.−0.4 eV) calculated by the Rhem–Weller’s eqn (6)19 where E0red* is the reduction potential of 1DCA* and E0ox is the oxidation potential of the benzyl alcohols 1a–d (1.52–1.59 V vs. SCE).20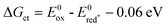 | (6) |
The product analysis showed higher reactivity in CH3CN with respect to the less polar CH2Cl2, which is in agreement with a reduced Coulombic barrier for the separation of the initially formed intermediates, the radical anion/radical cation pairs, in the more polar solvent.21 In fact, irradiation of DCA in CH2Cl2 in the presence of 1a–d leaves the alcohol almost unreacted, while in CH3CN, there was considerable conversion of 1a–c into the corresponding carbonyl products. Only alcohol 1d was practically unreactive in the latter solvent. From these experimental results, the mechanism already proven for analogous alcohols (Scheme 1), but under different experimental conditions, seems to be operative. The radical cation, produced by electron transfer from the alcohol to the singlet excited state of DCA, deprotonates thus forming a benzyl radical, that probably reacts with dioxygen (path a) to give the final carbonyl product. Path b should not be thermodynamically favored because DCA (E0DCA/DCA˙ = −0.98 V vs. SCE)10 should not be able to oxidize the radical AnC˙ROH (E0ox = −0.2 ÷−0.3 V vs. SCE).22,23 The sensitizer is re-established by the fast reaction of DCA−˙ with O2, thus forming O2−˙, owing to the reduction potential of DCA being slightly more negative than that of O2 (E0O2/O2−˙ = −0.87 V vs. SCE).24 The low yield of 4-methoxybenzophenone formed by 1d cannot be justified by the mechanism proposed for 1a–c, because the benzyl hydrogens are missing in this compound; anyway, no further investigation was carried on this mechanism.
Despite the presence of O2−˙ that could react with the radical cation 1c++˙,25 the first-order decay kinetics obtained for 1c++˙ (k = 5.0 × 105 s−1)26 suggests that the deprotonation is mainly due to interactions with the solvent.
Reaction with NMQ+BF4−
The sensitizer N-methylquinolinium is more efficient than DCA in promoting an electron transfer reaction; in fact, in agreement with the higher reduction potential of 1NMQ+*, the quenching rate constants are slightly higher in the case of NMQ. The electron transfer from the substrate to singlet excited state of quinolinium produces a radical cation/neutral radical pair whose separation is facilitated by the lack of any electrostatic barrier, even in moderately polar solvents. In solvents of low-polarity, the return electron transfer reaction is slower which makes the electron transfer process more efficient.21 These properties of NMQ+BF4− explain the higher reactivity shown with this sensitizer in CH2Cl2 with respect to CH3CN (Table 2). As with DCA, smaller amount of NMQ+BF4− was used with respect to the substrate since it is known that molecular oxygen can generate NMQ+ by oxidation of NMQ˙ even if the reduction potential of NMQ+ is −0.85 V (vs.SCE) because the electrostatic factor (0.24 eV in CH2Cl2) is responsible for the slightly exoergonic free energy change.19Regarding the steady-state experiments, considerable yields of dibenzyl ethers were obtained by using NMQ+BF4− (also obtained for 1a and 1c in the presence of TPP+BF4− as sensitizer),8 together with the carbonyl compounds from alcohols 1a–c, while only styrene was formed in the reaction of 1d sensitized by NMQ+ (Table 3). The advantage of using NMQ+BF4− instead of TPP+BF4− in photoinduced electron transfer processes is related to its ground state absorption spectrum. In fact, while the bleaching of the NMQ+BF4− ground state does not significantly affect the time-resolved absorption spectra above 330 nm (molar absorption coefficient smaller than 5000 M−1 cm−1), in the presence of TPP+BF4− a large wavelength region (ca. 330–450 nm, where the molar absorption coefficient reaches also values of ca. 3.3 × 104 M−1 cm−1) is hidden by the ground state bleaching of the sensitizer. Therefore it is difficult to detect transients and analyze their decay kinetics in this spectral region.
