The hydration of glucose: the local configurations in sugar–water hydrogen bonds†
Teppei
Suzuki
*
Advanced Research Institute for Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku, Tokyo, 169-8555, Japan. E-mail: teppei_suzuki@moegi.waseda.jp; Fax: +81 (0)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32071488; Tel: +81 (0)352863378
32071488; Tel: +81 (0)352863378
First published on 19th November 2007
Abstract
The hydration of a simple sugar is an essential model for understanding interactions between hydrophilic groups and interfacial water molecules. Here I perform first-principles molecular dynamics simulations on a glucose–water system and investigate how individual hydroxyl groups are locally hydrated. I demonstrate that the hydroxyl groups are less hydrated and more incompatible with a locally tetrahedral network of hydrogen bonds than previously thought. The results suggest that the hydroxyl groups form roughly two hydrogen bonds. Further, I find that the local hydration of the hydroxyl groups is sensitively affected by seemingly small variations in the local electronic structure and bond polarity of the groups. My findings offer insight into an atomic-level understanding of sugar–water interactions.
Introduction
A detailed description of hydrogen bonds (H-bonds) is the key to understanding the properties of any H-bonded system. A perfect example is liquid water, which is held together by a three-dimensional, locally tetrahedral network of H-bonds.1–3 From a physicochemical standpoint, an H-bond can basically be viewed as a special kind of dipole–dipole interaction or as an interaction between bond dipoles.4,5 Hence, polar groups such as hydroxyl groups can readily form H-bonds with highly polar water molecules.Over the last half-century, hydroxyl groups of simple sugars, or monosaccharides, have generally been assumed to mimic the locally tetrahedral character of the H-bond network in liquid water with a little distortion because they contain a fragment of a water molecule.6–9 However, there are some computational studies suggesting that the hydroxyl groups are not as hydrated as they could potentially be.10–12
Here I perform ab initio molecular dynamics (MD) simulations13,14 on a glucose–water system, an essential model for molecular biology,15 to address this contradictory issue. While previous works focused on the overall hydration structure of glucose and provided many insights, the local H-bonding configurations in sugar–water H-bonds have been little addressed. My position, however, is that an accurate understanding of how individual hydroxyl groups are locally hydrated should precede a better understanding of the overall hydration structure. To put it another way, the microscopic hydration of glucose cannot be properly understood unless the local H-bonding interactions between the hydroxyl groups and water molecules are accurately understood.
Hence, my approach is entirely different from previous ones; the main objective of this work is to analyse the local interactions between the hydroxyl groups and water molecules instead of the overall hydration structure. For this reason, the present work focuses exclusively on the local H-bonding configurations and electronic properties.
By analysing the electronic and H-bonding properties, I provide a quantitative reason why the local hydration of the hydroxyl groups is not predominantly three-coordinated but roughly two-coordinated. I also show that the hydroxyl groups usually enhance the dipole moment of water molecules in the first hydration shell, but only when the groups form donor H-bonds. Together with this, my detailed analysis demonstrates that the local H-bonding properties alter more sensitively from one hydroxyl group to another than previously conceived. All this suggests that the hydroxyl groups are more incompatible with a locally tetrahedral H-bond network than previously thought.
Methods
The glucose–water system
The glucose–water system consisted of one glucopyranose molecule and 60 water molecules in a cubic supercell of side 12.54 Å (about 1 g cm−3 density) (Fig. 1a and 1b), with periodic boundary conditions.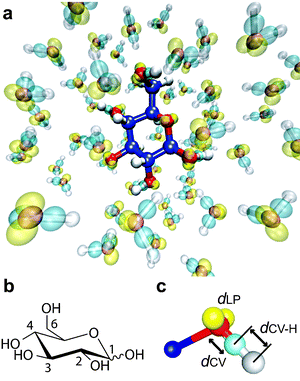 | ||
| Fig. 1 The glucose–water system. (a) A snapshot from an ab initio MD simulation on glucopyranose in aqueous solution. Carbon, oxygen and hydrogen atoms are represented by blue, red and white respectively. Transparent yellow and light blue represent the centres of localized Wannier functions associated with lone pairs and covalent OH bonds respectively. The molecular graphic was created by VMD.57 (b) The drawing of glucopyranose with the numbering scheme of its carbon atoms. The wavy bond at the anomeric site (C1) indicates either the α or β anomer. (c) A localized orbital description of the electronic properties of a hydroxyl group. The colour representation is the same as in (a). To evaluate the electronic properties, I used three distances: dLP, dCV and dCV–H. (The same definitions hold for water molecules.) The distance dLP is the average distance from the oxygen atom to the two centres of the lone pair Wannier orbitals. The distances dCV and dCV–H are the distances from the centre of the covalent OH-bond orbital to the oxygen atom and to the hydrogen atom respectively. The molecular graphic was created by gOpenMol.58 | ||
The number of water molecules in this system may seem small compared with the number that can readily be handled in classical force-field MD simulations. Nonetheless, this system size allows one to investigate important aspects of water molecules in the first hydration shell, as already proven by previous ab initio MD studies on simple sugars in aqueous solution with similar system sizes10,12,16,17 or smaller.18 In fact, Stubbs and Marx performed force-field MD simulations on the hydration of glucose by changing the number of water molecules to assess the effect of the system size,16 and they found that increasing the system size has little effect on the pair correlation function between a glucose oxygen-atom and water oxygen-atoms. The study suggested that 57 water molecules are enough to study the behaviour of water molecules in the first hydration shell using the ab initio MD method.
Note that investigating the behaviour of water molecules in the second and third hydration shells is beyond the scope of the present work. Instead, my focus is exclusively on H-bonds between the hydroxyl groups and water molecules in the first hydration shell.
