Potential shift correction in multivariate curve resolution of voltammetric data. General formulation and application to some experimental systems†
Arístides
Alberich
,
José Manuel
Díaz-Cruz
*,
Cristina
Ariño
and
Miquel
Esteban
Departament de Química Analítica, Facultat de Química, Universitat de Barcelona, Martí i Franquès 1-11, E-08028-Barcelona, Spain. E-mail: josemanuel.diaz@ub.edu; Fax: +34 934021233; Tel: +34 934039116
First published on 2nd November 2007
Abstract
A new mathematical algorithm is proposed to correct the progressive potential shift of some voltammetric signals that decrease the linearity of the data. The corrected data matrix can be further analysed by Multivariate Curve Resolution by Alternating Least Squares (MCR-ALS) and the vector including the potential shift corrections can be fitted to specific equations such as that by DeFord–Hume. A detailed discussion is given on the different cases of potential shift correction, and, in some of them, mathematical simulation is made or experimental systems [Cd(II)–glutathione and Zn(II)–glycine] are analysed.
1. Introduction
For many years, the application of Chemometrics to electroanalytical data had been quite scarce as compared to the case of spectroscopic techniques.1 Among the possible reasons for that, the traditional use of electrochemical hard modelling (i.e. the resolution of an a priori model in the form of a series of equations to be fitted to experimental data), a philosophy excessively different from most chemometrical methods, based on the a posteriori recognition of a model from the analysis of the data variation along the experiments (what some authors call electrochemical soft modelling) could be mentioned. Another reason could be the lack of linearity between many electrochemical signals and the bulk concentration of the electroactive species, as a consequence of different phenomena such as electrode saturation, fast interconversion of species during the measurement, adsorption on the electrode surface, capacitive currents, oxidation of the electrode material favoured by some species, etc. This lack of linearity, much more usual in electroanalytical data than in spectroscopic data, hinders the application of numerous chemometrical methods to certain electrochemical systems.Nevertheless, and despite these inherent problems, the number and variety of chemometrical applications to electrochemical data has increased considerably in the past few years.2,3 This is partially due to the need of reliable methods for multivariate analysis in electrochemical systems of increasing complexity like ultramicroelectrodes or arrays of electrochemical sensors4,5 (which are usually involved in the so-called electronic noses and electronic tongues). Additionally, the fast development of Artificial Neural Network (ANN) methods, which allow the study of non-linear systems, has expanded dramatically the range of electrochemical datasets susceptible to chemometrical analysis.2–5 These methods, however, suffer from a difficult and time-consuming calibration.
Multivariate Curve Resolution by Alternating Least Squares (MCR-ALS) is a powerful chemometrical method which allows the resolution of strongly overlapped signals with the application of a series of constraints that ensure some physicochemical meaning to the solution. It was originally developed for spectroscopy6 and later successfully applied to electroanalytical measurements.7 The implementation of new restrictions like peak shape, adapted to the special character of electrochemical signals, allowed the resolution of quite intricate metal complex systems.8 As for linearity, it was realised that the best results were obtained when MCR-ALS was applied to unequivocally linear systems like electrochemically inert complexes, i.e. these for which the electrochemical measurement is much faster than the association–dissociation reactions, so that every species contributes independently to the current with a magnitude that is proportional to its bulk concentration. In the case of electrochemically labile complexes, i.e. those with association–dissociation equilibria much faster than the measurement, a progressive potential shift of the free metal signal is observed as the bound metal fraction increases. This signal displacement has been shown to decrease the linearity of the data, although it is possible to obtain acceptable results for moderate potential shifts.8 In contrast, larger potential shifts usually produce too large errors or demand too high a number of components. Finally, in some systems, potential shifts that are too large make the use of MCR-ALS impossible. This is the case, for instance, of the movement along the potential axis of some irreversible reduction signals as a function of pH.
Thus, in order to expand the use of MCR-ALS (and, eventually, other chemometrical methods) to a wider range of electrochemical systems, it seems valuable to investigate suitable methods to overcome the problems derived from the lateral movement of the signals (along the potential axis in the case of voltammetric measurements).
Although signal shift correction is a practically unexplored subject in electroanalytical chemometrics, this is a very common practice in the analysis of data obtained by chromatography,9–13capillary electrophoresis (CE)14 or NIR13 and NMR15,16 spectroscopies. Such techniques produce a large number of narrow, overlapping signals that can be used in multivariate calibration or as a fingerprint in classification or pattern-recognition methods. The x-axis position of these signals can change in a quite unpredictable way with small variations of the experimental conditions (use of different apparatus, changes in temperature or mobile phase composition, degradation of the chromatographic column, etc.), so that in many cases the use of warping (or alignment) procedures to line up all spectra in a consistent data matrix is mandatory. Among the most popular warping methods we can mention dynamic time warping (DTW), which uses distance as a measure of the similarity of two signals13 and correlation optimised warping (COW), which aligns two signals by piece-wise linear stretching and compression, the optimal alignment being determined by correlation of the aligned fragments of signals.13 Anyway, more sophisticated methods can be applied as, for instance, in the use of a genetic algorithm (peak alignment by a genetic algorithm, PAGA).15
These warping techniques are very useful in the case of spectra with plenty of narrow, weakly overlapping signals and are essentially based on the philosophy of expanding, compressing or interpolating in a different manner the different sections of such long spectra by comparison with a reference. Thus, in the case of much separated peaks it is possible to shift them even in opposite directions; but, in contrast, very close or overlapping peaks are always shifted in a similar way. In electroanalysis this could be useful, for instance, to line up voltammograms obtained with different reference electrodes (overall shift of the potential values) or using different potential steps or starting potentials (compression/expansion of the potential scale). Nevertheless, the goal of this work is the correction of the lateral movement of peaks much wider than these of chromatography, CE or NMR and which are overlapping in a stronger way. Moreover, the usual situation in electroanalysis is the movement of only one out of several overlapping signals, while the rest remain in the same position and overlap with the moving signal in a different way. Most warping methods would move or compress all overlapping signals in the same direction, so that such methods do not appear to be suitable for this purpose.
In some cases a preliminary correction based on the theoretical knowledge of the system can be possible. A pioneering experience of this strategy has been carried out with good results in the MCR-ALS analysis of NMR spectra of acidic substances, where the signal of the chemical shift of the acidic proton changes as a function of pD.17 Then, a mathematical transformation previous to the MCR-ALS analysis distributes the height of the moving peak between the respective x-positions of both the acidic and basic forms of the molecule. Although this strategy could be applied to electrochemical systems, the large variety of possible situations and hard-modelling approaches suggest that it would be valuable to test a different option based on a preliminary determination of potential shifts and a further analysis of them in terms of electrochemical models.