The experimental evidence suggests that the particular reactivity of the NMQ+-sensitized photolysis of 1a–d is probably due to the involvement of the BF4− anion which can associate with the radical cation of the alcohols. This kind of interaction has already been proposed to take place between aromatic cation radicals and anions such as PF6−, BF4− and ClO4− in disproportionation equilibria.27
One of the main proofs here proposed in agreement with the formation of a (AnRR1OH+˙/BF4−) complex is the time-resolved absorption spectra recorded by irradiation of NMQ+ in the presence of 1c in aerated CH2Cl2 (Fig. 2).28 The species formed within the laser pulse together with NMQ˙ and absorbing at 350 and 420 nm was identified as the complex (1c++˙/BF4−). This assignment is confirmed by the semiempirical INDO/1-CI calculations which can reproduce the spectral changes due to the (1c++˙/BF4−) interaction. This interaction prevents the intramolecular charge resonance interactions between the neutral donor and charged acceptor rings of the radical cation, as confirmed by the lack of the NIR absorption in the presence of BF4−. The presence of a charge transfer complex is also in agreement with the solvent effect on reactivity. In fact, the higher reactivity noted in CH2Cl2 with respect to that obtained in CH3CN can be justified on the basis of a better separation of the radical cation/NMQ˙ pairs formed in the ET process in a less polar solvent. A further contribution could be due to the higher stability of the complex in CH2Cl2, since the ions were less solvated in this solvent.
The intense absorption of the ground state of NMQ+ below 330 nm prevented the investigation of this spectral region; in any case, the formation of the complex radical cation/BF4− is also extended in bona fides to alcohols 1a, 1b and 1d.
On the basis of experimental and theoretical evidence collected in this work, the mechanism reported in Scheme 5 is suggested for the NMQ+ photosensitized oxidation of alcohols 1a–d. With alcohols 1a, 1b and 1c, the complex radical cation/BF4− could react according to two paths. Firstly, according to path a (analogous to path a in Scheme 1 already noted in the DCA-photosensitized oxidation) the radical cation deprotonates to form the benzyl radical that evolves to the final carbonyl product by reacting with O2. The oxidation of the benzyl radical can be excluded because its oxidation potential is well above the reduction potential of NMQ+, as already seen in the case of DCA. Secondly, path b involves the formation of a cationic intermediate, probably assisted by O2−˙ (see below) that reduces the incipient hydroxyl radical (as indicated from the quantum mechanical calculations).29 The cation forms the symmetrical ether by reacting with the neutral alcohol (path c). There was direct evidence for the formation of the benzyl cation only with 1c (λmax = 510 nm, Fig. 2). With the other alcohols the absorption of the cations is hypsochromic, shifted out of the detectable spectral region, but the formation of symmetrical ethers in the product mixture suggests the involvement of this intermediate in the reaction.
 | ||
| Scheme 5 | ||
Considering the strong acidity of this kind of radical cation,30 path b should be a less important reaction pathway; this is confirmed by the low quantum yield (0.035) of the cation formed in the case of 1c; the value is about 6% of the quantum yield of radical cation (0.60). Instead, the formation yields of the ether (Table 4) were surprisingly high. The kinetics of 1a++˙, 1b++˙ and 1c++˙ follow first order decays (Table 5), as expected when the deprotonation process is the main decay path. The close values of the decay rate constants (of the order of 105 s−1, as already found for similar radical cations) can be justified with the formation of an early transition state of the deprotonation reaction of the alkylaromatic radical cations.6
Path a of Scheme 5 is excluded in the case of 1d because no benzyl hydrogens are present in the substrate; therefore, the formation of AnC+(CH3)2 is the only reactive way. The direct evidence of this intermediate in the absorption spectra (λmax = 370 nm, Fig. 3) supports such a hypothesis. Furthermore, the efficiency of the formation of 4-methoxycumil cation was high (0.62), close to the quantum yield of the corresponding radical cation (0.69) and in line with the high stability of the cation.31 This cation, in contrast with the others, loses one proton thus forming the corresponding styrene (Scheme 5, path d). When styrene is cumulated, its double bond is attacked by the cationic intermediate and gives the mixture of substituted p-methoxystyrenes (4f and 4g) reported in Table 3. The driving force of path c seems to be the stability of the substituted olefin; in fact, alcohol 1b that could lose the proton of the β carbon, does not form a detectable amount of styrene.