The ab initio MD method
All the ab initio MD simulations were performed using CPMD code.13,14,19 The electronic structure calculations were carried out in the framework of the Kohn–Sham formulation of density functional theory (DFT).20,21 The gradient-corrected BLYP functional was used.22,23 The valence–core interactions were treated by norm-conserving pseudopotentials by Goedecker, Teter and Hutter, which give the optimal efficiency in numerical calculations when plane waves are used as a basis set.24 Only the Γ point was used to sample the Brillouin zone. The Nosé–Hoover chain thermostat method25,26 was used to control the ionic temperature at about 300 K; and after this stage (about 3 ps), microcanonical simulations were then performed for data collection (6 to 10 ps; for more detail, see Table S1 in the ESI†). The trajectories were sampled every ten steps, and the maximally localized Wannier functions27,28 were also computed after every ten steps.Conformations
Carbohydrates generally have the potential to form multiple conformational families in aqueous solution. In this regard, simulating carbohydrates is a great challenge from a theoretical and computational standpoint—and this is especially true of ab initio MD simulations. In fact, in previous ab initio MD simulations on glucose in aqueous solution, conformers are relatively limited.10I performed seven simulations on the α anomer and seven on the β anomer, where the three staggered hydroxymethyl rotamers were taken into account. (Note that the aim of this study is not to explore fully the conformational space of glucose in aqueous solution.) These hydroxymethyl rotamers are usually characterized by the O5–C5–C6–O6 torsion angle (see Fig. S1 in the ESI†). The computational details of the simulations on the conformers are summarized in Table S1 in the ESI.† The results of the conformations in the hydroxymethyl group are summarized in Table S2 in the ESI.†
Data analysis
Since my main objective was to understand the basic differences between water–water H-bonds and sugar–water H-bonds, the differences in the local hydration structure among the conformers were beyond the scope of this work. When I calculated the electronic, the structural and the H-bonding properties, the data were first averaged for each simulation, and then they were again averaged over all the simulations.This procedure may seem to be a slightly simplified approach, since individual conformers exist in different population ratios; however, the procedure is still considered reasonable if the following points are taken into account. First, since some of the population ratios remain uncertain both experimentally and theoretically (e.g. the population ratios of hydroxymethyl rotamers are still somewhat controversial17), it would be inappropriate to add such uncertainty into the subsequent analysis. Second, the length of each simulation is unavoidably limited because of the high computational cost. Therefore, (i) the differences in the electronic properties of the hydroxyl groups among the conformers are difficult to assess accurately; (ii) each MD trajectory may not necessarily represent all the features of the hydration structure of individual conformers.
Because of all this, in the present context averaging over the simulations with equal weighting is considered to be practical. This procedure nonetheless allows the sampling of a variety of configurations in which individual hydroxyl groups were hydrated in different ways; it still ensures the statistical reliability for my main objective.
I also computed the dipole moments of water molecules in the first hydration shell using the method of maximally localized Wannier functions.27,28 (The averaging procedure was the same as I described above.) The dipole moments of water molecules were obtained using the following numbers of electronic configurations of water molecules: (i) water molecules forming acceptor H-bonds with the hydroxyl groups, about 474000 configurations; (ii) water molecules forming donor H-bonds, about 579000; (iii) water molecules that form H-bonds with water molecules, about 4960000; (iv) water molecules that form H-bonds with the individual hydroxyl groups, about 93000–98000 each.
A localized orbital approach
In modern ab initio MD simulations, the method of maximally localized Wannier functions, a localized orbital approach, plays an important role.27 This approach makes it possible to study the electronic properties of hydroxyl groups of glucose intuitively and accurately, even if the molecule is surrounded by water molecules (Fig. 1a). In this localized orbital picture, the chemical bonding of a hydroxyl group that is mainly relevant for H-bonding can be described by three localized orbitals: two lone pair orbitals (the acceptor side) and one orbital associated with the covalent OH-bond (the donor side) (Fig. 1c). I carefully investigated the two sides.Results and discussion
The acceptor side
How much does that shrinkage affect the local configurations on the acceptor side? To answer this question, I plotted the number of acceptor H-bonds (Nacc) as a function of dLP (Fig. 2a). Since the anomeric carbon (C1) is bonded to two oxygen atoms and is thereby located in a chemically different environment (Fig. 1b), I divided the data into two subgroups: one consisting of the data points from O2, O3, O4 and O6 (Fig. 2a, open circles), and the other from O1 (Fig. 2a, closed triangles). The correlation coefficient of the former was 0.87, whereas that of the latter was 0.88. In either case, Nacc was highly correlated with dLP [the correlation coefficient for the whole data set was 0.84 (Fig. 2a, solid line)]. The results demonstrate that the shrinkage of dLP is a good measure of how weakly the hydroxyl groups form acceptor H-bonds.
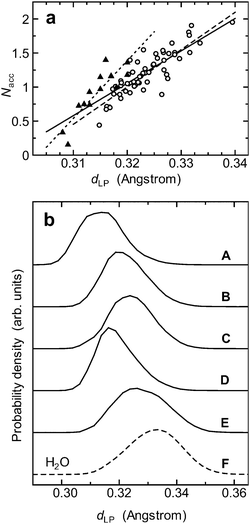 | ||
| Fig. 2 The electronic and H-bonding properties of the acceptor side of the hydroxyl groups. (a) The number of acceptor H-bonds (Nacc) is plotted as a function of the distance dLP. For the definition of dLP, see Fig. 1c. The criteria used for an H-bond are OA⋯H ≤ 2.4 Å and the OA⋯H–OD angle > 120°, where OA and OD–H are an H-bond acceptor and an H-bond donor respectively. An individual data point represents the average values for Nacc and dLP from a hydroxyl group in a simulation; hence, the plot consists of 70 data points (5 hydroxyl groups; 14 simulations). In this plot, the data are divided into two subgroups: one consists of the data points from O2, O3, O4 and O6 (open circles) and the other from O1 (closed triangles). The least-squares lines for the subgroups are shown by the dashed and the dotted lines respectively. The solid line is the least-squares line obtained using all the data points. (b) The site-dependent differences in the shrinkage of dLP: (A) OH1; (B) OH2; (C) OH3; (D) OH4; (E) OH6; (F) bulk water molecules (denoted by the dashed curve). | ||
This seemingly small shrinkage of dLP plays a key role in the local H-bonding configurations on the acceptor side. As the distance dLP shrinks from 0.34 to 0.32 Å, the number Nacc roughly decreases from 2 to 1 (Fig. 2a, solid line); that is, a 6% shrinkage of dLP leads to a 50% drop in Nacc. This is basically due to a contact-like character of H-bonding interaction that is turned on when lone pairs touch a hydrogen atom of a water molecule but is turned off as soon as the contact is broken.30 In the language of hybridization, one could say that upon the shrinkage of dLP, the lone pairs of the hydroxyl groups have more s character and less p character than those of a water molecule.31
To be more precise, however, H-bonding originates from various kinds of physical interactions; it is a complex interaction that can arise from electrostatic interactions (or electrostatic plus polarization interactions), charge transfer, exchange repulsion and dispersion.4,32 That said, there are basically two ways to view H-bonding.33 On the one hand, H-bonding is attributed to purely electrostatic interactions or electrostatic plus polarization interactions; on the other hand, H-bonding is considered to be stabilized largely by charge transfer—the interaction between a lone pair orbital of one molecule and an antibonding covalent orbital of another.33,34
Because of such complex nature of H-bonding, its precise definition remains a matter of debate,32–38 and quantifying how much each of the above components contributes to a given H-bond is an intricate task in general. There is also a report suggesting that the component which is dominant for a given H-bonded system alters from one species to another.39 Hence, a straightforward, single explanation for why the acceptor side of the hydroxyl groups forms weaker H-bonds would probably be difficult.