In this paper, a procedure is presented for the analysis of voltammetric data which is based on a preliminary empirical correction of the potential shifts in comparison to some reference signals. The resulting set of corrected voltammograms, which is expected to be linear, can be further analysed by MCR-ALS and, depending on the case, the potential shifts detected in the correction can be also analysed by means of electrochemical hard modelling.18
2. Theory
2.1. The MCR-ALS procedure
Fig. 1a summarises the usual MCR-ALS method when applied to voltammetric linear data. Voltammograms are arranged in a matrix I, where every element Iij corresponds to the current measured along the scan i at the potential j. The number of components linearly contributing to the signal can be determined in different ways, i.e. by singular value decomposition, SIMPLISMA,19,20 or visual inspection of the data matrix.21 Then, I is factorised as the product of a C matrix containing the concentration profiles (i.e. the evolution of the concentrations of the components along the different scans) and a V matrix containing the pure signal of each component k (i.e. its contribution to the signal per unit of concentration) plus an error matrix X. This is done by means of an iterative process (ALS) which requires an initial estimation of either the C or V matrix. In the usual case of peak-shaped signals, the home-made program peakmaker21 can be used to estimate V (see Experimental section). Finally, along ALS iterations, several constraints can be applied, such as non-negativity, closure, selectivity or signal shape (see refs 8,21,22 for more information).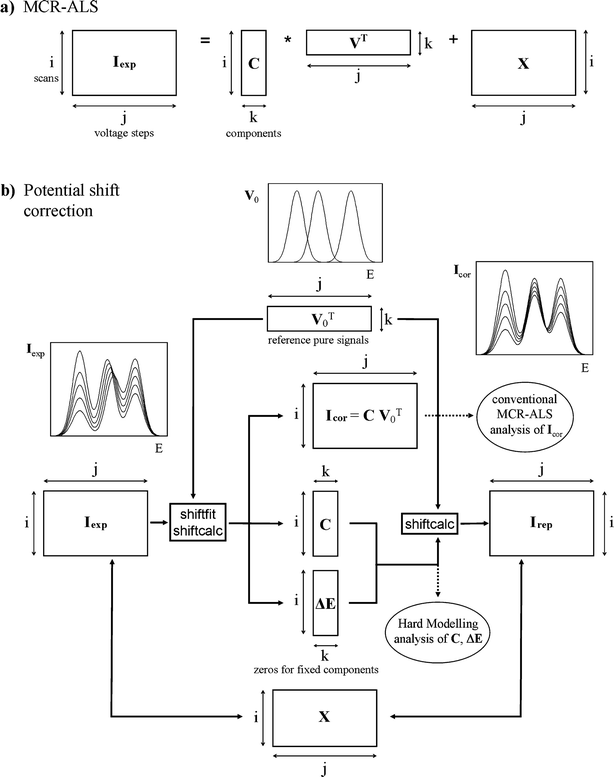 | ||
| Fig. 1 Flowcharts of the standard MCR-ALS method (a) and the potential shift correction algorithm proposed in this work (b), as applied to a current data matrix Iexp. In the first case, a concentration matrix C, a pure voltammogram matrix V and an error matrix X are obtained. In the second case, the application of shiftfit and shiftcalc programs to Iexp with a predefined reference signal matrix V0 produces a movement-corrected matrix (Icor), a concentration matrix C and a potential shift matrix ΔE. The inverse process by means of the shiftcalc function yields the reproduced matrix Irep that allows computation of the error matrix in the form X = Iexp – Irep. Subscripts i, j and k denote the voltammogram, potential and component number, respectively. The transposed matrix is denoted by ‘T’. | ||
2.2. The potential shift correction: general overview
The general method proposed to analyse matrices with laterally moving signals is summarised in Fig. 1b and is described in more detail in Fig. 2 and 3. It basically consists of the alignment of all moving signals to the same reference position given by the matrix of reference pure signals V0, by means of an iterative algorithm which uses the home-made Matlab23 functions shiftcalc and shiftfit.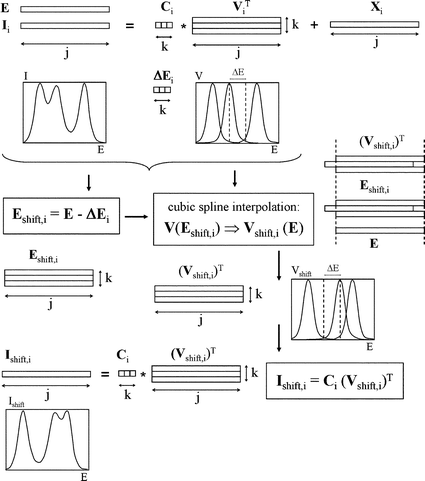 | ||
| Fig. 2 Flowchart for the potential shift implementation by means of the shiftcalc program. Subscripts i, j and k denote the voltammogram, potential and component number, respectively. Transposed matrices are denoted by ‘T’. | ||
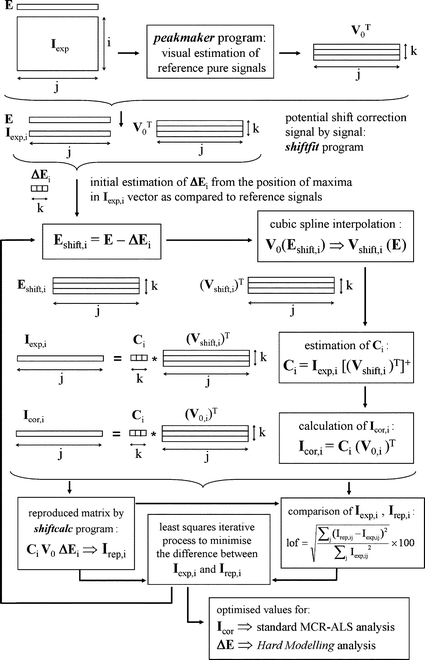 | ||
| Fig. 3 Flowchart for the potential shift correction by means of the shiftcalc and shiftfit programs. Subscripts i, j and k denote the voltammogram, potential and component number, respectively. Transposed and pseudo-inverse matrices are denoted by ‘T’ and ‘+’, respectively. | ||
Fig. 1b shows that the combined application of shiftfit and shiftcalc to an experimental matrix Iexp affected by lateral movement of one or more signals produces:
(i) A corrected matrix Icor where every moving signal has been aligned to a fixed (by V0 matrix) reference potential.
(ii) A matrix ΔE containing the evolution of all potential shifts (as compared to the reference potentials) along the different scans. In the case of signals which are not moved, the corresponding elements of ΔE equal zero.
(iii) A matrix C containing a rough estimation of the concentration profiles of the components considered.
The Icor matrix is expected to be linear, so it can be analysed by MCR-ALS in the standard way using V0 or any other estimation of pure voltammograms to obtain optimised C and V matrices. The ΔE matrix can be used, depending on the case, to fit electrochemical hard-modelling equations to determine parameters like stability constants or proton-to-electron ratios. In a further discussion, some specific cases are analysed in detail. On the other hand, the potential shift correction can be inverted from V0, C and ΔE matrices by means of shiftcalc to obtain a reproduced matrix Irep that can be compared to Iexp yielding the error matrix X (= Iexp – Irep).