Two things suggest the involvement of O2−˙ in the formation of the cation by Scheme 5, path b: (i) the very high reactivity of the hydroxyl radical that makes the monomolecular process of cation formation too energy-expensive and (ii) the second-order decay of 1d+˙ which suggests a reaction with a species at a similar concentration, like O2−˙. A reaction between OH˙ and O2−˙, which could favor the formation of the cation, has already been reported in the literature.32 The higher reactivity of the complex (1d++˙/BF4−) in comparison with the “free”1d++˙ radical cation is in line with the increased spin density in the O atom which reaches a value of 30% in the complex.
The quantum yield of ether formation, at least for 1b and 1c (i) is much larger than that of the cation, (ii) depends on the irradiation time and (iii) becomes larger than unity (Table 4). The findings suggest the involvement of a thermal chain pathway in the formation of the final products. Scheme 6 depicts a probable reaction pathway where the cation interacts with the neutral alcohol, bonded by a hydrogen bond with another alcohol molecule (a), and releases the ether and a protonated alcohol molecule (b). The cation can then be re-generated by the loss of a water molecule from the protonated alcohol molecule.
 | ||
| Scheme 6 | ||
In contrast, the final product of 1d was 4-methoxystyrene, with a quantum yield less than one, independent of the irradiation time. This suggests that for this compound the deprotonation of the cation, probably assisted by the hydroxyl anion formed in path b of Scheme 5, is much faster than the interaction with the neutral alcohol.
The results of this investigation show that the previously reported mechanism (Scheme 3),8 in which the benzyl cations, formed through deprotonation of the radical cations by interaction with the neutral alcohols, are precursors of the final ethers, is not applicable for several reasons: (i) the reactivity of the radical cation is different when sensitized with different sensitizers, (ii) the decay rate constant of the radical cation and the growth of the cation (Fig. S4 of ESI)† are independent of the ground state concentration of the alcohol and (iii) the formation of the cation from compound 1d could not be explained.
Experimental
Materials
4-Methoxybenzyl alcohol (1a), 4-methoxy-α-methylbenzyl alcohol (1b), and 4,4′-dimethoxydiphenylmethanol (1c) were commercial samples (Aldrich). N-Methylquinolinium tetrafluoroborate was prepared according to a literature procedure.339,10-Dicyanoanthracene (Kodak) was recrystallized from pyridine. CH2Cl2 (Carlo Erba, HPLC grade) was purified by letting it stand with CaH2 and distilling as needed. CH3CN (Fluka for UV spectroscopy) and toluene (Carlo Erba, RPE) were used as received.
4-Methoxy-α,α′-dimethylbenzyl alcohol (1d) was prepared by reacting 4-methoxyacetophenone with Grignard reactant CH3MgI, obtained by adding CH3I to a Mg suspension in anhydrous ethyl ether. After workup, the crude was purified by column chromatography (neutral alumina, eluent n-hexane/ethyl ether 6:4) and identified by 1H NMR34 and GC-MS; m/z (70 eV, EI): 166 (M+, 21%), 151 (100), 135 (7), 121 (4), 109 (7), 91 (3), 77 (12), 65 (5), 51 (3).
Bis(4-methoxy-α-methylbenzyl) ether (3b) was prepared by reacting alcohol 1b in dimethyl sulfoxide at 175 °C for 15 min.35 The reaction mixture was chromatographed on a silica gel column (eluent n-hexane/ethyl ether 9:1). The product was a mixture of two diastereomers (meso and dl isomers) in different amounts. They were identified by 1H NMR and GC-MS; δH(200 MHz, CDCl3, Me4Si) of diastereomer A: 7.27 (4 H, d, Ar), 6.84 (4 H, d, J = 8.7 Hz, Ar), 4.47 (2 H, q, J = 6.6 Hz, CH), 3.81 (6 H, s, OCH3), 1.45 (6 H, d, J = 6.4 Hz, CH3); diastereomer B: 7.27 (4 H, d, Ar), 6.92 (4 H, d, J = 8.7 Hz, Ar), 4.18 (2 H, q, J = 6.6 Hz, CH), 3.84 (6 H, s, OCH3), 1.36 (6 H, d, J = 6.4 Hz, CH3). Two distinguishable peaks were obtained by GC-MS analysis of the two diastereomers, which corresponded to the same mass spectrum. m/z (70 eV, EI): 286 (M+, 2%), 207 (1), 178 (7), 151 (4), 135 (100), 121 (13), 105 13), 91 (17), 77 (20), 65 (8), 51 (4).
