Enzyme-catalyzed signal amplification for electrochemical DNA detection with a PNA-modified electrode
Byoung Yeon
Won
a,
Hyun C.
Yoon
b and
Hyun Gyu
Park
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology, 373-1 Guseong-dong, Yuseong-gu, Daejeon 305-701, Republic of Korea. E-mail: hgpark@kaist.ac.kr; Fax: +82 42 869 3910; Tel: +82 42 869 3932
bDepartment of Molecular Science & Technology, Ajou University, San 5 Wonchun-dong, Yeongtong-gu, Suwon 443-749, Republic of Korea
First published on 16th October 2007
Abstract
The signal amplification technique of peptide nucleic acid (PNA)-based electrochemical DNA sensor was developed in a label-free and one-step method utilizing enzymatic catalysis. Electrochemical detection of DNA hybridization on a PNA-modified electrode is based on the change of surface charge caused by the hybridization of negatively charged DNA molecules. The negatively charged mediator, ferrocenedicarboxylic acid, cannot diffuse to the DNA hybridized electrode surface due to the charge repulsion with the hybridized DNA molecule while it can easily approach the neutral PNA-modified electrode surface without the hybridization. By employing glucose oxidase catalysis on this PNA-based electrochemical system, the oxidized mediator could be immediately reduced leading to greatly increased electrochemical signals. Using the enzymatic strategy, we successfully demonstrated its clinical utility by detecting one of the mutation sequences of the breast cancer susceptibility gene BRCA1 at a sample concentration lower than 10–9 M. Furthermore, a single base-mismatched sample could be also discriminated from a perfectly matched sample.
1. Introduction
There are many kinds of techniques for the detection of target biomolecules such as fluorescence,1,2 colorimetric methods,3,4 Raman spectroscopy,5 surface plasmon resonance (SPR),6 quartz crystal microbalance (QCM),7 electrochemical methods8 and so on. Among these, the electrochemical method has many advantageous features including rapid process time, simplicity of the process, and cost effectiveness.9 Particularly, owing to the fact that it does not require any complicated equipment and it generates a numerical signal without computational data analysis, the electrochemical method has been a good candidate for a miniaturized or portable biosensor.There have also been several reports on genetic analysis based on the electrochemical detection strategy,10–13 and fairly recently an idea for the electrochemical DNA detection with the concept of ion-channel sensing using the charge properties of peptide nucleic acid (PNA) was reported.14–17PNA is the mimic of DNA and has a peptide backbone instead of a phosphate backbone, providing it with electrical neutrality as compared with the negatively charged DNA molecule.18 The neutral property of the PNA was efficiently employed to achieve a PNA-based electrochemical sensor for the detection of DNA hybridization.
One main disadvantage of the electrochemical method, however, is that the detection sensitivity is not high enough as desired when compared with commonly used fluorescence-based methods. Therefore, a signal amplification strategy could be demanded to achieve enough sensitivity with the electrochemical method, and several strategies have been developed utilizing enzymatic electrocatalysis,19 precipitation brought about by an enzyme,20 or impedance spectroscopy.21 To amplify the electrochemical signal, enzyme-mediated electrocatalytic reactions can be employed22 and glucose oxidase (GOX) is a good candidate enzyme due to its high reactivity and stability.23 We have also been very successful in the achievement of electrochemical signal amplification using GOX.24 With the amplification method, the oxidized mediator was able to be immediately reduced by the enzymatic catalysis, consequently leading to the greatly enhanced signal magnitude.
With an effort to develop efficient methods for genetic diagnosis,25–27 we herein report a signal-amplified DNA sensing strategy on a PNA-modified electrode surface. Onto the fabricated electrode, we employed negatively charged mediator such that the hybridization of the negatively charged target DNA sample causes the electrochemical signal to be suppressed. Utilizing GOX as a signal generator, we could achieve significant signal amplification and successfully demonstrated its clinical utility by detecting one of the mutation sequences of BRCA1 with highly improved sensitivity.
