Application of furyl-stabilized sulfur ylides to a concise synthesis of 8a-epi-swainsonine†
Jie
Bi
and
Varinder K.
Aggarwal
*
Cantock's Close, School of Chemistry, University of Bristol, UK BS8 1TS. E-mail: V.Aggarwal@Bristol.ac.uk; Fax: +44 117 929 8611; Tel: +44 117 954 6315
First published on 17th October 2007
Abstract
The total synthesis of 8a-epi-swainsonine has been achieved in 20% overall yield from R-glyceraldehyde dimethylacetonide 3 through epoxidation with the achiral furyl-substituted sulfonium ylide 2d as one of the key steps.
Swainsonine 1 (Fig. 1), a naturally occurring polyhydroxyindolizidine, was first isolated from the fungus Rhizoctonia leguminicola in 19731 and has since attracted much attention, primarily because of its significant biological activity. For example, it was found to be an effective inhibitor of α-D-mannosidases,2 including the glycoprotein-processing enzyme mannosidase II.3 It also exhibits important antimetastatic,4 antitumor-proliferative,5 anticancer,6 and immunoregulating activities.7Swainsonine was also the first inhibitor to be selected for testing as an anticancer drug, reaching phase II clinical trials in the US.8 Furthermore, considerable structural variants (alternative epimers, other structural analogues) have also been prepared in order to try to improve the biological activity and/or the selectivity of the natural compound.9
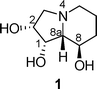 | ||
| Fig. 1 Structure of swainsonine. | ||
Its considerable biological activity and interesting structure has made swainsonine a popular target amongst synthetic chemists.9,10 Our own analysis of a potential route to this compound evolved from our work on sulfur ylides.11 In early studies, we had shown that the phenyl-stabilized ylide 2a reacted with glyceraldehyde dimethylacetonide 3 to give the cisepoxide 4a (Scheme 1).12 In this kinetically controlled reaction, the C1 stereochemistry is controlled by the chiral sulfur ylide (reagent control) and the C2 stereochemistry is controlled by the substrate. We reasoned that if we could effect the same type of reaction with the related furyl-stabilized ylide 2b to give epoxide 5a, then following ring opening by NH3 and application of the Achmatowicz reaction we should arrive at piperidine7a (Scheme 2). Piperidine7a is just a few functional group interconversions away from swainsonine.
 | ||
| Scheme 1 Reaction of chiral sulfur ylide with chiral aldehyde . | ||
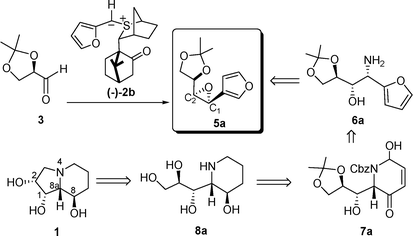 | ||
| Scheme 2 Proposed route to swainsonine. | ||
Realization of this strategy, however, quickly revealed a significant obstacle. Although the furyl-stabilized ylide 9 could be generated from the corresponding tosylhydrazone salt 10 and reacted with PhCHO (as shown previously),13 the related reaction with glyceraldehyde dimethylacetonide 3 under the same conditions failed (Scheme 3). This is presumably due to the instability of the aldehyde at 40 °C over a prolonged period.