Bis(4-methoxybenzyl) ether (3c) was prepared by performing the NMQ+BF4− photosensitized reaction of alcohol 1c in a preparative scale. The product was isolated by column chromatography (silica gel, eluent n-hexane/ethyl ether 9:1) and identified by 1H NMR and GC-MS.36
4-Methoxy-α-methylstyrene (4e) was prepared by reacting alcohol 1d with HClO4 in CH3CN for 5 min. After workup, the crude product was isolated by column chromatography (neutral alumina, hexane as eluent) and identified by 1H NMR and GC-MS.372,4-Bis(4-methoxyphenyl)-4-methyl-1-pentene (4f) and E-2,4-bis(4-methoxyphenyl)-4-methyl-2-pentene (4g) were obtained as a mixture by column chromatography (neutral alumina, n-hexane/ethyl ether 9:1) in the preparation of 4e. They were identified by 1H NMR, 13C-NMR and GC-MS; δH(200 MHz, CDCl3, Me4Si) of 4f: 7.15 (2 H, d, Ar), 7.08 (2 H, d, Ar), 6.70 (4 H, d, Ar), 5.01 (1 H, d, CH), 4.63 (1 H, m, CH), 3.70 (6 H, s, OCH3), 2.68 (2 H, s, CH2), 1.12 (6H, s, CH3); 4g: 7.25 (4 H, d, Ar), 6.79 (2 H, d, Ar), 6.75 (2 H, d, Ar), 5.96 (1 H, m, CH), 3.73 (6 H, s, OCH3), 1.55 (3 H, s, CH3), 1.48 (6 H, s, CH3); δC(200 MHz, CDCl3) of 4f: 158.4 157.1, 135.8, 135.3 (ipso, Ar), 127.0, 126.7 and 113.3 and 112.8 (CH, Ar), 142.7 (CH styrene), 115.4 (CH2styrene), 55.2 e 55.1 (OCH3), 38.0 (quaternary styrene), 28.8 (CH3); 4g: 158.4 157.1, 135.8, 135.3 (ipso, Ar), 127.0, 126.7 and 113.3 and 112.8 (CH, Ar), 135.3 (quaternary), 128.2 (CH styrene), 55.1 (OCH3), 29.6 (quaternary), 31.6 (CH3); m/z (70 eV, EI) of 4f: 296 (M+, 3%), 175 (3), 149 (100), 133 (3), 121 (10), 109 (7), 91 (5), 77 (3), 65 (2), 51 (2); 4g: 296 (M+, 93%), 281 (100), 265 (10), 227 (5), 188 (7), 173 (67), 158 (17), 135 (17), 121 (18), 91 (8), 77 (7), 65 (3), 51 (1).
Steady state photolysis
Photooxidation reactions were carried out in an Applied Photophysics photoreactor equipped with 6 lamps of 15 W each (310 ± 20 nm with NMQ+BF4−, 410 ± 20 nm with DCA). A solution containing the alcohol (2.0 × 10−2 M) and the sensitizer (NMQ+BF4−, 1.0 × 10−3 M or DCA, 3.0 × 10−4 M) in air-saturated solvent (CH3CN or CH2Cl2) was irradiated in a cylindrical flask provided with a water cooling jacket. The mixture was analyzed after adding an internal standard (bibenzyl) by GC, GC-MS and 1H-NMR and all the products were identified by comparison with authentic specimens.Product analysis was carried out on a HP 6890-2 gas chromatograph (capillary column, 30 m), on an HP 6890 gas chromatograph equipped with a MSD-HP 5973 mass selective detector and on a Bruker AC 200-NMR spectrometer. The material balance was always satisfactory (>90%). The sensitizer analysis was performed by optical density measurements on a HP-8451 diode array spectrophotometer. No product was formed in a blank experiment, carried out in the dark condition or by irradiating the solutions in the absence of the sensitizer.
The amount of H2O2 was quantitatively determined by titration with iodide anion. The water solution, obtained from the reaction mixture workup, was treated, after dilution, with an excess of KI and a few drops of AcOH. The amount of I3− formed was determined by spectrophotometric analysis (ε = 25000 M−1 cm−1 at λmax = 361 nm).38 No H2O2 was formed in blank experiments performed in the dark.