Finally, it must be remarked that the arbitrary and invariant character of the pure voltammograms used in matrix V0 (which remains constant along all iterations) can be refined if the optimised unit signals obtained with MCR-ALS are used again as the V0 matrix for a better movement correction of the original matrix Iexp prior to a new application of MCR-ALS. In this way, an alternating iterative process MCR-ALS/movement correction can be carried out until the overall lack of fit does not change appreciably.
2.3. The potential shift correction: the algorithm in detail
To understand how the functions shiftcalc![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) /shiftfit work, one must consider that: (i) shiftcalc can displace every signal in every voltammogram of a matrix I a given potential shift ΔE to produce the matrix Ishift, and (ii) shiftfit can iteratively optimise the values of ΔE that have to be ‘discounted’ from a matrix Iexp (by means of shiftcalc) so that all signals in the new matrix Icor remain at the fixed potentials stated in the V0 matrix.
/shiftfit work, one must consider that: (i) shiftcalc can displace every signal in every voltammogram of a matrix I a given potential shift ΔE to produce the matrix Ishift, and (ii) shiftfit can iteratively optimise the values of ΔE that have to be ‘discounted’ from a matrix Iexp (by means of shiftcalc) so that all signals in the new matrix Icor remain at the fixed potentials stated in the V0 matrix.
Essentially, shiftcalc displaces the E-axis a magnitude ΔEik which is different for each scan i and each moving component k and is zero for ‘immobile’ components (Fig. 2). The full set of ΔE values can be arranged in a ΔE matrix, with as many rows i as original voltammograms and as many columns k as components accepted for the I matrix. For every original voltammogram, every pure signal is moved according to the corresponding ΔEij value using cubic spline interpolation to change from the E-axis to a new E – ΔEij axis. This means that using the new potential axis (E – ΔEj), every column in the new pure voltammogram will correspond to a different potential as compared to the original E-axis, so that the new currents that are going to occupy these columns have to be interpolated for the new potential values by using the closest current vs. potential points in the original pure voltammogram. Then, every moved voltammogram can be obtained as the sum of the products of concentration and moved pure signal for each component.
Concerning to the program shiftfit (Fig. 3), it is basically a non-linear least squares iterative algorithm to optimise the set of ΔE values that, applied to the reference pure signals in V0 by means of the function shiftcalc, generates a reproduced matrix Irep as close as possible to the experimental data matrix Iexp. This operation is independently applied to each individual voltammogrami of the Iexp matrix and can be summarised in the following steps:
(i) For each moving signal (or component) k, a potential window is defined where the corresponding pure current in V0 (i.e. V0,k) is higher than 20% of the maximum current of the peak in V0,k. Inside this region, a local maximum is searched (in the form of an absolute maximum whose second derivative, estimated by the Savitzky–Golay method,23,24 is negative). The difference between the x-coordinates of the maximum found in this way and the maximum in V0,k is kept as an initial estimation of the potential shift of component k in the experiment (or voltammogram) i:
| ΔE0,i,k = E (local max in Iexp,i) – E (max in V0,k) | (1) |
(ii) A matrix Eshift,i is made by applying for every k component the transformation:
| Eshift,i,k = E – ΔE0,i,k | (2) |
(iii) Cubic splines are used to interpolate from each set of V0,k data (referred to as E potentials) the values that would correspond to the new Eshift,i,k axis. This yields the Vshift,i matrix of ‘moved’ pure voltammograms.
(iv) An initial estimation of the concentration profiles for the experiment i is generated in the form:
Ci = Iexp,i![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) [(Vshift,i)T]+ [(Vshift,i)T]+ | (3) |
(v) A dataset that we can denote as ‘xdata’ is made by combination of Ci and V0 (taking advantage of the fact that the vector and the matrix have the same number of columns k). Additionally, a function F is defined which, from the set of ‘xdata’ of the experiment i and depending on the values of the ΔEi parameters, produces a set of ‘reproduced ydata’ which are the currents of the vector Irep,i and are compared to the ‘ydata’ of the experimental vector Iexp,i. This F function can be computed, indeed, by using the shiftcalc program previously described. Then, the ΔEi values can be optimised to achieve a maximum agreement between reproduced and experimental currents by means of a non-linear least squares minimisation procedure that can be summarised by the condition:
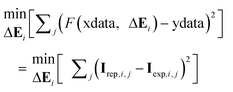 | (4) |
(vi) If convergence is achieved, the potential-shift-corrected vector for the experiment i, Icor,i, can be obtained in the form:
| Icor,i = Ci(V0,i)T | (5) |
If these steps are applied independently to each one of the ivoltammograms of the data matrix Iexp, the resulting Ci, ΔEi, Icor,i, Irep,i vectors can be column-wise integrated into the overall matrices C, ΔE, Icor, Irep.
In order to quantify the goodness of the fitting, the percentage of lack of fit (lof) is used, which is defined in the same way as for standard MCR-ALS analysis:
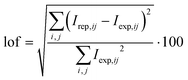 | (6) |
It must be pointed out that in signal-moving systems a first lof can be computed for the movement correction and a second lof for the MCR-ALS process applied to the corrected matrix. However, more interesting than these separated values is the overall lof, which compares the original data matrix with that reproduced after both the CVT product of the matrices optimised by MCR-ALS and the subsequent application of the optimised potential shift. Although such an overall lof value is expected to be larger than the partial ones (since it involves both steps), in practice it should not be much larger, since the increase in the error produced by the application of a second procedure (MCR-ALS) is counterbalanced by the improvement in the fitting of the pure voltammograms, which in the movement correction step were not optimised.
2.4. The electrochemical interpretation of ΔE values
In the following, different typical situations in voltammetric analysis which produce lateral movement of the signals will be discussed.In any of these circumstances, the potential correction is used just to group the moving signals at the same reference potential to decrease the non-linearity promoted by their lateral movement, so that the full data matrix can be resolved by MCR-ALS. As a result, a concentration profile (C matrix) is obtained for the corresponding moving component which summarises the evolution of its contribution along the experiment and, eventually, can be associated to a species concentration (e.g. some anodic signals are proportional to the ligand concentration). Concerning the values in the ΔE matrix, they can be regarded as practically useless from a quantitative point of view.
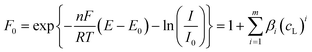 | (7) |
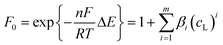 | (8) |
When the stability constants of the successive complexes are not too large, the potential shifts are small and the voltammetric data remain quite close to linearity, thus MCR-ALS can reasonably explain the lateral movement with the progressive overlapping of a group of contiguous pure signals.8,30 However, large values of the stability constants could produce a dramatic decrease of linearity and hinder the analysis by MCR-ALS. In this case, the free metal ion and all their successive labile complexes can be grouped together in a unique component and the lateral movement can be corrected by means of the proposed method taking as a reference the signal of the free metal ion. The corrected current matrix Icor can be analysed then by MCR-ALS and, in the particular case of the grouped labile component, the concentration profile obtained in the C matrix will indicate the evolution of the total concentration of the labile fraction. As for the ΔE matrix resulting from shiftfit, the column associated with the mobile component, jointly by the corresponding ligand concentrations, can be fitted to eqn (8) to optimise the values of the successive stability constants.