 | ||
| Scheme 3 Attempts to use the catalytic sulfur ylide reaction. PTC = phase transfer catalyst. | ||
Application of the stoichiometric sulfur ylide reaction was therefore explored.14 However, all attempts to form simple furfuryl sulfonium salt 2c led to polymers or very low yields of products (Scheme 4).15 We therefore attempted to stabilize the sulfonium salt by blocking the 5-position on the furyl ring with a suitable electron-withdrawing group. Of the substrates explored, only the chloro and sulfonyl groups led to stable intermediates and of these only the sulfonyl substituted salt 2d reacted with glyceraldehyde dimethylacetonide 3. This furnished a mixture of epoxides (6 : 83 : 11) in which 11b predominated. The stereochemical outcome at C2 (11a + 11b : 11c) is in keeping with a polar Felkin–Anh controlled addition of the ylide to the chiral aldehyde . Interestingly, use of either of the two chiral sulfonium salts (+)-2e and (–)-2e gave similar results, indicating that the ylide reaction was now under substrate control rather than reagent control. This implies that betaine formation (which would be expected to be controlled by the reagent) must be reversible, and the epoxide selectivity is determined by the equilibrium ratios of the betaine intermediates and their rates of ring closure. Thus, the introduction of the sulfonyl group on the furyl ring, necessary to stabilize the sulfonium salt, has changed the nature of the ylide from a semi-stabilized ylide that would otherwise react non-reversibly, to a stabilized ylide that reacts reversibly. In fact, the level of selectivity observed is similar to that reported for amide-stabilized sulfur ylides (0 : 84 : 16; a : b : c) which are also believed to react reversibly.16
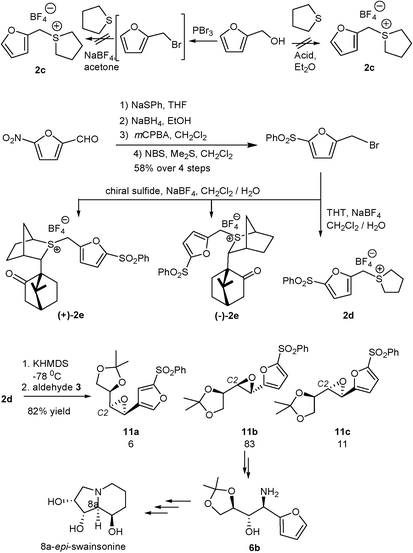 | ||
| Scheme 4 Synthesis and reactions of furyl-substituted ylides . THT = tetrahydrothiophene. | ||
With a scalable synthesis of transepoxide 11b in hand we continued our synthesis, the next step of which involved ring opening of the epoxide with an appropriate nitrogen nucleophile . If this occurred with retention of configuration this would lead to swainsonine, whilst inversion would lead to 8a-epi-swainsonine. Direct aminolysis with aqueous ammonia smoothly ring-opened the oxirane with inversion to give the anti amino alcohol subunit,17 from which the sulfone group was subsequently removed with sodium amalgam to give the amino alcohol 6b (Scheme 5). Attempts to effect ring opening of the epoxide with a retention of configuration were not successful. Use of TMSN318 only returned starting material, presumably because the sulfonyl group destabilized carbocation formation adjacent to the furyl group. Furthermore, removal of the sulfonyl group using Na/Hg amalgam could not be effected without destroying the epoxide . A double inversion strategy,19 using MgBr2 followed by NaN3, ultimately gave the same major isomer as that obtained from direct aminolysis, indicating that the initial ring opening had occurred with retention in this case! Presumably, the bromohydrin was formed under thermodynamic control through multiple attacks by the bromide ion.
 | ||
| Scheme 5 Completion of the synthesis of 8a-epi-swainsonine. | ||
Access to multigram quantities of 6b nevertheless allowed us to potentially obtain 8a-epi-swainsonine and so we next considered the oxidative ring rearrangement—the aza-Achmatowicz reaction.20 A key issue in this regard was the choice of the protecting groups for the amino alcohol subunit to advance our synthesis. Despite the common use of a sulfonamide N-protecting group in the aza-Achmatowicz reaction due to its compatibility with the acidic reaction conditions,21 we decided to carry forward a Cbz-protecting group due to the potential for a one-pot reduction–deprotection at the end of our total synthesis.22 We were pleased to find that when Cbz-protected α-hydroxy amine 12 was subjected to the oxidative reaction conditions (using anhydrous mCPBA),22 the ring expanded dihydropyridinone7b was obtained in 72% yield. Key to the success of this reaction was the use of anhydrous mCPBA and avoidance of an aqueous work-up. Evidently the dihydropyridinone7b was quite water soluble.