Fluorescence quenching
Measurements were carried out on a Spex Fluorolog F112AF spectrofluorometer. Relative emission intensities at 400 nm (NMQ+BF4− emission maximum) or 434 nm (DCA emission maximum) were measured by irradiating at 316 nm (NMQ+BF4− absorption maximum) or 400 nm (DCA absorption maximum) a solution containing the sensitizer (1 × 10−5 M) with the substrate at different concentrations (from 2 × 10−3 to 1.5 × 10−2 M) in air-saturated CH2Cl2. The error estimated on the Stern–Volmer constants (KSV) was ±5%.Laser flash photolysis
An excitation wavelength of 355 nm (from a Nd:YAG laser, Continuum, third harmonic, pulse width ca. 7 ns and energy <3 mJ per pulse) was used in nanosecond flash photolysis experiments.39 The transient spectra were obtained by a point-to-point technique, monitoring the change of absorbance (ΔA) after the laser flash at intervals of 5–10 nm over the spectral range 300–900 nm, averaging at least 10 decays at each wavelength. The lifetime values (the time at which the initial signal is reduced to 1/e) are reported for the transients showing first-order decay kinetics. A 2 ml solution containing the substrate (1.0 × 10−2 M), the sensitizer (NMQ+BF4−, 2.5 × 10−3 M or DCA, 2.0 × 10−4 M) and the cosensitizer (1 M toluene with NMQ+BF4− and 0.5 M biphenyl with DCA) was flashed in a quartz photolysis cell, while nitrogen or oxygen was bubbling through them. The experimental error was ±10%.Transient quantum yields (ϕTr) were obtained by using the relationship (eqn 8) between quantum yields, absorption coefficient (ε), and changes of absorbance (ΔA), measured at the corresponding absorption maxima of the transient (Tr) and the reference (ref.), namely triplet benzophenone (ϕT = 1 and εT = 6500 M−1 cm−1 at 520 nm).40 The experimental error on ϕTr was ±15%. All measurements were carried out at 22 ± 2 °C.
 | (8) |
Quantum yields
A 2 mL solution of NMQ+BF4− (3.9 × 10−4 M) and 1b–d (1.0 × 10−2 M) in air-saturated CH3CN was placed in a quartz cell and irradiated at 313 nm, selected with a Balzer interference filter by a high pressure Hg lamp. The photoproducts were quantified by GC and 1H-NMR analysis, by using bibenzyl as internal standard. The light intensity (ca. 1 × 1015photons s−1) was measured by potassium ferric oxalate actinometry.Quantum mechanical calculations
The lowest excited state energies, electronic structures and transition probabilities of 1c++˙ and its complex with the anion BF4− were investigated by semi-empirical computational methods. The geometry of the substrates was optimized using the PM3 model,41 while the transition energies and oscillator strengths were calculated by the INDO/1-CI model42 (ZINDO program of CAChe 7.05 package). The configuration interaction (CI) calculations included the singly excited configurations built on the 15 highest occupied (HOMO) and 15 lowest virtual (LUMO) molecular orbitals. In order to investigate the effect of BF4− on the charge and spin-density distributions of the radical cations of the substrates, ab initio calculations were performed on 1d++˙ at a DFT level (B3LYP/6-311G(d)) with Gaussian 0343 after optimization of the molecular structures with the same method.Conclusions
The photooxydation of the methoxybenzyl alcohols 1a–d, sensitized by 9,10-dicyanoanthracene (DCA) and N-methylquinolinium tetrafluoroborate (NMQ+BF4−) produced the radical cation of the alcohol and the corresponding reduced form of the sensitizer within the laser pulse.When the sensitizer was DCA, the back electron transfer process was operative in CH2Cl2; all the substrates were left unreacted. Alcohol 1d was unreacted also in CH3CN. For 1a–c in CH3CN, where the benzyl hydrogen is present, the radical cation was able to deprotonate and then form the corresponding carbonyl compounds. The same behavior was found when compounds 1a–c were sensitized by NMQ+BF4− in both CH2Cl2 and CH3CN.
A parallel decay path of the 1a–d radical cations with formation of the AnC+RR1 cation was operative in both CH2Cl2 and CH3CN when the sensitizer was NMQ+BF4−. The cation was a precursor of ethers (1a–c) and styrene (1d) (see Scheme 5, path b).