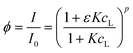 | (9) |
It must be noted that pH studies yield valuable information about the stoichiometry of the complexes. Thus, if we consider the scheme:
| ML + mH + ne ⇄ M(Hg) + HmL | (10) |
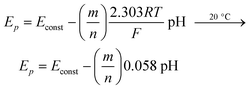 | (11) |
However, if a noticeable complex dissociation takes place during the voltammetric measurement, the signal of the free metal is increased and moved to more negative potentials as compared to the totally inert situation. As for the signal of the complex, it stays at the same potential but its current is decreased.36–40 As a consequence of the movement of the free metal signal, the partial dissociation of inert complexes (quasi-inert situation) causes a decrease of the data linearity that in extreme cases can hinder their MCR-ALS analysis.
The proposed method for potential shift correction is able to produce a corrected matrix which allows the resolution of the free metal ion and the complex. Nevertheless, the concentration profiles obtained will not be totally reliable unless the change in the currents caused by complex dissociation is also taken into account. A rigorous solution for this problem is not easy, since it requires considering the dissociation kinetics of the complex. Alternatively, a reasonable improvement of the results could be obtained by assuming that, in intermediate situations between labile and inert behaviour, eqn (7) compensates the negative potential shift of the free metal signal due to dissociation with the corresponding current increase.40 Such approximation, which produced acceptable results in the Cd(II)–NTA system,40 can be used in two ways: (i) by applying eqn (7) directly to the peak potentials and currents measured for the signal of the free metal in the same way as for a totally labile system, and (ii) by modifying the signal heights of the free metal and the complex in the shiftfit-corrected matrix as a function of the observed potential shift and further applying MCR-ALS. For this purpose, let us assume that eqn (7) holds for both inert and quasi-inert situations. Then, it is possible to write:
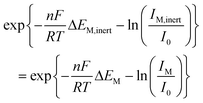 | (12) |
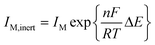 | (13) |
Finally, it must be noted that some aspects of this method for the M/ML case can be extended to the situation of different successive complexes which (i) are all inert or (ii) some of them are inert and some of them are labile.
3. Experimental and data treatment
3.1. Chemicals and instrumentation
Glutathione (GSH), in the reduced form, was provided by Merck and glycine (Gly) by Sigma, both with a purity greater than 99%. All other reagents used were Merck and Sigma analytical grade. Cd(II) and Zn(II) stock solutions were prepared by dissolving Cd(NO3)2·4H2O and Zn(NO3)2·4H2O in water and standardised complexometrically.41Tris(hydroxymethyl)aminomethane–HNO3buffer solutions of pH 7.5 were used for pH control. Ultrapure filtered water (Milli-Q plus 185, Millipore) was employed in all experiments.
Voltammetric measurements by differential pulse polarography (DPP) were carried out with a 757 VA Computrace (Metrohm). The working, reference, and auxiliary electrodes were a static mercury drop electrode (SMDE) with a drop area of 0.6 mm2, Ag/AgCl/KCl (3 mol L–1), and a glassy carbon electrode, respectively. Instrumental parameters were as follows: pulse amplitude of 0.05 V, pulse time of 0.04 s, drop time of 1 s; voltage step was 4 mV for the titrations of the Cd(II)–GSH system and 3 mV for the titrations of the Zn(II)–Gly system.
Experiments were performed at a controlled room temperature of 20 °C and under a purified N2 atmosphere.
The measurements of pH values during the experiments were carried out by means of a Crison micropH2000 pH meter.
3.2. Procedures
For titrations of glutathione with cadmium, 20 mL of GSH (2 × 10–5 mol L–1) were placed into the cell and deaerated with nitrogen for 20 min. Then the DPP curve was recorded. After that, successive additions of 8 × 10–4 mol L–1Cd(II)-ion solution were made. After each addition and deaeration for 1 min, voltammograms were recorded. The peptide solution was freshly prepared before each experiment by dissolving the peptide in a previously deaerated buffer solution. All solutions were prepared in 0.05 mol L–1 of TRIS.The experiments to study Zn(II)-glycinate stability constants were carried out placing 20 mL of 1 × 10–5 mol L–1Zn(II) solution into the cell and deaerated with nitrogen for 30 min. Then, several DPP curves were recorded each 5 min in order to make certain of the absence of adsorption onto the cell walls. After that, aliquots of 1.67 mol L–1glycine solution (0.01 mol L–1glycinate) were added and DPP curves recorded. Between additions, solutions are purged and mechanically stirred for 1 min. All solutions were freshly prepared in 0.05 mol L–1 KNO3/0.01 mol L–1 TRIS.
3.3. Data simulation for a simple labile system
A preliminary test of the proposed method has been carried out on simulated data. The system considered was relatively simple: two successive labile metal complexes with stability constants log β1 = 4.50 and log β2 = 8.00 and with diffusion coefficients equal to that of the free metal ion. The individual signals were constructed according to the theoretical expression for the difference δ between the currents before and after the pulse application in differential pulse polarography (DPP):42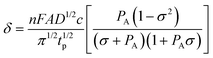 | (14) |
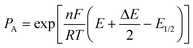 | (15) |
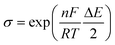 | (16) |
3.4. Initial estimation of pure voltammograms using peakmaker function
Peakmaker is a home-made Matlab function21 that generates Gaussian peaks according to the equation: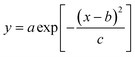 | (17) |
(i) The program plots the data matrix I in a 2D graph (currents versus potentials).
(ii) The user moves the cursor with the mouse to place it on the top of each maximum of the graph susceptible to be a component and then presses the mouse button. The program determines the height (parameter a) and the x-position (parameter b) of every peak, which are given by the y- and x-coordinates, respectively, of the cursor every time the button is pressed.
(iii) With the I matrix still visible, the program draws one Gaussian peak at each position previously determined by the mouse with a default width (parameter c = 10).
(iv) By following the same order as in the graphical input, the program draws a horizontal line at half of the height of each peak. Then, the user selects, by comparison with the I matrix in the background, the left or right x-position at half of the peak height (x1/2) and presses the button. Using this value, the program computes parameter c of the Gaussian peak in the form:
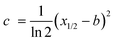 | (18) |
(v) All individual peaks are integrated into the V matrix.
The program also allows one to generate asymmetric peaks by selecting two different half-peak widths (right and left). Then, the peak is defined by a different Gaussian equation at each side of the maximum.