Nevertheless, I point out three physical interactions that are presumably relevant to the lesser ability of the hydroxyl groups to form acceptor H-bonds. For one thing, the lesser polarity of the O–C bonds perhaps contributes to a weaker electrostatic attraction. For another thing, the shrinkage of dLP is considered to have more direct effects; it probably leads to (i) a decrease in the local polarization between the oxygen nucleus and the lone pairs and (ii) a decrease in the overlap of lone pair orbitals of the hydroxyl groups and antibonding OH orbitals of water molecules (i.e. a decrease in the charge transfer), resulting in a lesser energetic stabilization.33,34
All this may still sound like a chicken-and-egg situation: does a decrease in the number of acceptor H-bonds merely result in a decrease in dLP? Or does a decrease in dLP fundamentally cause a decrease in the number of acceptor H-bonds?
One way to answer this question is to consider the difference between a water molecule and a hydroxyl group from a chemical standpoint. An oxygen atom of a water molecule can pull the bonding electrons equally from both sides; however, an oxygen atom of a hydroxyl group cannot. This is because the electronegativity of carbon is slightly larger than that of hydrogen, and thus the oxygen atom can no longer pull the OH bonding electrons from the carbon-side as easily as from the hydrogen-side.
With this in mind, it seems easier for the oxygen atom to pull the lone pairs toward itself than to pull the bonding electrons. My analysis suggests that this is actually the case. The main reason is that, as can be seen from Table 1 (and as will be discussed later in the analysis on the donor side), the oxygen atom in a hydroxyl group usually fails to pull the OH bonding electrons effectively (on average only a 0.2% decrease). Instead, to compensate for the “loss” of electrons, the oxygen atom tends to pull exclusively the lone pair electrons toward itself (a 3.2% decrease), which results in the shrinkage of dLP. All this suggests that the hydroxyl groups are less hydrated because of the shrinkage of dLP.
| The acceptor side | The donor side | |||||
|---|---|---|---|---|---|---|
| Hydroxyl group | d LP /Å (%) | N acc | d OH /Å (%) | d CV /Å (%) | d CV–H/Å (%) | N dnr |
| a In the columns for dLP, dOH, dCV and dCV–H, values in parentheses are the deviations (%) from the corresponding values for water molecules in the liquid. [In the case of dCV–H, in evaluating the deviations (%) I used dOH−dCV≈dCV–H.] For the definitions for dLP, dCV and dCV–H, see Fig. 1c. In my calculations, the electronic and structural properties of water molecules in the liquid were the following: dLP, 0.3324 Å; dOH, 1.0106 Å; dCV, 0.5102 Å. Nacc and Ndnr are the average number of acceptor H-bonds and that of donor H-bonds respectively. For the criteria for H-bonding I used, see Fig. 2 caption. In short, the results indicate weaker acceptor H-bonds and stronger donor H-bonds of the hydroxyl groups than water–water H-bonds. The table also shows that the average number of H-bonds per hydroxyl group is 2.083. | ||||||
| OH1 | 0.3144 (−5.40) | 0.928 | 1.0195 (+0.88) | 0.5039 (−1.24) | 0.5171 (+3.05) | 0.967 |
| OH2 | 0.3225 (−2.97) | 1.166 | 1.0126 (+0.20) | 0.5112 (+0.18) | 0.5031 (+0.22) | 0.918 |
| OH3 | 0.3245 (−2.35) | 1.267 | 1.0123 (+0.18) | 0.5105 (+0.06) | 0.5034 (+0.30) | 0.943 |
| OH4 | 0.3196 (−3.86) | 0.987 | 1.0127 (+0.21) | 0.5107 (+0.09) | 0.5035 (+0.33) | 0.917 |
| OH6 | 0.3280 (−1.32) | 1.395 | 1.0130 (+0.24) | 0.5096 (−0.12) | 0.5047 (+0.60) | 0.935 |
| Average | 0.3218 (−3.18) | 1.147 | 1.0140 (+0.34) | 0.5092 (−0.20) | 0.5064 (+0.90) | 0.936 |
This is the reason why my argument is unlikely to be a chicken-and-egg situation. Certainly, the degree to which a hydroxyl group is hydrated can cause an increase or decrease in dLP for a short period of time. However, in the long run, the decrease of dLP causes the decrease in the number of acceptor H-bonds. That is, I suggest that the decrease in dLP is not the accidental result of the simulations, but the main cause of the decrease in the number of acceptor H-bonds.
Now, given that the average distance dLP in the hydroxyl groups was 0.322 Å, the results indicate that on average an individual hydroxyl group of a monosaccharide does not form two acceptor H-bonds, but forms only about one acceptor H-bond (Fig. 2a). Actually, in my simulations, the average number of acceptor H-bonds per hydroxyl group was 1.15, and the average number of H-bonds per hydroxyl group was 2.08 (see Table 1). The feature of this kind was in fact observed in previous classical11 and ab initio10,12 MD studies on monosaccharides, but the reason for it has long been elusive. Now my result gives a quantitative reason for why the local hydration of hydroxyl groups of a monosaccharide is roughly two-coordinated.