Acknowledgements
Thanks are due to the Ministero dell’Università e della Ricerca and the University of Perugia (Italy) for financial support.References
- (a) S. Fukuzumi, K. Yasui, T. Suenobu, K. Ohkbo, M. Fujitsuka and O. Ito, J. Phys. Chem., 2001, 105, 10501–10510 Search PubMed; (b) S. Fukuzumi, S. Itoh, T. Komori, T. Suenobu, A. Ishida, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2000, 122, 8435–8443 CrossRef CAS; (c) W. Tong, H. Ye, H. Zhu and V. T. D’Souza, J. Mol. Struct. (THEOCHEM), 1995, 333, 19–27 CrossRef CAS.
- (a) T. Del Giacco, C. Rol and G. V. Sebastiani, J. Phys. Org. Chem., 2000, 13, 745–751 CrossRef CAS; (b) T. Del Giacco, M. Ranchella, C. Rol and G. V. Sebastiani, J. Phys. Org. Chem., 2003, 16, 127–132 CrossRef CAS; (c) S. Vijaikumar, N. Somasundaram and C. Srinivasan, Appl. Catal., A, 2002, 223, 129–135 CrossRef CAS.
- E. A. Mayeda, L. L. Miller and J. F. Wolf, J. Am. Chem. Soc., 1972, 94, 6812–6816 CrossRef CAS.
- E. Baciocchi, S. Belvedere, M. Bietti and O. Lanzalunga, Eur. J. Org. Chem., 1999, 5, 1785–1793 CAS.
- E. Baciocchi, M. Bietti and S. Steenken, Chem.–Eur. J., 1999, 5, 1785–1793 CrossRef CAS.
- M. Bietti and O. Lanzalunga, J. Org. Chem., 2002, 67, 2632–2638 CrossRef CAS.
- M. Martiny, E. Steckhan and T. Esch, Chem. Ber., 1993, 126, 1671–1682 CAS.
- B. Branchi, M. Bietti, G. Ercolani, M. A. Izquierdo, M. A. Mirando and L. Stella, J. Org. Chem., 2004, 69, 8874–8885 CrossRef CAS.
- The lifetime of DCA singlet state is 13.1 μs in nitrogen degassed CH2Cl2. A. F. Olea, D. R. Worrall, R. Wilkison, S. L. Williams and A. A. Abdel-Shafi, Phys. Chem. Chem. Phys., 2002, 4, 161–167 Search PubMed.
- J. Eriksen and C. S. Foote, J. Phys. Chem., 1978, 82, 2659–2662 CrossRef CAS.
- K. P. Dockery, J. P. Dinnocenzo, S. Farid, J. L. Goodman, I. R. Gould and W. P. Tood, J. Am. Chem. Soc., 1997, 119, 1876–1883 CrossRef CAS.
- G. Guirado, C. N. Fleming, T. G. Ligenfelter, M. L. Williams, H. Zuilhof and J. P. Dinnocenzo, J. Am. Chem. Soc., 2004, 126, 14086–14094 CrossRef CAS.
- J. Bartl, S. Steenken, H. Mayr and R. A. MeClelland, J. Am. Chem. Soc., 1990, 112, 6918–6928 CrossRef CAS.
- F. Cozens, V. M. Kanagasabapathy, R. A. McClelland and S. Steenken, Can. J. Chem., 1999, 77, 2069–2082 CrossRef CAS.
- I. R. Gould, D. Ege, J. E. Moser and S. Farid, J. Am. Chem. Soc., 1990, 112, 4290–4301 CrossRef CAS.
- G. A. Olah, C. U. Pittman, R. Waack and M. Doran, J. Am. Chem. Soc., 1966, 88, 1488–1495 CrossRef CAS.
- R. A. McClelland, C. Chan, F. Cozens, A. Madro and S. Steenken, Angew. Chem., Int. Ed. Engl., 1991, 30, 1337–1339 CrossRef.
- Handbook of Photochemistry, ed. S. L. Murov, M. Dekker, New York, 1973 Search PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CAS.
- Measured by Dr Marta Bettoni by cyclic voltammetry in CH3CN.