4. Results and discussion
4.1. Analysis of simulated data
Fig. 4 and Table 1 summarise the analysis of the data simulated as described in Section 3.3 by using the standard MCR-ALS procedure and by means of MCR-ALS with a previous potential shift correction. In the first case, four components have been used to describe the system: one for the metal ion, two for the successive complexes and one for the free ligand. As Table 1 shows, the lack of linearity of the system produces an excessive lof (10.3% in the case including 0.1% of Gaussian noise) and values of the stability constants somewhat lower (0.25 and 0.32 log units) to these applied in the simulation.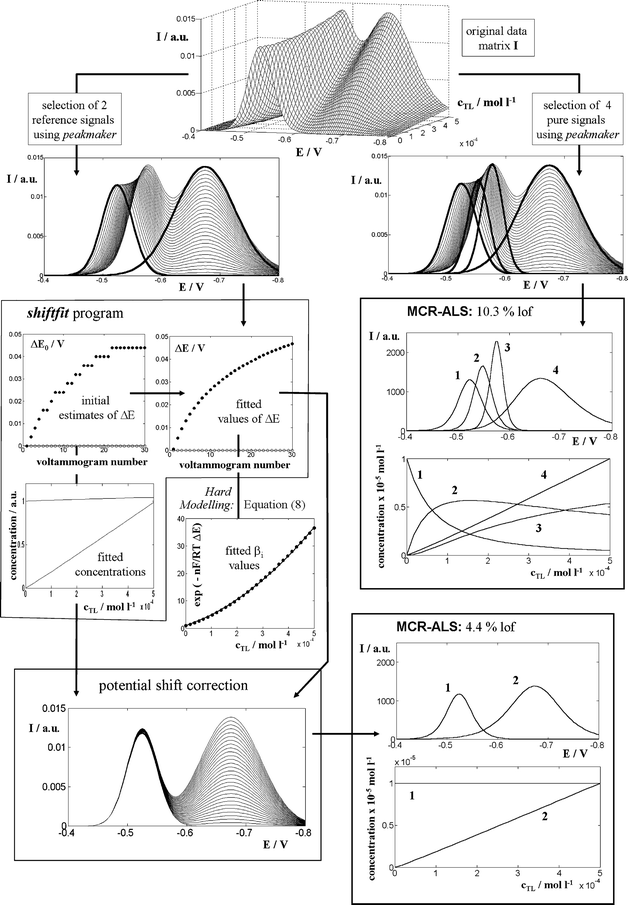 | ||
| Fig. 4 Comparison of the standard MCR-ALS procedure and MCR-ALS with a preliminary potential shift correction (by means of the shiftfit program), when applied to signals simulated with a 0.1% of Gaussian error (expressed as the percentage of the standard deviation with respect to the maximum current of the matrix). | ||
| Method | % noise | % lof shiftfit | % lof MCR-ALS | % lof overall | log β1 | log β2 |
|---|---|---|---|---|---|---|
| Standard MCR-ALS | 0.1 | — | 10.3 | 10.3 | 4.25 (0.02) | 7.68 (0.02) |
| Non-negativity, signal shape and equilibrium constraints | ||||||
| Potential shift correction and MCR-ALS | 0 | 2.53 | 4.38 | 3.24 | 4.492 (0.007) | 7.912 (0.007) |
| Non-negativity, signal shape and closure for metal species | 0.1 | 2.54 | 4.38 | 3.25 | 4.491 (0.007) | 7.912 (0.007) |
| 0.5 | 2.79 | 4.38 | 3.45 | 4.484 (0.007) | 7.917 (0.006) | |
| 1 | 3.43 | 4.40 | 4.03 | 4.48 (0.01) | 7.91 (0.01) | |
| 2 | 5.30 | 4.44 | 5.66 | 4.53 (0.01) | 7.90 (0.01) | |
| 5 | 17.9 | 4.71 | 18.1 | 4.42 (0.05) | 7.97 (0.04) | |
In contrast, the data previously corrected for potential shifts can be analysed by MCR-ALS considering two components only (free metal + metal complexes on one hand and free ligand on the other) and they produce better values of lof (4.38% in the same case as before). Table 1 shows that these values do not increase significantly when noise increases, which suggests that a large portion of noise is ‘filtered’ by the shiftfit program. Finally, the fitting of eqn (8) to the optimised ΔE values computed by shiftfit produces quite precise values for the stability constants which are very close to the values used for simulation (discrepancies between 0.01 and 0.10 log units).
It must be pointed out, however, that the lof of standard MCR-ALS (10.3% in the previous case) cannot be strictly compared to that obtained after a previous potential shift correction (4.38%), since this previous operation is also contributing to the final error (in the case considered, shiftfit produces a lof of 2.54%). A more realistic calculation should be done as indicated in Section 2.2. Anyway, the resulting overall lof in this case (3.25%) is still better than the value obtained using the standard MCR-ALS procedure.
4.2. The Cd(II)–GSH system
Fig. 5a shows the current data matrix obtained in the DPP titration of glutathione (GSH) with a Cd(II) solution at pH 7.5 in 0.05 mol L–1 TRIS. As Cd(II) is being added, the initial anodic signal of GSH (signal 2 in Fig. 5) starts to decrease and new signals develop, which can be attributed to an inert electroactive Cd(II)–GSH complex (signal 4) of 1 : 2 stoichiometry34 and to an anodic signal of the mentioned complex (signal 1). Finally, at large enough concentrations of added cadmium, the peak of free Cd(II)-ion starts a fast increase (signal 3). A visual inspection of the experimental matrix evidences an important movement of signal 1, a very slight shift of signal 4 and no shift at all for signals 2 and 3. This is consistent with the inert character of the complex, except for the slight movement of signal 4, which could be due to different reasons as, for instance, a decrease of the electrochemical reversibility of complex reduction. Anyway, the movement is so small that it can hardly affect the linearity of the system. A different question is the wide displacement of signal 1, which is likely to decrease linearity. Indeed, when standard MCR-ALS is applied with four components (Fig. 5e,f) a too large lof is observed (14.5%), as well as a certain ambiguity of the signals along the iterations which required the use of selectivity constraints. The successive application of shiftfit, to correct the movement of signal 1, and MCR-ALS for the analysis of the corrected matrix (Fig. 5b–d) improve the lof considerably (8.2 and 3.1% respectively). Fig. 6 shows the good reproduction of the experimental matrix by both consecutive procedures (i.e. as the product CVT of the matrices obtained by MCR-ALS shifted by the shiftcalc program by using the optimal ΔE values). The overall lof (9.3%) is slightly higher than that of shiftfit and is still much better than that of conventional MCR-ALS.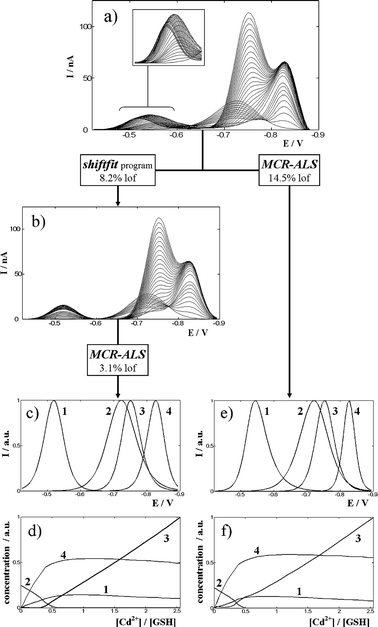 | ||
| Fig. 5 Analysis of the experimental data matrix (a) containing the differential pulse polarograms measured for a GSH solution 2 × 10–5 mol L–1 titrated with Cd(II) at pH 7.5 in a medium 0.05 mol L–1 in TRIS buffer. The application of the shiftfit program produces a corrected matrix (b) that is analysed by MCR-ALS with the constraints of non-negativity and signal shape to obtain pure signals (c) and concentration profiles (d). The results obtained by direct MCR-ALS analysis of the experimental matrix using the same constraints (plus selectivity for signal 1) are also shown (e,f). The inset in (a) shows a magnification of the region where signal 1 is moving. The lack of fit of every operation is also indicated. | ||
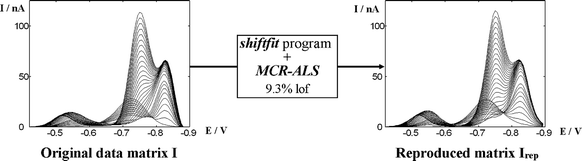 | ||