Predictably, these agreed with the results of the number Nacc. O6 formed the greatest number of acceptor H-bonds (1.40), followed by O3 (1.30), and O4 and O1 formed less than one acceptor H-bond (0.99 and 0.93). All this suggests that O6 is the best H-bond acceptor, followed by O3, and that O1 is the poorest acceptor, followed by O4.
I note that subtle variations in the O–C bond polarity in the hydroxyl groups probably play an additional role in determining these local H-bonding configurations via a kind of dipole–dipole interaction. Since H-bonding can be regarded as a special case of dipole–dipole interaction,5 an H-bond between the acceptor side of a hydroxyl group and the donor side of a water molecule can be viewed as an interaction between an O–C bond dipole and an O–H bond dipole. In this respect, how weakly the individual hydroxyl groups form acceptor H-bonds not only results from subtle variations in the shrinkage of dLP, but may also be affected by those in the O–C bond dipoles.
Generally speaking, bond polarity arises when a pair of electrons is shared unequally by two atoms of a covalent bond; hence, measuring how unequally the electron pair is shared by the two atoms is the key to understanding the bond dipole.
A simple and quick way to estimate this unequal sharing is to calculate a ratio, the O–WFC distance/(the O–WFC distance + the WFC–C distance), where WFC denotes the centre of the covalent C–O bond orbital. Since the electronegativity of the O atom is larger than that of the C atom, the ratio is smaller than 50%. As a general rule, the closer to 50% the ratio is, the less polar the O–C bond will be; in other words, the smaller the ratio is, the more polar the O–C bond will become.
I calculated the ratio for each O–C bond. The ratio for the C6–O6 bond was the smallest (38.34%), whereas that of the C1–O1 bond was the largest (38.79%). More specifically, the order of the ratio magnitudes was as follows: C6–O6 (38.34%) < C3–O3 (38.49%) < C2–O2 (38.66%) ≈ C4–O4 (38.68%) < C1–O1 (38.79%). This order agrees with that of the number of acceptor H-bonds, Nacc (see Table 1). At the same time, I found that the correlation between the ratio and Nacc was in the middle range (−0.53). Hence, subtle variations in the O–C bond polarity in the hydroxyl groups probably play an intermediate role.
Where do the site-dependent differences come from? Why does the degree to which dLP decreases depend on the individual sites? To understand this, let me take a closer look at two cases: O1 and O6. To start with, I explain the case of O1 because the anomeric site is a special case. Unlike other carbon atoms, C1 is bonded to two oxygen atoms, rather than one. This makes C1 relatively electron-poor compared with other carbon atoms; therefore, O1 cannot effectively pull the bonding electrons from C1. To compensate for this, O1 pulls its lone pairs more strongly toward itself, which leads to the largest decrease in dLP (Table 1). This explains why dLP in O1 is the shortest, and that results in the smallest number of acceptor H-bonds.
On the other hand, C6 is relatively electron-rich compared to other carbon atoms since it is bonded to two hydrogen atoms. Because of this, O6 can pull the bonding electrons relatively easily from C6. Thus, O6 does not have to pull its lone pairs as tightly as other oxygen atoms do, which leads to the smallest decrease of dLP (Table 2). This explains why dLP in O6 is the longest; and that results in the largest number of acceptor H-bonds.
| Hydroxyl group | R(OD⋯OW)a/Å | μ b/Debye |
|---|---|---|
| a OD denotes the oxygen atom of the donor side of a hydroxyl group OD–H; and OW denotes an acceptor oxygen atom of a water molecule. b Dipole moments of water molecules that formed acceptor H-bonds with the individual hydroxyl groups. | ||
| OH1 | 2.7891 | 3.153 |
| OH2 | 2.8638 | 2.996 |
| OH3 | 2.8438 | 3.026 |
| OH4 | 2.8484 | 3.086 |
| OH6 | 2.8374 | 3.149 |
These are two examples but in general, since individual glucose carbon atoms are in different chemical environments, the O–C local bond dipoles are slightly different from each other. That is, the unequal sharing of the electron pair along the C–O bond slightly alters from one hydroxyl group to another, which in turn affects the degree to which dLP decreases. All this contributes to the local H-bonding configurations on the acceptor side.
The donor side
The results indicated that the hydroxyl groups form stronger H-bonds than bulk water molecules. The average distances dCV–H stretched by 0.9% from that of bulk water molecules, and the distance dCV shrank by 0.2% (but see below). Also, the average OH distance (dOH) stretched by 0.3% (Table 1). This suggests that the bare protons of the hydroxyl groups tend to be more exposed than those of water molecules.
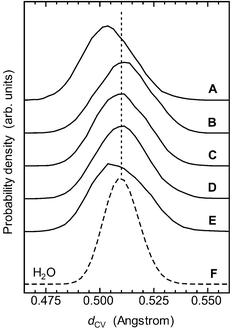 | ||
| Fig. 3 The electronic properties of the donor side of the hydroxyl groups. Shown are the individual distributions of the distance dCV: (A) OH1; (B) OH2; (C) OH3; (D) OH4; (E) OH6; (F) bulk water molecules (denoted by the dashed curve). The average value for the distance dCV of bulk water molecules is also indicated by the dotted line. For the definition of dCV, see Fig. 1c. The distance dCV in OH1 and that in OH6 are both shorter than that of bulk water molecules, indicating stronger donor H-bonds of these groups than those in the bulk. On the other hand, though their distributions are broader than that of the bulk, the maximum values for OH2, OH3 and OH4 are almost equal to that of the bulk. The results indicate that OH1 and OH6 are stronger H-bond donors than OH2, OH3 and OH4. | ||
Another way to characterize how strongly the donor side forms H-bonds is to calculate the oxygen–oxygen distance. I calculated the distance R(OD⋯OW), where OD denotes the oxygen atom of the donor side of a hydroxyl group OD–H and OW is an acceptor oxygen atom of a water molecule. The distance R(O1D⋯OW) was the shortest (2.7891 Å), followed by R(O6D⋯OW) (2.8374 Å) (Table 2); they are shorter by 0.071 Å and 0.023 Å respectively than the distance R(O⋯O) in the liquid (2.8601 Å).