2. Experimental
2.1. Chemicals and reagents
The PNA (N-CTTCTTAATATT-C) was purchased from Panagene® (Daejeon, Korea) and its N-terminal end was thiolated with mercaptoundecanoic acid. The target DNA oligonucleotides were synthesized by Genotech® (Daejeon, Korea) (perfect target from the 3459 position of BRCA1exon 11: 5′-CAAGAATATTAAGAAGTA-3′, single base-mismatched target: 5′-CAAGAATATGAAGAAGTA-3′, non-complementary target: 5′-ATGATCATTTTTGCCTCT-3′). The synthesized products were purified by HPLC and their identities were confirmed by MALDI-TOF. Glucose oxidase (GOX, from Aspergillus niger) and β-D-glucose were purchased from Sigma. Ferrocenemethanol (Fc-MeOH), ferrocenedicarboxylic acid [Fc(COOH)2] and potassium ferricyanide [K3Fe(CN)6] were purchased from Aldrich. 50 mM PBS (pH 7.2) was prepared with phosphate buffered saline pack (Pierce) and 1× SSPE (saline sodium phosphateEDTA) buffer was prepared by dilution of 20× SSPE (Sigma). All other materials used were of the highest quality available and doubly distilled water was used with a specific resistance over 18 MΩ cm.2.2. Instruments
All electrochemical analysis was performed with a CH instrument 620B electrochemical analyzer (Austin, TX) coupled with a desktop computer for data acquisition. The sensing well was composed of a gold working electrode, a platinum counter electrode and a silver/silver chloride reference electrode. All electrolytes were used after nitrogen-purging.2.3. PNA-modified electrode fabrication
The coating of titanium (20 nm) on a Si wafer (100) was followed by Au (99.999%) thin layer formation (200 nm) by an e-beam evaporator. The Au-coated electrode surface was immersed into piranha solution (H2SO4 : H2O2 = 4 : 1) for 5 min. CAUTION: Piranha solution reacts violently with most organic materials and must be handled with extreme care. The surfaces were washed with PBS thoroughly, and immersed into 1 µM aqueous PNA solution for 2 h. After washing with PBS, the surfaces were moved into 1 mM mercaptohexanol solution for 30 min. Finally, the fabricated electrode surfaces were washed with PBS.2.4. Electrochemical detection of DNA hybridization on the PNA-modified electrode
Prior to the target hybridization , the PNA-modified electrodes were pre-treated with the hybridization buffer (1× SSPE) for 15 min. 100 µM of target DNA were dropped onto the electrode surfaces, and allowed to hybridize with the PNA probe on the electrode surface at 25 °C for 30 min in a humidity chamber. After hybridization on the PNA-modified sensing surface, cyclic voltammetry was carried out with Ferrocenemethanol, potassium ferricyanide, or ferrocenedicarboxylic acid. The concentration of all mediators was 0.1 mM and the scan rate was 100 mV s–1. The potential sweep ranges were 0–0.5 V (vs. Ag/AgCl) for Ferrocenemethanol and 0–0.6 V (vs. Ag/AgCl) for ferrocenedicarboxylic acid and potassium ferricyanide.2.5. Enzymatic signal amplification for the detection of DNA