| Fig. 6 Comparison of the original and reproduced data matrices after the overall process of shiftfit and MCR-ALS in the case of the voltammograms shown in Fig. 5. | ||
At this point, it is interesting to notice that the corrected matrix (Fig. 5b) does not present the slight movement of component 4, even when we have not forced the shiftfit program to correct it. This is an intrinsic characteristic of the method: as all compounds have to be reproduced in the form CkV0,kT (see Fig. 3), all of them will remain at a fixed potential, independently of their movements. The main difference, however, between the forced and unforced movement correction is that the former optimises a ΔE value, so that in the reproduced matrix (to be compared to the experimental one in the lof) ΔE values recover the movement and the reproduction is good, whereas the latter yields ΔE = 0, so that the movement is not reproduced and the lof increases. These statements are confirmed when shiftfit is required for simultaneously moving components 1 and 4 and MCR-ALS is applied to the corrected matrix. Then, the overall lof changes from 9.3% (only signal 1 corrected) to 8.1% (signals 1 and 4 corrected). Such decrease evidences a small contribution of signal 4 to non-linearity that, anyway, is not large enough to justify the correction of their small potential shifts.
Finally, the ΔE values obtained by means of shiftfit do not require a further treatment, since the most important information concerning to anodic signal 1 is the evolution of the concentration profile, which is very similar to that of the complex, thus reinforcing the consistency of the data. At this point it must be noted that an alternative solution for the movement of signal 1 would be cutting off the initial part of the matrix (ca. until –0.65 V) in order to remove component 1. This option, however, does not allow the calculation of a valuable concentration profile (that of signal 1) which confirms the nature and evolution of component 4. In situations where the moving signal was more strongly overlapped, an effective cutting would be simply impossible.
4.3. The Zn(II)–glycine system
The Zn(II)–glycine system has been studied at pH 7.5 in 0.05 mol L–1 KNO3 and 0.01 mol L–1 TRIS in the glycinate concentration range 0–1.8 × 10–4 mol L–1. The concentration of deprotonated ligand (glycinate) has been computed using the acidity constants pK1 = 2.36 and pK2 = 9.57.43,44 The same literature provides values for the Zn(II)-glycinate stability constants (Table 2) which ensure that, under the experimental conditions of the present work, only ML and ML2 complexes are expected.| Experimental technique | Experimental conditions | Additional information | % lof | log β1 | log β2 | Ref. |
|---|---|---|---|---|---|---|
| DPP | 20 °C | Standard MCR-ALS | 15.7 | 4.78 (0.03) | 8.94 (0.03) | This work |
| 0.05 M KNO3–0.01 M TRIS | Non-negativity, signal shape and equilibrium constraints | |||||
| DPP | 20 °C | Shiftfit program | 6.7 | 5.2 (0.1) | 9.88 (0.02) | This work |
| 0.05 M KNO3–0.01 M TRIS | ||||||
| Calorimetry | 25 °C | IUPAC tentative values | — | 5.06 | 9.44 | 43 |
| 0.05 M KCl | ||||||
| Glass electrode | 25 °C | IUPAC recommended values | — | 5.03 | 9.23 | |
| 0.15 M KCl | ||||||
| Glass electrode | 25 °C | — | — | 4.96 ± 0.01 | 9.19 ± 0.1 | 44 |
| 0.1 M ionic strength | ||||||
| Glass electrode | 25 °C | — | — | 5.38 ± 0.1 | 9.81 ± 0.2 | |
| 0 M, ionic strength | ||||||
As Fig. 7a shows, the addition of glycinate causes both a progressive decrease and a shift to negative potentials of the peak of the free metal ion. Additionally, there is a small peak increasing and further decreasing at ca. –1.15 V and a third one monotonically increasing at ca. –1.27 V.
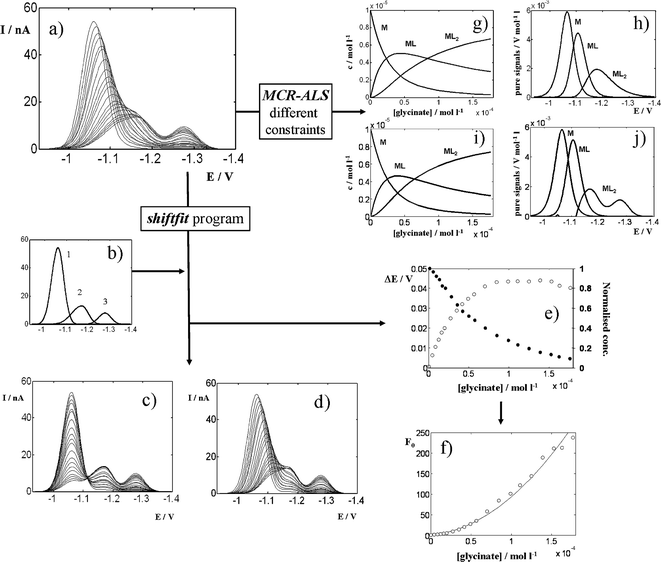 | ||
| Fig. 7 Analysis of the experimental data matrix (a) containing the differential pulse polarograms measured for a Zn(II) solution 1 × 10–5 mol L–1 titrated with glycine at pH 7.5 in a medium 0.05 mol L–1 in KNO3 and 0.01 mol L–1 in TRIS buffer. The application of the shiftfit program using the reference signals shown in (b) produces the reproduced matrix (d), the corrected matrix (c) and a series of potential shifts ΔE and concentration/current decreases (e) which are integrated into the Leden F0 function (f), which can be fitted to eqn (7) to yield a curve (denoted with a solid line) and the stability constants summarised in Table 2. The application of MCR-ALS with non-negativity, signal shape and equilibrium constraints for all three components allows one to obtain concentration profiles (g) and pure signals (h) for M, ML and ML2, as well as the fitted values of the stability constants shown in Table 2. Fig. 7i,j show the results obtained when the signal shape constraint is applied only to the species M and ML. | ||
From a qualitative point of view, this can be explained by assuming that both ML and ML2 complexes are electroactive and partially inert. The first signal is decreased because, as the ligand is being added, an increasing fraction of the free metal ions initially present are reduced in the form of ML (second signal) or ML2 (third signal) complexes. If the complexes were totally inert, the first signal would remain at the same potential and the system would be linear from a chemometrical point of view. Thus, the observed progressive shift due to the partial dissociation of both complexes can introduce in the system an important source of non-linearity. In Section 2.4.5 an approximation is proposed to compensate potential shifts and current decreases due to partial complex dissociation in order to apply the DeFord–Hume method [eqn (7)] in these circumstances.