Consistently, the distributions of R(OD⋯OW) for the individual hydroxyl groups suggest that OH1 and OH6 are better H-bond donors (Fig. 4). Further, the results show that the distributions for OH1 and OH6 are significantly asymmetrical (Fig. 4, curves A and E), which indicates that water molecules were strongly attracted to the donor side of OH1 and OH6. In general, the shorter the overall oxygen–oxygen distance, the stronger the charge transfer.33 Hence, all this suggests that the charge-transfer interaction probably plays a role in these stronger H-bonds.
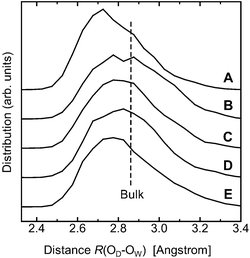 | ||
| Fig. 4 The distributions of the distance R(OD⋯OW). OD denotes the oxygen atom of the donor side of a hydroxyl group OD–H; and OW denotes an acceptor oxygen atom of a water molecule in the first hydration shell for the individual hydroxyl groups: (A) OH1; (B) OH2; (C) OH3; (D) OH4; (E) OH6. The results show that the distributions for OH1 and OH6 are significantly asymmetrical, indicating that water molecules were strongly attracted to the donor side of OH1 and OH6. The dashed line indicates the average distance in the liquid waterR(O⋯O). | ||
The order of R(OD⋯OW) was as follows: R(O1D⋯OW) < R(O6D⋯OW) < R(O3D⋯OW) < R(O4D⋯OW) < R(O2D⋯OW); this order is basically consistent with that of the dCV–H distance (Table 1) and with that of the dipole moments of water molecules forming acceptor H-bonds with the hydroxyl groups (Table 2; see also the next subsection for more detail). On the other hand, this order did not agree perfectly with that of the number of donor H-bonds, Ndnr (Table 1). This is probably because the criteria for H-bonding is generally arbitrary. Nonetheless, considering all the available data presented here, the relative strength of the individual hydroxyl groups to form donor H-bonds is considered to be the following order: OH1 > OH6 > OH3 ≈ OH4 > OH2.
The dipole moments of interfacial water molecules
Calculating the dipole moment of water molecules in the first hydration shell is another important way to investigate sugar–water H-bonds and to clarify my findings. Thanks to the method of maximally localized Wannier functions, today we can do such calculations more easily and accurately than ever before because the electronic contribution to the dipole moment is directly computed from the electronic structure within DFT calculations.41The results suggested that the dipole moment increases only when water molecules act as H-bond acceptors, which in turn supports the idea that the hydroxyl groups form stronger donor H-bonds. The average dipole moment of water molecules forming acceptor H-bonds with the hydroxyl groups increased by 2%, or 0.060 Debye (Fig. 5a, solid line), from that of the bulk, 3.026 Debye (Fig. 5, dashed line). By contrast, the average dipole moment of water molecules forming donor H-bonds decreased marginally by 0.2%, or 0.006 Debye, but remained almost unchanged (Fig. 5a, dotted curve) (the average dipole moment of bulk water molecules agrees well with a previous ab initio MD study41). This is consistent with another previous ab initio MD study on ribose in aqueous solution.12
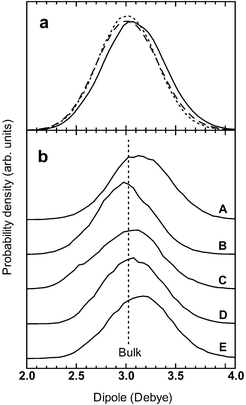 | ||
| Fig. 5 The dipole moments of water molecules in the first hydration shell. (a) The dipole moments of water molecules forming acceptor H-bonds (solid curve) and donor H-bonds (dotted curve) with the hydroxyl groups are compared with that of the bulk (dashed curve). The former shows a 2% increase compared with that of the bulk, whereas the latter does not. This suggests stronger donor H-bonds of the hydroxyl groups. (b) The dipole moments of water molecules forming acceptor H-bonds with the individual hydroxyl groups: (A) OH1; (B) OH2; (C) OH3; (D) OH4; (E) OH6. When water molecules form acceptor H-bonds with OH1 or OH6, the dipole moment rises by 4%, or 0.125 Debye, from that of the bulk (indicated by the dashed line). The result is consistent with that shown in Fig. 3 and 4. | ||
Let me take a closer look at the mechanism of the enhancement. There are two major origins that can affect the dipole in the system. One is how weakly or strongly the acceptor or donor side of the hydroxyl groups can interact with water molecules. As I have already shown, the acceptor side and the donor side of the hydroxyl groups interact more weakly and more strongly respectively with water molecules than bulk water molecules.
The other is how sensitively the dipole moment of a water molecule responds to a local change of its molecular dipole. To understand quantitatively which side of a water molecule (the donor side or the acceptor side) more directly influences its molecular dipole moment, I calculated correlation coefficients between the absolute value of the dipole moment and some of the electronic or structural parameters, using 4.96 × 107 electronic configurations of bulk water molecules. The parameters I chose were the following: the LP–O–LP angle (where LP denotes the centre of a lone pair orbital); the CV–O–CV angle (where CV denotes the centre of a covalent OH bond); the H–O–H angle; the distance dLP; the distance dCV; and the OH distance dOH.
The correlation coefficients for the above parameters were the following: the LP–O–LP angle, −0.44; the CV–O–CV angle, −0.08; the H–O–H angle, −0.29; dLP, 0.73; dCV, −0.53; dOH, 0.40. (The averaging procedure was the same as described in the Methods.) Hence, the distance dLP is highly correlated with the absolute value of the dipole moment, followed by the distance dCV. Since dLP is associated with the enhancement of the dipole on the acceptor side whereas dCV is associated with that on the donor side, a simple physical explanation of this is that lone pairs can move more easily than a hydrogen atom does and thus can respond more sensitively to surrounding positively charged species. This in turn means that a local change on the acceptor side has a more direct effect on the dipole moment.
All this explains why the dipole moment responds sensitively to a subtle increase in the exposure of the protons of the hydroxyl groups. When water molecules act as H-bond donors, the dipole moment remains almost unchanged because (i) hydroxyl groups form weaker acceptor H-bonds and (ii) the dipole moment correlates less highly with the distance dCV. On the other hand, when water molecules act as H-bond acceptors, the dipole moment increases more readily because (i) hydroxyl groups form stronger donor H-bonds and (ii) the dipole moment correlates highly with the distance dLP.