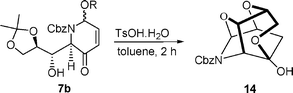
We proposed to protect the hemi-aminal 7b with the internal hydroxyl group, leading to bicycle 13, prior to the regio- and diastereoselective reduction of the carbonyl group. This anhydro-bridged structure would not only simultaneously protect both functionalities (the alcohol and the hemi-aminal ) but would also effectively block the Re face of the enone, and thus should result in high diastereoselectivity in attack of sodium borohydride from the less hindered Si (lower) face.
Thus, treatment of 7b with TsOH·H2O, in toluene in the presence of 4 Å molecular sieves23,24 gave the anhydro-bicycle 13 in 82% yield. As expected, Luche reduction25 gave essentially one diastereomer, 15, and in high (95%) yield.26 Subsequent hydrogenation with Pd/C simultaneously effected reduction of the alkene , N,O-acetal and Cbz cleavage to furnish the free amine 19. The synthesis was completed by first deprotection of the acetonide to the free tetrahydroxy amine 8b under standard catalytic acidic conditions, followed by an intramolecular N-alkylation under Appel conditions,27 which finally gave 8a-epi-swainsonine in the form of its hydrochloride salt 20. An X-ray structure of 20 confirmed its relative (and absolute) stereochemistry (see ESI).†‡
In conclusion we have achieved a concise synthesis of 8a-epi-swainsonine in an overall yield of 20% from R-glyceraldehyde by applying our epoxidation reaction with furyl-substituted sulfur ylides. The synthesis is also noteworthy for its brevity, minimal use of protecting groups, and for demonstrating the latent functionality inherent in furyl epoxides.
We thank the ORS and University of Bristol for a Scholarship (JB) in support of this work and Dr J. P. H. Charmant and Mr Saowanit Saithong for X-ray analysis. VKA thanks the Royal Society for a Wolfson Research Merit Award and EPSRC for a Senior Research Fellowship.
Notes and references
- F. P. Guengerich, S. J. DiMari and H. P. Broquist, J. Am. Chem. Soc., 1973, 95, 2055 CrossRef CAS.
- P. R. Dorling, C. R. Huxtable and S. M. Colegate, Biochem. J., 1980, 191, 649 CAS.
- A. D. Elbein, P. R. Dorling, K. Vosbeck and M. Horisberger, J. Biol. Chem., 1982, 257, 1573 CAS.
- D. A. Winkler and G. Holan, J. Med. Chem., 1989, 32, 2084 CrossRef CAS.
- J. W. Dennis, Cancer Res., 1986, 46, 5131 CAS.
- M. J. Humphries, K. Matsumoto, S. L. White and K. Olden, Cancer Res., 1986, 46, 5215 CAS.
- H. Motohiro, N. Kunio, T. Hiroshi, H. Junji, K. Masanobu, A. Hatsuo and I. Hiroshi, Chem. Abs., 1984, 101, 28283x Search PubMed.
- P. C. Das, J. D. Roberts, S. L. White and K. Olden, Oncol. Res., 1995, 7, 425 CAS.
- For reviews see: (a) S. G. Pyne, Curr. Org. Synth., 2005, 2, 39 Search PubMed; (b) A. E. Nemr, Tetrahedron, 2000, 56, 8579 CrossRef CAS.
- For recent examples see: A. J. Murray, P. J. Parsons and P. Hitchcock, Tetrahedron, 2007, 63, 6485 Search PubMed; (a) J. Ceccon, A. E. Greene and J. F. Poisson, Org. Lett., 2006, 8, 4739 CrossRef CAS; (b) N. S. Kumar and B. M. Pinto, J. Org. Chem., 2006, 71, 1262 CrossRef CAS; (c) C. W. G. Au and S. G. Pyne, J. Org. Chem., 2006, 71, 7097 CrossRef CAS; (d) H. Guo and G. A. O'Doherty, Org. Lett., 2006, 8, 1609 CrossRef CAS.