To check the suitability of such approximation, the data matrix in Fig. 7a has been analysed in two ways: (i) by means of MCR-ALS with non-negativity, signal shape and equilibrium constraints; and (ii) by fitting eqn (7) to the ΔE and current decrease values obtained by applying the shiftfit program to the experimental data matrix. The results of both methodologies are summarised in Table 2 and Fig. 7.
It can be seen that an appropriate choice of the reference signals (Fig. 7b) produces a set of potential shifts and current decreases (Fig. 7e) with a good reproduction of the original data matrix (Fig. 7d), confirmed by a reasonable lof (6.7%). The application of eqn (12) leads to an F0 function (Fig. 7f) that allows the fitting of eqn (7) to yield the log β values which appear in Table 2. In contrast, the application of MCR-ALS (Fig. 7g,h) produces quite unrealistic pure signals, especially that of ML2, which appears at ca. 0.1 V less negative potentials than the third signal in the data matrix. This results in a too high lof (15.7%) and in a set of stability constants notoriously lower than these obtained by using shiftfit (Table 2). If the signal shape constraint is only applied to M and ML, the lof improves slightly (10.5%), similar concentration profiles are obtained (Fig. 7i) and the signal of ML2 is split in two parts to explain the dissociated and undissociated fractions of the complex (Fig. 7j). As compared to literature data, the values obtained by MCR-ALS are lower and these from shiftfit are higher but somewhat closer to them, especially considering literature data at the lowest ionic strengths (0 and 0.05). These facts confirm a significant improvement of the MCR-ALS data analysis when using the shiftfit approach.
5. Conclusions
In voltammetric techniques, the relationship between the kinetics of chemical/electrochemical reactions and the time window of the measurement can dramatically affect the linearity of the data. For fast enough techniques, systems become inert and signals usually appear at the same potential, which ensures data linearity and allows a successful application of MCR-ALS. In contrast, slower techniques or faster kinetics produce signals which in fact average contributions of species that are converting into each other during the measurement time. This usually produces signals moving along the potential axis in intermediate positions between the pure signals of the interconverting species. Also, the change in the measurement speed and/or in the experimental conditions (e.g. pH, ionic strength…) can modify the kinetics of electrochemical reductions, which can produce a potential shift and occasionally a broadening of the reduction signals. These facts cause a decrease of linearity which, depending on the magnitude of the potential shift, can still be afforded by MCR-ALS or, in many cases, hinders MCR-ALS analysis (too large lof values are registered unless an unrealistically high number of components is used to explain non-linearity).For non-overlapping signals, electrochemical hard modelling provides a wide variety of methods for the study of labile or kinetically-governed systems. However, they are based on peak (or half-wave) potentials, peak (or limiting) currents or even on the full signal shape, but in all cases free of overlapping with other signals. The potential shift correction method by means of the shiftfit program allows one to virtually extract the non-linearity of the data in the form of a potential shift matrix that can be further analysed by hard modelling as in the case of non-overlapping signals. Moreover, shiftfit produces a corrected, linear matrix that can be easily analysed by MCR-ALS. Indeed, the proposed method is a combination of soft- and hard-modelling approaches which takes advantages from different aspects of both strategies.
In the present work, the analysis of simulated and experimental data has shown the reliability of the potential shift method in some of the situations described in the theoretical formulation, mostly those concerning non-negligible complex dissociation or the presence of moving anodic signals caused by the ligand. The results of such studies, as well as some preliminary tests made in more complex systems, allow us to summarise some general considerations about the proposed method:
• In the potential shift correction, an appropriate selection of the reference signals is critical: small variation in that can seriously increase the lof. For this purpose, signals of the isolated species (e.g. free metal ion) can be used or, if not available, the peakmaker program or the SIMPLISMA method can be applied.
• All moving signals should be corrected simultaneously: shiftfit is based on reproducing all non-moving signals as a product of C-like and V-like matrices, so that a remaining moving signal would not be properly reproduced and this can generate a large error in the computation of the corrected matrix that cannot be further corrected by a new application of shiftfit.
• The position of the reference signal of the moving peak is totally arbitrary and does not affect the performance of shiftfit. It just states the origin from which all ΔE values are measured.
• From a data matrix containing a set of moving signals, shiftfit can usually provide a matrix of potential shifts and a linear corrected matrix. However, the use of that corrected matrix only will be reliable as far as there is an electrochemical explanation for the existence of such potential shift. Otherwise, shiftfit could introduce artifacts in the data analysis. In some cases, the explanation also makes possible the calculation of significant parameters by hard modelling.
• The use of shiftfit may require the integration of different species in a single component.
• The proposed method can only linearise data if the moving signals maintain their shape along the whole experiment. A change in the shape of a unit voltammogram (typically, the broadening caused by a progressive decrease of electrochemical reversibility) causes a dramatic increase of non-linearity that cannot be corrected by shiftfit.
At present, additional work is being carried out in our research group to apply the proposed method to experimental systems representative of other situations mentioned in Section 2.4. If these studies are successful, shiftfit could become a very useful tool to extend chemometrical applications to many electrochemical systems that by now are practically forbidden or restricted to ANN or PLS with an unrealistically high number of latent variables. Furthermore, it would be interesting to test the applicability of the algorithm to other linearity-based chemometrical methods like the already mentioned PLS for multivariate calibration or other multivariate curve resolution methods like Iterative Target Transformation Factor Analysis (ITTFA), which has many similarities to MCR-ALS and has already been applied to electrochemical systems.45–47 It would also be desirable to test the method with data obtained with other experimental techniques (e.g. spectroscopic ones) but equally affected by signal movement along x-axis.
Acknowledgements
The authors acknowledge support of the Spanish Ministry of Education and Science (Project CTQ2006-14385-C02-01/BQU) and from the Generalitat of Catalonia (project 2005SGR00186).References
- S. D. Brown and R. S. Bear, Jr., Crit. Rev. Anal. Chem., 1993, 24, 99 CAS and refs cited therein.