A further investigation revealed that the enhancement depends on which hydroxyl group the water molecules form acceptor H-bonds with. More specifically, water molecules forming acceptor H-bonds with OH1 or OH6 mainly accounted for the enhancement. The dipole moments of these water molecules rose by 4%, to 3.15 Debye, showing the greatest increase (Table 2 and Fig. 5b, curves A and E). On the other hand, the dipole moment of water molecules forming acceptor H-bonds with OH2 decreased slightly by 1%, to 2.996 Debye (Table 2 and Fig. 5b, curve B). When water molecules formed acceptor H-bonds with OH3 or OH4, the dipole moment remained almost unchanged but increased marginally (Fig. 5b, curves C and D). The results agree with the localized orbital analysis on the donor side of the hydroxyl groups (Fig. 3) and the oxygen–oxygen distance (Table 2).
Accuracy of plane-wave DFT calculations
At this point, I should mention the basic accuracy of plane-wave DFT calculations. The accuracy of plane-wave DFT calculations within the Car–Parrinello MD (CPMD) method is a fundamental issue, and several studies have been dedicated to this subject.42–45 In general, the performance of the plane-wave DFT calculations depends on different computational details. Examples of these are the length of simulations, the choice of the exchange–correlation functional, initial configurations, the cell size, the pseudopotential approximation, cut-off energy, the fictitious electronic mass, the system on which simulations are performed, and so on. In the following, I particularly mention the system size and the exchange–correlation functional.Grossman et al. pointed out that the influence of the cell size is relatively minor compared with the choice of the fictitious mass.43 In addition, the cell size probably has little effect on the centres of the maximally localized Wannier functions,41 which indicates that the electronic properties and the dipole moments are relatively reliably discussed. Considering all this, the cell size appears to have little or only a small effect, at least on the electronic and H-bonding properties.
Furthermore, I want to mention current practices in CPMD simulations of aqueous systems. In 1993, a CPMD simulation was performed on a system consisting of 32 water molecules in a cubic cell.46 10 years later, CPMD studies of liquid water are still performed mostly on a system consisting of 32 or 64 water molecules in a cubic cell.43–45
CPMD simulations were also performed on the hydration of the smallest organic molecules such as uracil,47ribose12 and glucose,16,17 and the number of water molecules was 60 or smaller. As I already wrote earlier, this system size nonetheless allows us to investigate important aspects of water molecules in the first hydration shell. While this point needs to be handled in the future, at present using this kind of system size is the most practical way to investigate the H-bonding properties between glucose and water reasonably in atomic detail.
First, while studies suggest that care should be taken when one uses the BLYP functional, these works are mainly concerned with the first peak in the pair correlation function and the dynamic properties.42–45 In this paper, however, I mainly discuss the electronic and H-bonding properties. While I sometimes discuss the structural properties, my focus is not on the dynamic properties such as the self-diffusion coefficient or the mean-square displacement. The above studies are of great importance, but their findings are not necessarily relevant to my main objective.
Second, whether or not an H-bond network is overstructured is considered relatively irrelevant to the behaviour of the dipole moment of individual water molecules. Thus, the main findings in my analysis on the dipole moment of water molecules will remain unchanged, at least on a qualitative level.
Third, the trend shown in Fig. 2a is likely to remain unchanged, regardless of different computational details within the framework of the CPMD methodology. That being the case, this still suggests that the H-bonding properties are sensitive to seemingly small changes in the electronic and structural parameters, if a contact-like character of H-bonding interaction is taken into account.
Fourth, while the BLYP functional tends to make the first peak in the pair correlation function sharper,44,45 the coordination number does not significantly change. This is mainly because the coordination number is an accumulated property. With this in mind, my suggestion that the acceptor side is less hydrated than previously thought is likely to hold. Several computational points being addressed, in the next subsection I will discuss how the hydroxyl groups are locally hydrated.
The microscopic hydration of glucose: how the hydroxyl groups are locally hydrated
Before I discuss and characterize how glucose is hydrated in atomic detail, I briefly mention the local structure of liquid water. The local structure of the first coordination shell of a water molecule in the liquid has been under intense scrutiny for the past three years. Recent X-ray absorption and X-ray Raman experiments together with theoretical calculations challenged the traditional view, i.e. the view that water is a locally tetrahedral liquid. According to the report, most water molecules in the liquid form only two H-bonds: one strong donor and one strong acceptor.48 That report being published, several studies addressed this issue, and a number of computational studies still supported the traditional view.2,49–52In particular, a recent ab initio MD study showed that available experimental data and its first-principles calculations of X-ray absorption spectra are consistent with the conventional picture of a locally tetrahedral character of the liquid.2 Considering all this, although many structural aspects may remain subject to debate, in the present work I regard water as a locally tetrahedral liquid.
Later on, classical9,53 and ab initio10 MD studies pointed out that this picture based on an ice-like lattice is irrelevant, and, in fact, demonstrated computationally that there is no clear evidence for such an ice-like structure. Since then, computational studies focused instead on the stereochemical or topological aspects associated with the molecular structures of monosaccharides.9,53 This anisotropic-structuring picture rather emphasizes that the hydration structure varies from one monosaccharide to another.
This picture has important aspects in itself; however, the classic picture still has an appealing point. There is a similarity between the OH bond in a hydroxyl group and that in a water molecule. That is, a hydroxyl group has a fragment of a water molecule. Thus, it could be reasoned that hydroxyl groups of a monosaccharide are highly compatible with a locally tetrahedral network of H-bonds with a little or no distortion.
Logically speaking, describing an overall structure around a monosaccharide from the viewpoint of the molecular structure is one thing, but analysing how individual hydroxyl groups are locally hydrated is another. In this sense, the two pictures described above are not so mutually exclusive as one might think. Actually, while the anisotropic-structuring picture emphasizes that the hydration of monosaccharides can be distorted by their molecular structures one way or another, it also assumes that individual hydroxyl groups remain highly compatible with a locally tetrahedral structure of H-bonds.9 Hence, in either picture, the compatibility of the hydroxyl groups with a locally tetrahedral network has been directly or indirectly assumed.