The target DNA solutions were prepared in a concentration range from 1 nM to 100 µM in 1× SSPE buffer. The procedure for target DNA hybridization was the same as describe above, but 1 mg mL–1glucose oxidase and 10 mM glucose were included in the electrolyte containing 0.1 mM ferrocenedicarboxylic acid. In this study, the potential sweep range was 0.2–0.7 V (vs. Ag/AgCl) and the scan rate was 2 mV s–1.3. Results and discussion
3.1. Overall scheme for electrochemical DNA detection on a PNA electrode with enzymatic signal amplification
Fig. 1 illustrates the overall strategy of the DNA detection. Basically, the current is generated by the diffusion of the mediators to the PNA-modified electrode surface followed by their oxidation and reduction at the electrode surface. Without hybridization , the negatively charged mediator can freely approach the neutral PNA-modified surface leading to the subsequent redox reaction. After hybridization of the complementary DNA sample, however, hybridized targets on the surface prevent the mediator from accessing the surface due to the charge repulsion between the phosphate backbone of the bound DNA and the mediator, both having a negative charge. This hybridization -induced reduction in the electrochemical signal can be used to determine the presence of the target DNA sequences.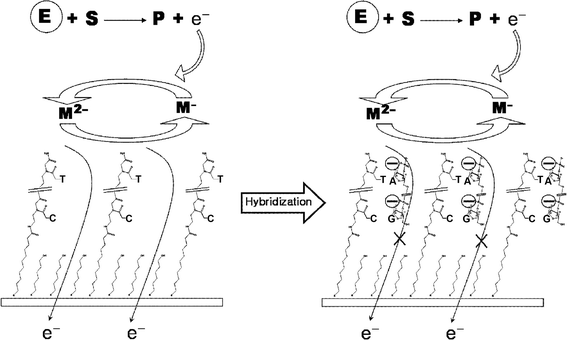 | ||
| Fig. 1 Electrochemical signal amplification strategy of PNA-based DNA sensing surface using enzymatic catalysis. The letters E, S, P and M mean enzyme, substrate, product and mediator, respectively. | ||
To achieve signal amplification with this strategy, GOX was employed as a signal generator. Basically, the anodic potential sweep induces the oxidation of the mediators leading to the generation of the anodic current. The GOX-based amplification process can follow this normal electrochemical signaling, which begins with the binding of glucose to flavin adenine dinucleotide (FAD) in the enzyme. The bound glucose was oxidized to gluconolactone with a concomitant reduction of the FAD to FADH–. In the presence of this FADH–, the oxidized mediators in the electrolyte are temporally moved to the enzyme followed by their immediate reduction. The reduced mediators can be re-oxidized generating an additional anodic current. In this way, the signal could be continuously amplified with the electrochemical DNA sensor.
Employing a gold surface as the working electrode, the sensing surface was fabricated with the method of Aoki et al.14 By varying the concentration of thiolated-PNA and the immobilization time, we investigated appropriate extent of the PNA SAM and the optimal immobilization time was determined to be 2 h with 1 µM PNA solution. Longer immobilization than 2 h caused the electrode surface to be covered too much with the PNA probe, which leads to insulation of the whole electrode surface. Consequently, the electrochemical flow of the mediator seemed to be hampered resulting in the reduced signal. Mercaptohexanol was employed as a surface-blocking reagent to remove unbound PNA and fill the pinholes on the surface to prevent direct adsorption of target DNA to the gold surface.