- V. K. Aggarwal and C. L. Winn, Acc. Chem. Res., 2004, 611 CrossRef CAS.
- V. K. Aggarwal and J. Bi, Beilstein J. Org. Chem., 2005, 1 DOI:10.1186/1860-5397-1-4.
- V. K. Aggarwal, E. Alonso, I. Bae, G. Hynd, K. M. Lydon, M. J. Palmer, M. Patel, M. Porcelloni, J. Richardson, R. A. Stenson, J. R. Studley, J.-L. Vasse and C. L. Winn, J. Am. Chem. Soc., 2003, 125, 10926 CrossRef CAS.
- V. K. Aggarwal, I. Bae, H. Y. Lee, J. Richardson and D. T. Williams, Angew. Chem., Int. Ed., 2003, 42, 3274 CrossRef CAS.
- Furfuryl sulfonium salts have been obtained in low yield from the corresponding alcohol and HPF6: S. Zhang, D. Marshall and L. S. Liebeskind, J. Org. Chem., 1999, 64, 2796 Search PubMed.
- (a) M. Valpuesta, P. Durante and F. J. Lopez-Herrera, Tetrahedron, 1993, 49, 9547 CrossRef; (b) M. Valpuesta, P. Durante and F. J. Lopez-Herrera, Tetrahedron, 1990, 46, 7911 CrossRef CAS.
- (a) M. Valpuesta, P. Durante and F. J. Lopez-Herrera, Tetrahedron Lett., 1995, 36, 4681 CrossRef; (b) B. Olofsson and P. Somfai, J. Org. Chem., 2002, 67, 8574 CrossRef CAS.
- B. Alcaide, C. Biurrun, A. Martinez and J. Plumet, Tetrahedron Lett., 1995, 36, 5417 CAS.
- P. Lupattelli, C. Bonini, L. Caruso and A. Gambacorta, J. Org. Chem., 2003, 68, 3360 CrossRef CAS.
- (a) Z. M. Wang and W. S. Zhou, Tetrahedron, 1987, 43, 2935 CrossRef CAS; (b) M. D. Burke, E. M. Berger and S. T. Schreiber, J. Am. Chem. Soc., 2004, 126, 14095 CrossRef CAS.
- C. F. Yang, Y. M. Xu, L. X. Liao and W. S. Zhou, Tetrahedron Lett., 1998, 39, 9227 CrossRef CAS.
- M. H. Haukaas and G. A. O'Doherty, Org. Lett., 2001, 3, 401 CrossRef CAS.
- In the absence of 4 Å sieves the polycyclic ether 14 was obtained via hydrolysis of the acetonide followed by addition.
. - J. Ostrowski, H. J. Altenbach, R. Wischnat and D. J. Brauer, Eur. J. Org. Chem., 2003, 1104 CrossRef CAS.
- J. L. Luche, J. Am. Chem. Soc., 1978, 110, 2226 CrossRef.
- In the absence of protection of the alcohol, Luche reduction on enone 16 did not give the expected allylic alcohol 17, but instead formed bicycle 18. It is believed that the free hydroxyl group adds to the enone (perhaps catalyzed by CeCl3) prior to reduction of the carbonyl group. Protection of the hydroxyl group was therefore required.
. - Y. W. Chen and P. Vogel, J. Org. Chem., 1994, 59, 2487 CrossRef CAS.

Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and X-ray structure of 20. See DOI: 10.1039/b713447a |
| ‡ CCDC 659348. For crystallographic data in CIF format see DOI: 10.1039/b713447a |
| This journal is © The Royal Society of Chemistry 2008 |