The results of the present work suggest that the hydroxyl groups are less hydrated (on average, 2.08 H-bonds per hydroxyl group) and rather more incompatible with a locally tetrahedral network of H-bonds than previously thought. On average, glucose formed 11.15 H-bonds with water molecules in the first hydration shell; it formed 4.68 donor H-bonds and formed 6.47 acceptor H-bonds (in which the average number of acceptor H-bonds for the ring oxygen was 0.73). (However, I note that these numbers can alter to some extent or other, depending on the definition of H-bonding.)
Further, my analyses on the acceptor and the donor sides suggest that the local H-bonding configurations vary more specifically and sensitively from one hydroxyl group to another than previously conceived (Fig. 2, 3 and 4). My analyses provide detailed knowledge about how glucose is locally hydrated: (i) the anomeric site (OH1) is the best donor, but the poorest acceptor among the hydroxyl groups; (ii) OH2 is a slightly poorer donor; (iii) OH3 is a better acceptor; (iv) OH4 is a poorer acceptor; and (v) OH6 is the best acceptor and a better donor as well.
Such detailed knowledge could lead to a deeper understanding of (i) diverse roles of oligosaccharides composed of glucose units, (ii) the transport of glucose across membranes,54 and (iii) (solvent-assisted) chemical reactions or molecular recognitions that involve sugars.
Conclusions
In conclusion, the present study suggests that the hydroxyl groups form roughly two H-bonds: one weaker acceptor H-bond and one stronger H-bond, with different variations that are sensitive to individual chemical environments of the groups. To my knowledge, such detailed analysis has been missing from virtually all previous works for about 50 years.The analysis points to a breakdown of a locally tetrahedral character at the sugar–water interface and thereby explains why, though being hydrophilic and polar, hydroxyl groups of simple sugars are less hydrated and more incompatible with a locally tetrahedral network of H-bonds than previously thought. Rather, the present study implies that the local H-bonding arrangements around monosaccharides (and perhaps those around oligosaccharides) are far more complex and substantially different.12 This aspect, for example, is considered responsible for the unusual dynamic properties of sugar–water solutions.55,56
A fundamental implication of this study is that a locally tetrahedral network of H-bonded water molecules is perhaps delicately maintained by the unique electronic structure and molecular dipole of the water molecules; and this work shows that, because of a contact-like character of H-bonding interaction, the local configurations in sugar–water H-bonds are likely to be affected by seemingly small changes in the local electronic structure and bond polarity of the hydroxyl groups. The present work provides valuable insights into carbohydrate chemistry, molecular biology and interactions between hydrophilic groups and interfacial water molecules.
Acknowledgements
All the ab initio MD simulations were performed on the Hitachi SR8000 supercomputers of the University of Tokyo.References
- P. Ball, Life’s Matrix: A Biography of Water, University of California Press, Berkeley and Los Angles, 2001, ch. 6 Search PubMed.
- D. Prendergast and G. Galli, Phys. Rev. Lett., 2006, 96, 215502 CrossRef.
- The local structure of the first coordination shell of a water molecule in the liquid has been under intense scrutiny for the past three years. For reasons discussed later in this paper, however, I traditionally classify water as a locally tetrahedral liquid.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- P. Y. Bruice, Organic Chemistry, Prentice Hall, Upper Saddle River, NJ, 5th edn, 2007, ch. 2 Search PubMed.
- M. A. Kabayama and D. Patterson, Can. J. Chem., 1958, 36, 563 CAS.
- D. T. Warner, Nature, 1962, 196, 1055 CAS.
- F. Franks, Pure Appl. Chem., 1987, 59, 1189 CrossRef CAS.
- B. Leroux, H. Bizot, J. W. Brady and V. Tran, Chem. Phys., 1997, 216, 349 CrossRef CAS.
- C. Molteni and M. Parrinello, J. Am. Chem. Soc., 1998, 120, 2168 CrossRef CAS.
- K. S. Sidhu, J. M. Goodfellow and J. Z. Turner, J. Chem. Phys., 1999, 110, 7943 CrossRef CAS.
- T. Suzuki and T. Sota, J. Phys. Chem. B, 2005, 109, 12603 CrossRef CAS.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471 CrossRef CAS.
- D. Marx and J. Hutter, in Modern Methods and Algorithms of Quantum Chemistry Proceedings, ed. J. Grotendorst, John von Neumann Institute for Computing, Forschungszentrum Julich, Germany, 2nd edn, 2000, NIC series, vol. 3, pp. 329–477 Search PubMed.
- J. Maddox, Nature, 1993, 364, 669.
- J. M. Stubbs and D. Marx, Chem.–Eur. J., 2005, 11, 2651 CrossRef CAS.
- T. Suzuki, H. Kawashima and T. Sota, J. Phys. Chem. B, 2006, 110, 2405 CrossRef CAS.
- X. Qian, M. R. Nimlos, M. Davis, D. K. Johnson and M. E. Himmel, Carbohydr. Res., 2005, 340, 2319 CrossRef CAS.
- CPMD, Version 3.7, MPI für Festkoerperforschung and IBM Zürich Research Laboratory, 1997–2001; http://www.cpmd.org Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B, 1996, 54, 1703 CrossRef CAS.
- G. J. Martyna, M. L. Klein and M. E. Tuckerman, J. Chem. Phys., 1992, 97, 2635 CrossRef.
- M. E. Tuckerman and M. Parrinello, J. Chem. Phys., 1994, 101, 1302 CrossRef CAS.
- (a) N. Marzari and D. Vanderbilt, Phys. Rev. B, 1997, 56, 12847 CrossRef CAS; (b) N. Marzari, I. Souza and D. Vanderbilt, in An Introduction to Maximally-Localized Wannier Functions, http://quasiamore.mit.edu/wannier/papers/MSVpsik.pdf (accessed June 2007) Search PubMed.
- G. Berghold, C. J. Mundy, A. H. Romero, J. Hutter and M. Parrinello, Phys. Rev. B, 2000, 61, 10040 CrossRef CAS . There are basically three types of spread functionals for Wannier functions: the Resta-type functional, the Vanderbilt-type functional and the Silvestrelli-type functional. The Vanderbilt-type functional was used in this work. However, Berghold et al. showed that the differences among the results obtained from these functionals are very small. Further, from a theoretical point of view, there is no fundamental reason to choose one definition of the spread over another. They concluded that these functionals are numerically equivalent.