3.2. Electrochemical detection of DNA hybridization on PNA-modified electrode
The detection principle of this study is based on the change of net charge of the PNA-modified sensing surface. Although there are reports about this sensing strategy,14–17 the evidence of the charge repulsion between the mediator and the electrode surface has not been clearly demonstrated. Therefore, to elucidate the charge interaction-based detecting strategy, Fc-MeOH, Fc(COOH)2 and K3Fe(CN)6 were tested as mediators for signaling. The sequence of the PNA capture probe was selected to detect one of the key Korean-specific mutations in the BRCA1 gene, which is at position 3459 of exon 11. The mutant sample has thymine at this mutation site instead of guanine in a wild-type sample. The length of the capture PNA was determined to be a 12-mer because it showed enough binding affinity with the complementary target DNA due to the PNA's higher affinity for its complementary DNA , which was clearly demonstrated by our previous work.25 On the PNA-modified electrode, perfectly matched target DNA was reacted according to the procedures described in the Experimental section and cyclic voltammetry (CV) was performed on the sensing surface by using the three different mediators in the electrolyte. As shown in Fig. 2, there were significant signal changes caused by the hybridization in the cases of Fc-(COOH)2 and K3Fe(CN)6 while no measurable signal change was detected with the Fc-MeOH mediator. In aqueous solution, Fc-(COOH)2 and K3Fe(CN)6 are ionized to Fc(COO–)2 and Fe(CN)63– respectively, and these act as negatively charged mediators. With the negatively charged mediators, the hybridization of the negatively charged DNA sample suppressed access of the mediators to the electrode surface, leading to the decreased CV signals. No significant signal change with Fc-MeOH indicates that the diffusion of the mediator was not affected by hybridization of the negatively charged target DNA . Moreover, the peak signal magnitude decreased in proportion to the concentration of the hybridized target DNA on the electrode surface in the two cases of the negatively charged mediators. In our preliminary study, we performed the same experiment under the same conditions (hybridization buffer, electrolyte, etc.) but without the target probe and there was no significant signal change observed, indicating that the signal change depended exclusively on the presence of the hybridized target DNA . Based on these results, it is clear that the electrochemical signal was decreased by hybridization of the negatively charged target DNA molecule only when negatively charged mediators like Fc-(COOH)2 and K3Fe(CN)6 were used. Furthermore, since the extent of the signal decrease depends on the concentration of the target sample, the strategy could be efficiently utilized for the detection of DNA hybridization . | ||
| Fig. 2 Calibration curves of peak current and target DNA concentration using various mediators such as ferricyanide (●), ferrocenedicarboxylic acid (■) and Ferrocenemethanol (○). | ||
3.3. Signal-amplified electrochemical detection of DNA hybridization based on PNA-modified electrode
Fig. 3A and 3B show the CV signal before and after enzyme (GOX) addition using ferrocenedicarboxylic acid as the mediator. In our preliminary study, 1 mg mL–1 enzyme and 10 mM glucose were determined to be sufficient concentrations to generate the reliable electrocatalytic signal under the fixed 0.1 mM mediator concentration. Therefore, those concentrations were employed throughout this study. Upon enzyme addition, the amplified peak difference between 1 nM and 100 µM target DNA became about three times larger than that before addition of the enzyme, enabling more sensitive detection for the complementary DNA molecules. When the same experiment was conducted with another negatively charged mediator, ferricyanide, the signal amplification through bioelectrocatalysis was not observed at a reasonable scan rate. This different behavior for ferricyanide may be ascribed to its much higher oxidizing capability. More specifically, the oxidizing tendency of ferricyanide is very fast compared with the reducing power of the electrons generated by the GOX-mediated catalysis, such that the accelerating effect by the electrons does not contribute much to the final electrochemical signal. To make it work, a very slow scan rate must be adopted, which is not desirable to achieve rapid sensing. For this reason, ferrocenedicarboxylic acid was used as the mediator in further experiments for electrocatalytic signal amplification.![Cyclic voltammetric patterns before (A) and after (B) enzyme addition using ferrocenedicarboxylic acid as the mediator. Two different concentrations [1 nM (—) and 100 µM (⋯)] of the target DNA samples were used. (C) Calibration curves of the CV peak current before (○) and after (●) signal amplification.](/image/article/2008/AN/b712638g/b712638g-f3.gif) | ||
| Fig. 3 Cyclic voltammetric patterns before (A) and after (B) enzyme addition using ferrocenedicarboxylic acid as the mediator. Two different concentrations [1 nM (—) and 100 µM (⋯)] of the target DNA samples were used. (C) Calibration curves of the CV peak current before (○) and after (●) signal amplification. | ||
Next, we investigated the GOX-mediated signal amplification by varying the concentrations of the target DNA sample. The peak currents from the amplified CVs also showed the tendency to decrease in proportion to the target DNA concentration. From registered data in CVs, the calibration curves before and after the signal amplification were made (Fig. 3C). Before signal amplification, the current change was from about 2.1 to 2.4 µA for target DNA concentrations between 1 nM and 100 µM. This signal pattern was not sufficient to distinguish the target concentration level. For example, in the target DNA concentration range from 0.1 to 100 µM, there were no big differences between the currents corresponding to the different concentrations and they were just little more than the common error range. On the other hand, when the enzyme-mediated signal amplification method was applied, the peak current range was widened remarkably (1.9–2.65 µM), and the slope was evidently increased as well manifested in comparison to the calibration curve before amplification. Therefore, the signal amplification strategy enabled us to distinguish the target concentration level more clearly with greatly increased sensitivity. Although high signals could be also obtained with a high concentration of the mediator, the resulting signals simply became higher rather than more distinguishable (data not shown).