- P. W. Todd, J. P. Dinnocenzo, S. Farid, J. L. Goodman and I. R. Gould, J. Am. Chem. Soc., 1991, 113, 3601–3602 CrossRef CAS.
- T. Lund, D. D. M. Wayner, M. Jonsson, A. G. Larsen and K. Daasbjerg, J. Am. Chem. Soc., 2001, 123, 12590–12595 CrossRef CAS . Indeed, the potentials are referred to unsubstituted ring, anyway a methoxy group can not increase the oxidation potential values up to the reduction potential DCA23.
- B. A. Sim, P. H. Milne, D. Griller and D. D. M. Wayner, J. Am. Chem. Soc., 1980, 102, 6635–6638.
- D. T. Sawyer, G. Chiericato, Jr, C. T. Angelis, E. J. Nanni, Jr and T. Tsuchiya, Anal. Chem., 1982, 54, 1720–1724 CrossRef CAS.
- Hydrogen peroxide was revealed at the end of the photolysis by iodometric titration, hence some acidity is finally neutralized by O2−˙.
- This value is similar to that already reported: E. Baciocchi, T. Del Giacco and F. Elisei, J. Am. Chem. Soc., 1993, 115, 12290–12295 Search PubMed.
- E. E. Bancroft, J. E. Pemberton and H. N. Blount, J. Phys. Chem., 1980, 84, 2557–2560 CrossRef CAS.
- For instance, the effect of salt addition on absorption spectra is known for aromatic radical cations forming the contact ion pairs with the anion PF6−. Y. Yamamoto, T. Aoyama and K. Hayashi, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2209–2214 Search PubMed.
- H. Ronsen and S. J. Klebanoff, J. Exp. Med., 1979, 149, 27–39 CrossRef.
- E. Baciocchi, Acta Chem. Scand., 1990, 44, 645–652 CrossRef CAS.
- J.-L. M. Abbound, I. Alkorta, J. Z. Davalos, P. Müller, E. Quintanilla and J.-C. Rossier, J. Org. Chem., 2003, 68, 3786–3796 CAS.
- NMQ˙ should be a possible reactant, but the reaction with O2 is faster (τ = 0.32 μs in air-equilibrated CH2Cl2) than that of the radical cation decay (τ = 2.2 μs).
- P. F. Donovan and D. A. Conley, J. Chem. Eng. Data, 1966, 11, 614–615 CrossRef CAS.
- R. Jacquesy and J. F. Patoiseau, Bull. Soc. Chim. Fr., 1978, 255–262 CAS.
- J. Emert, M. Goldenberg and G. L. Chiu, J. Org. Chem., 1977, 42, 2012–2013 CrossRef CAS.
- C. Henneuse, K. Gillard, O. Noiset and J. Marchand-Brynaert, Bull. Soc. Chim. Fr., 1995, 132, 333–336 CAS.
- D. Shukla, C. Lu, N. P. Schepp, W. C. Bentrude and L. J. Johnston, J. Org. Chem., 2000, 65, 6167–6172 CrossRef CAS.
- S. Fukuzumi, S. Kuroda and T. Tanaka, J. Am. Chem. Soc., 1985, 107, 3020–3027 CrossRef CAS.
- (a) A. Romani, F. Elisei, F. Masetti and G. Favaro, J. Chem. Soc., Faraday Trans., 1992, 88, 2147–2154 RSC; (b) H. Görner, F. Elisei and G. G. Aloisi, J. Chem. Soc., Faraday Trans., 1992, 88, 29–34 RSC.
- I. Carmichael and G. L. Hug, J. Phys. Chem. Ref. Data, 1986, 15, 1–250.
- (a) J. J. P. Stewart, J. Comput. Chem., 1989, 10, 209–220 CrossRef CAS; (b) J. J. P. Stewart, J. Comput. Chem., 1989, 10, 221–264 CrossRef CAS.
- M. C. Zerner, Semiempirical molecular orbital methods, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, VCH, New York, 1991, vol. 2, pp. 313–365 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford, CT, 2004.
Footnote |
| † Electronic supplementary information (ESI) available: Stern–Volmer plots of DCA and NMQ+BF4− fluorescence by 1a–d, time-resolved absorption spectra of 1a, growth of An2CH at different concentrations of 1c. See DOI: 10.1039/b711541e |
| This journal is © the Owner Societies 2008 |