- By bulkwater molecules, I refer to water molecules forming H-bonds only with water molecules (including water molecules forming H-bonds with first-hydration-shell water molecules). I compared the dipole moment of such water molecules with that of water molecules that formed H-bonds neither with the glucose molecule nor with first-hydration-shell water molecules. The two distributions were almost identical (Fig. S2 in the ESI†). For this reason, I conveniently refer to those water molecules which form H-bonds only with water molecules as bulk water molecules.
- P. Atkins and J. Paula, Physical Chemistry, Oxford University Press, New York, 8th edn, 2006, ch. 18 Search PubMed.
- The shrinkage of dLP leads to an increase in the lone pair angle because the two lone pairs tend to exert more electron repulsion between each other. In general, the wider the lone pair angle, the more s character the lone pair has.
- H. Umeyama and K. Morokuma, J. Am. Chem. Soc., 1977, 99, 1316 CrossRef CAS.
- R. Ludwig, Angew. Chem., Int. Ed., 2001, 40, 1808 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- E. D. Isaacs, A. Shukla, P. M. Platzman, D. R. Hamann, B. Barbiellini and C. A. Tulk, Phys. Rev. Lett., 1999, 82, 600 CrossRef CAS.
- A. Hellemans, Science, 1999, 283, 614 CrossRef CAS.
- T. K. Ghanty, V. N. Staroverov, P. R. Koren and E. R. Davidson, J. Am. Chem. Soc., 2000, 122, 1210 CrossRef CAS.
- A. H. Romero, P. L. Silvestrelli and M. Parrinello, J. Chem. Phys., 2001, 115, 115 CrossRef CAS.
- J. J. Dannenberg, L. Haskamp and A. Masunov, J. Phys. Chem. A, 1999, 103, 7083 CrossRef CAS.
- The shift of the centres of the covalent OH1 and OH6 bond orbitals toward their oxygen nuclei (Fig. 3) indicates that their electronic charge densities shifted toward the oxygen nuclei along the bonds. This probably reduces the repulsive overlap interactions between the bond orbitals and lone pairs of water molecules, leading to an enhancement of the H-bonding (ref. 34). Further, strong charge-transfer interactions tend to lengthen the O–H bond distance and to reduce the overall oxygen–oxygen distance (ref. 33). Actually, this tendency was observed for the OH1 and OH6 bonds (Tables 1 and 2).
- (a) P. L. Silvestrelli and M. Parrinello, Phys. Rev. Lett., 1999, 82, 3308 CrossRef CAS; (b) P. L. Silvestrelli and M. Parrinello, J. Chem. Phys., 1999, 111, 3572 CrossRef CAS.
- A. Kohlmeyer, in Effects of System Size and Time Scales in Molecular Dynamics Simulations of Bulk Water, http://www.theochem.ruhr-uni-bochum.de/~axel.kohlmeyer/files/talk-trieste2004-water.pdf (accessed August 2007) Search PubMed.
- J. C. Grossman, E. Schwegler, E. W. Draeger, F. Gygi and G. Galli, J. Chem. Phys., 2004, 120, 300 CrossRef CAS.
- I.-F. W. Kuo, C. J. Mundy, M. J. McGrath, J. I. Siepmann, J. VandeVondele, M. Sprik, J. Hutter, B. Chen, M. L. Klein, F. Mohamed, M. Krack and M. Parrinello, J. Phys. Chem. B, 2004, 108, 12990 CrossRef CAS.
- J. VandeVondele, F. Mohamed, M. Krack, J. Hutter, M. Sprik and M. Parrinello, J. Chem. Phys., 2005, 122, 014515 CrossRef.
- K. Laasonen, M. Sprik, M. Parrinello and R. Car, J. Chem. Phys., 1993, 99, 9080 CrossRef CAS.
- M.-P. Gaigeot and M. Sprik, J. Phys. Chem. B, 2003, 107, 10344 CrossRef CAS.
- Ph. Wernet, D. Nordlund, U. Bergmann, M. Cavalleri, M. Odelius, H. Ogasawara, L. Å. Näslund, T. K. Hirsch, L. Ojamäe, P. Glatzel, L. G. M. Pettersson and A. Nilsson, Science, 2004, 304, 995 CrossRef CAS.
- A. K. Soper, J. Phys.: Condens. Matter, 2005, 17, S3273 CrossRef CAS.
- T. Head-Gordon and M. E. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7973 CrossRef CAS.
- J. D. Smith, C. D. Cappa, B. M. Messer, W. S. Drisdell, R. C. Cohen and R. J. Saykally, J. Phys. Chem. B, 2006, 110, 20038 CrossRef CAS.
- R. L. C. Wang, H. J. Kreuzer and M. Grunze, Phys. Chem. Chem. Phys., 2006, 8, 4744 RSC.
- (a) R. K. Schmidt, M. Karplus and J. W. Brady, J. Am. Chem. Soc., 1996, 118, 541 CrossRef CAS; (b) Q. Liu and J. W. Brady, J. Am. Chem. Soc., 1996, 118, 12276 CrossRef CAS.
- In Cells, ed. B. Lewin, L. Cassimeris, V. R. Lingappa and G. Plopper, Jones & Bartlett, Sudbury, MA, 2007, pp. 63–70 Search PubMed.
- C. J. Roberts and P. G. Debenedetti, J. Phys. Chem. B, 1999, 103, 7308 CrossRef CAS.
- L. J. Smith, D. L. Price, Z. Chowdhuri, J. W. Brady and M.-L. Saboungi, J. Chem. Phys., 2004, 120, 3527 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef.
- L. Laaksonen, J. Mol. Graphics, 1992, 10, 33 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1, computational details; Table S2, the conformational properties of the hydroxymethyl groups in my ab initio molecular dynamics simulations; Fig. S1, three staggered hydroxylmethyl rotational conformations; Fig. S2, the dipole moment of water molecules in the first hydration shell. See DOI: 10.1039/b708719e |
| This journal is © the Owner Societies 2008 |