However, it should be noted that this GOX-catalyzed reaction is always expected to increase the electrochemical signal by providing the additional reducing power to the oxidized mediator. As shown in Fig. 3C, under low concentrations of the target DNA below 0.1 µM, the amplified signals were higher than the unamplified signals. However, with the target concentrations higher than 0.1 µM, the amplified signals were unexpectedly lower than the original signals and this interesting result was reproducibly observed. This can be partly explained in that the negatively charged enzyme in the electrolyte condition becomes less accessible to the PNA surface more highly hybridized with the DNA molecules, thus greatly diminishing the amplification effect. However, to fully understand the signal decreasing effect caused by the electrocatalysis, further intensive studies are needed.
Finally, using the amplified strategy, we examined its clinical utility by detecting the Korean-specific BRCA1 mutation sequence. As shown in Fig. 4, the application of the perfectly matched DNA sample resulted in a signal decrease due to its hybridization to the capture PNA on the electrode, while there was no significant signal decrease detected upon application of the non-complementary DNA sample. Furthermore, the amounts of the decreased signals were closely dependent on the applied concentrations. These results indicate that the complementary target sample can be well distinguished from the non-complementary sample through its hybridization , and the resulting signal decrease can be used for the quantitative detection of target DNA . Based on the results in Fig. 4, the target DNA sample is considered to be detected up to the 10–9 M concentration level. Next, we checked if it is possible to discriminate the perfectly matched sample from only a single base-mismatched sample using our strategy. Although this system was not intended to distinguish the single base-mismatched target, we were also able to detect the single base-mismatch of the target samples in concentrations higher than 0.01 µM, which is again a remarkable benefit provided by the signal amplification strategy.
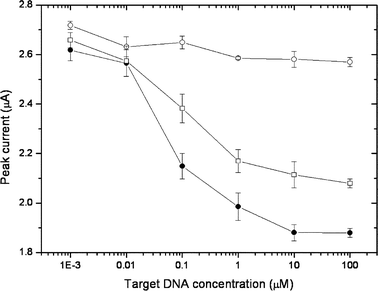 | ||
| Fig. 4 Electrochemical detection of DNA hybridization using enzyme-catalyzed signal amplification: non-complementary target (○), perfectly matched target (●), single base-mismatched target (□). | ||
4. Conclusion
We have successfully incorporated the enzyme-catalyzed signal amplification method to the novel PNA-based DNA detection strategy and demonstrated that the strategy could be efficiently used to diagnose a BRCA1 mutation site with greatly enhanced sensitivity. The signal amplification was easily achieved by simply adding the additional enzyme and its substrate into the electrolyte solution. This PNA-based detection method does not require any sample pre-treatment step like labeling or additional steps such as reaction with secondary probes, which are typical steps required by conventional methods. Furthermore, the entire detecting process takes less than 40 min and an automated detection kit for various genetic diagnoses could be rapidly achieved based on this work.Acknowledgements
This work was supported by the Brain Korea 21 (BK21) program and the Centre for Ultramicrochemical Process Systems.References
- W. C. Lee, B. S. Chun, B. K. Oh, W. H. Lee and J. W. Choi, Biotechnol. Bioprocess Eng., 2004, 9, 241 Search PubMed.