- E. Richards, C. Bessant and S. Saini, Electroanalysis, 2002, 14, 1533 CrossRef CAS and refs cited therein.
- M. Esteban, C. Ariño and J. M. Díaz-Cruz, TrAC, Trends Anal. Chem., 2006, 25, 86 CrossRef CAS and refs cited therein.
- R. I. Stefan, J. F. van Staden and H. Y. Aboul-Enein, Crit. Rev. Anal. Chem., 1999, 29, 133 CrossRef CAS and refs cited therein.
- V. Pravdová, M. Pravda and G. G. Guilbault, Anal. Lett., 2002, 35, 2389 CrossRef CAS.
- R. Tauler, A. Smilde and B. R. Kowalski, J. Chemom., 1995, 9, 31 CAS.
- J. M. Díaz-Cruz, R. Tauler, B. S. Grabaric, M. Esteban and E. Casassas, J. Electroanal. Chem., 1995, 393, 7 CrossRef.
- M. Esteban, C. Ariño, J. M. Díaz-Cruz, M. S. Díaz-Cruz and R. Tauler, TrAC, Trends Anal. Chem., 2000, 19, 49 CrossRef CAS.
- J. H. Christensen, G. Tomasi and A. B. Hansen, Environ. Sci. Technol., 2005, 39, 255 CAS.
- A. M. van Nederkassel, M. Daszykowski, P. H. C. Eilers and Y. vander Heyden, J. Chromatogr., A, 2006, 1118, 199 CrossRef CAS.
- W. Yao, X. Yin and Y. Hu, J. Chromatogr., A, 2007, 1160, 254 CrossRef CAS.
- S. F. Moller and B. M. Jorgensen, J. Chromatogr. Sci., 2007, 45, 169 CAS.
- V. Pravdová, B. Walczak and D. L. Massart, Anal. Chim. Acta, 2002, 456, 77 CrossRef CAS.
- E. Szymańska, M. J. Markuszewski, X. Capron, A. M. van Nederkassel, Y. vander Heyden, M. Markuszewski, K. Krajka and R. Kaliszan, Electrophoresis, 2007, 28, 2861 CrossRef CAS.
- J. Forshed, I. Schuppe-Koistinen and S. P. Jacobsson, Anal. Chim. Acta, 2003, 487, 189 CrossRef CAS.
- J. Forshed, R. J. O. Torgrip, K. M. Åberg, B. Kalberg, J. Lindberg and S. P. Jacobsson, J. Pharm. Biomed. Anal., 2005, 38, 824 CrossRef CAS.
- J. Jaumot, M. Vives, R. Gargallo and R. Tauler, Anal. Chim. Acta, 2003, 490, 253 CrossRef.
- D. R. Crow, Polarography of Metal Complexes, Academic Press, London, 1969 Search PubMed.
- W. Windig and J. Guilment, Anal. Chem., 1991, 63, 1425 CrossRef CAS.
- W. Windig and S. Markel, J. Mol. Struct., 1993, 292, 161 CrossRef CAS.
- M. J. López, C. Ariño, S. Díaz-Cruz, J. M. Díaz-Cruz, R. Tauler and M. Esteban, Environ. Sci. Technol., 2003, 37, 5609 CrossRef CAS.
- J. M. Díaz-Cruz, J. Agulló, M. S. Díaz-Cruz, C. Ariño, M. Esteban and R. Tauler, Analyst, 2001, 126, 371 RSC.
- Matlab version 7.3.0.267, Mathworks Inc., Natick, MA, USA, 2006 Search PubMed.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627 CrossRef CAS.
- T. F. Coleman and Y. Li, SIAM J. Optimization, 1996, 6, 418 Search PubMed.
- T. F. Coleman and Y. Li, Math. Program., 1994, 67, 189 CrossRef.
- K. Levenberg, Q. Appl. Math., 1944, 2, 164 Search PubMed.
- D. Marquardt, SIAM J. Appl. Math., 1963, 11, 431 Search PubMed.
- D. D. DeFord and D. N. Hume, J. Am. Chem. Soc., 1951, 73, 5321 CrossRef CAS.
- M. Fernández, C. Ariño, J. M. Díaz-Cruz, R. Tauler and M. Esteban, J. Electroanal. Chem., 2001, 505, 44 CrossRef CAS.
- H. G. de Jong, H. P. van Leeuwen and K. Holub, J. Electroanal. Chem., 1987, 234, 1 CrossRef CAS.
- H. G. de Jong and H. P. van Leeuwen, J. Electroanal. Chem., 1987, 234, 17 CrossRef CAS.
- H. G. de Jong and H. P. van Leeuwen, J. Electroanal. Chem., 1987, 235, 1 CrossRef CAS.
- M. S. Díaz-Cruz, J. Mendieta, R. Tauler and M. Esteban, J. Inorg. Biochem., 1997, 66, 29 CrossRef CAS.
- J. Heyrovsky and J. Kuta, Principles of Polarography, Academic Press, Publishing House of the Czechoslovak Academy of Sciences, New York, Prague, 1966, p. 161 Search PubMed.
- J. Koryta, Collect. Czech. Chem. Commun., 1959, 24, 3057 CAS.
- N. E. Schmidt, E. E. Mercer and R. H. Philp, Jr., J. Electroanal. Chem., 1993, 359, 115 CrossRef CAS.
- M. M. Correia dos Santos, M. L. Simões Gonçalves and J. C. Romaõ, J. Electroanal. Chem., 1996, 413, 97 CrossRef.
- M. Torres, J. M. Díaz-Cruz, C. Ariño, B. S. Grabaric, R. Tauler and M. Esteban, Anal. Chim. Acta, 1998, 371, 23 CrossRef CAS.
- M. Torres, J. M. Díaz-Cruz, C. Ariño, B. S. Grabaric and M. Esteban, Electroanalysis, 1999, 2, 93 CrossRef.
- A. I. Vogel, Textbook of Quantitative Chemical Analysis, Longman, London, 5th edn, 1989 Search PubMed.
- A. M. Bond, Modern Polarographic Methods, M. Dekker, New York, 1980, p. 249 Search PubMed.
- IUPAC Stability Constants Database, v. 5.16, Academic Software, 2001.
- A. M. Martell and R. M. Smith, Critical Stability Constants: Amino Acids v. 1, Plenum Press, New York, 1974 Search PubMed.
- Y. Ni, S. Kokot, M. Selby and M. Hodgkinson, Anal. Chim. Acta, 1995, 316, 233 CrossRef CAS.
- Y. Ni and L. Jin, Chemom. Intell. Lab. Syst., 1999, 45, 105 CrossRef CAS.
- U. Evans, O. Soyemi, M. S. Doescher, U. H. F. Bunz, L. Kloppenburg and M. L. Myrick, Analyst, 2001, 126, 508 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: details of the peakmaker, shiftcalc, shiftfit and shiftlof programs. See DOI: 10.1039/b715667g |
| This journal is © The Royal Society of Chemistry 2008 |