- H. G. Park, J. Y. Song, K. H. Park and M. H. Kim, Chem. Eng. Sci., 2006, 61, 954 CrossRef CAS.
- Y. K. Jung, H. G. Park and J. M. Kim, Biosens. Bioelectron., 2006, 21, 1536 CrossRef CAS.
- N. Y. Lee, Y. K. Jung and H. G. Park, Biochem. Eng. J., 2006, 29, 103 CrossRef CAS.
- J. W. C. Cao, R. Jin and C. A. Mirkin, Science, 2002, 297, 1536 CrossRef CAS.
- X. Su, Y. J. Wu, R. Robelek and W. Knoll, Langmuir, 2005, 21, 348 CrossRef CAS.
- A. E. Gerdon, D. W. Wright and D. E. Cliffel, Anal. Chem., 2005, 77, 304 CrossRef CAS.
- R. M. Umek, S. W. Lin, J. Vielmetter, R. H. Terbrueggen, B. Irvine, C. J. Yu, J. F. Kayyem, H. Yowanto, G. F. Blackburn, D. H. Farkas and Y. P. Chen, J. Mol. Diagn., 2001, 3, 74 Search PubMed.
- E. Bakker, Anal. Chem., 2004, 76, 3285 CrossRef CAS.
- J. J. Gooding, Electroanalysis, 2002, 14, 1149 CrossRef CAS.
- J. Wang, Anal. Chim. Acta, 2002, 469, 63 CrossRef CAS.
- J. Wang, Anal. Chim. Acta, 2003, 500, 247 CrossRef CAS.
- J. Wang, Analyst, 2005, 130, 421 RSC.
- H. Aoki, P. Bühlmann and Y. Umezawa, Electroanalysis, 2000, 12, 1272 CrossRef CAS.
- H. Aoki and Y. Umezawa, Electroanalysis, 2002, 14, 1405 CrossRef CAS.
- H. Aoki and Y. Umezawa, Analyst, 2003, 128, 681 RSC.
- Y. Umezawa and H. Aoki, Anal. Chem., 2004, 76, 320A CAS.
- H. P. Vernille, L. C. Kovell and J. W. Schneider, Bioconjugate Chem., 2004, 15, 1314 CrossRef.
- F. Patolsky, Y. Weizmann and I. Willner, J. Am. Chem. Soc., 2002, 124, 770 CrossRef CAS.
- F. Patolsky, E. Katz and I. Willner, Angew. Chem., Int. Ed., 2002, 41, 3398 CrossRef CAS.
- A. Bardea, F. Patolsky, A. Dagan and I. Willner, Chem. Commun., 1999, 21 RSC.
- H. C. Yoon and H.S. Kim, Anal. Chem., 2000, 72, 922 CrossRef CAS.
- A. E. G. Cass, G. Davis, G. D. Francis, H. A. O. Hill, W. J. Aston, I. J. Higgins, E. V. Plotkin, L. D. L. Scott and A. P. F. Turner, Anal. Chem., 1984, 56, 667 CrossRef CAS.
- B. Y. Won, H. G. Choi, K. H. Kim, S. Y. Byun, H. S. Kim and H. C. Yoon, Biotechnol. Bioeng., 2005, 89, 815 CrossRef CAS.
- J. Y. Song, H. G. Park, S. O. Jung and J. C. Park, Nucleic Acids Res., 2005, 33, e19 CrossRef.
- H. G. Park, H. O. Ham, K. H. Kim and N. Huh, Biosens. Bioelectron., 2005, 21, 637 CrossRef CAS.
- S. C. Yim, H. G. Park, H. N. Chang and D. Y. Cho, Anal. Biochem., 2005, 337, 332 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |