The development of an immobilized enzyme reactor containing glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma cruzi: the effect of species' specific differences on the immobilization
Carmen Lúcia Cardosoa, Marcela Cristina de Moraesb, Rafael Victorio Carvalho Guidoc, Glaucius Olivac, Adriano Defini Andricopuloc, Irving William Wainerd and Quezia Bezerra Cass*b
aDepartamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, 14040-901, São Paulo, Brazil
bDepartamento de Química, Universidade Federal de São Carlos, Cx. Postal 676, São Carlos, 13565-905, São Paulo, Brazil. E-mail: quezia@dq.ufscar.br; Fax: +55-16-3351-8350; Tel: +55-16-3351-8087
cCentro de Biotecnologia Molecular Estrutural – CBME, Instituto de Física de São Carlos, Universidade de São Paulo, São Carlos, São Paulo, Brazil
dNational Institute of Aging, National Institutes of Health, Baltimore, MD, USA
First published on 8th October 2007
Abstract
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) plays an important role in the life cycle of the Trypanosoma cruzi, and an immobilized enzyme reactor (IMER) has been developed for use in the on-line screening for GAPDH inhibitors. An IMER containing human GAPDH has been previously reported; however, these conditions produced a T. cruzi GAPDH-IMER with poor activity and stability. The factors affecting the stability of the human and T. cruzi GAPDHs in the immobilization process and the influence of pH and buffer type on the stability and activity of the IMERs have been investigated. The resulting T. cruzi GAPDH-IMER was coupled to an analytical octyl column, which was used to achieve chromatographic separation of NAD+ from NADH. The production of NADH stimulated by D-glyceraldehyde-3-phosphate was used to investigate the activity and kinetic parameters of the immobilized T. cruzi GAPDH. The Michaelis–Menten constant (Km) values determined for D-glyceraldehyde-3-phosphate and NAD+ were Km = 0.5 ± 0.05 mM and 0.648 ± 0.08 mM, respectively, which were consistent with the values obtained using the non-immobilized enzyme.
Introduction
Chagas' disease, also known as American trypanosomiasis, is an infection caused by the protozoan parasite Trypanosoma cruzi. Chagas' disease is endemic in 15 countries of Latin America, and, according to the World Health Organization the disease affects 16–18 million people, about 40 million people are at risk and 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases are registered each year.1,2 The drugs currently available for the treatment of this disease (e.g. benznidazole and nifurtimox) are inadequate and their use is limited by serious side effects, toxicities, and ineffectiveness. Therefore, the discovery of new drugs with different mechanisms of action for the treatment of Chagas' disease is a critical need and a challenging task.3
000 new cases are registered each year.1,2 The drugs currently available for the treatment of this disease (e.g. benznidazole and nifurtimox) are inadequate and their use is limited by serious side effects, toxicities, and ineffectiveness. Therefore, the discovery of new drugs with different mechanisms of action for the treatment of Chagas' disease is a critical need and a challenging task.3One promising approach to accomplish this task is the selective inhibition of enzymes that participate in the glycolytic pathway of the parasite. The trypanosomatids are highly dependent on glycolysis for ATP production,4,5 and the reaction catalyzed by glycosomal glyceraldehyde-3-phosphate dehydrogenase (GAPDH) plays a central role in controlling ATP production in pathogenic parasites.6,7 GAPDH (EC 1.2.1.12) catalyzes the oxidative phosphorylation of D-glyceraldehyde-3-phosphate into 1,3-diphosphateglycerate in the presence of NAD+ and inorganic phosphate.
Crystallographic studies showed that GAPDH from T. cruzi and human GAPDH differ by a substitution of Asp210 (T. cruzi) by Leu194 (human).8 Based on this difference, it is possible that a selective inhibitor of T. cruzi GAPDH could be developed to treat Chagas' disease.9 The discovery and development of a selective T. cruzi GAPDH inhibitor is a challenging task, which requires the development of methods to rapidly identify lead compounds in complex chemical and biological mixtures, and to assess the specificity for GAPDH of the target (T. cruzi) relative to the host (human). One such approach is on-line screening using an immobilized enzyme reactor (IMER).
IMERs have been prepared from a wide variety of enzymes and have been used in high performance liquid chromatographic systems for carrying out on-line synthesis, in the study of enzyme kinetics for the determination Michaelis–Menten constant and in the identification of enzyme inhibitors. The development and use of IMERs have recently been reviewed.10 As part of our program to develop new treatments for Chagas' disease, this laboratory initially developed an IMER containing human GAPDH immobilized within a fused silica capillary.11 However, when the same immobilization procedure was followed using T. cruzi GAPDH in place of human GAPDH, the resulting IMER had poor enzymatic activity and stability. This work reports the results from a systematic study of factors, such as pH and buffer type, that affect T. cruzi GAPDH stability and activity. The optimized conditions were used to prepare a T. cruzi GAPDH-IMER, which was placed in a multidimensional high performance chromatographic system, and the resulting system was used to characterize the activity of the immobilized enzyme. The data from this study demonstrate that subtle changes in protein structure can require significant alterations in the procedures required to immobilize the protein in a chromatographic environment.
Experimental
Reagents and materials
D,L-Glyceraldehyde-3-phosphate free acid (GA3P), β-nicotinamide adenine dinucleotide reduced form (NADH), and β-nicotinamide adenine dinucleotide (NAD+) were purchased from Sigma Aldrich (St. Louis, MO, USA). Buffer components and all chemical materials used during the immobilization procedure were of analytical grade and were supplied by Sigma or by Merck (Darmstadt, Germany). All solvents were HPLC grade and were purchased from J.T. Baker (Phillipsburg, USA). Water was purified with a Millipore Milli-Q system (Millipore, São Paulo, Brazil) and was used for all experiments. The mobile phases were prepared daily. The fused silica capillary for electrophoresis (0.375 mm × 0.10 mm) used to immobilize the enzyme and to prepare the IMERs was purchased from Polymicro Technologies (Phoenix, AZ, USA). The Luna® octyl silica (10.0 µm, 100 Å) was supplied by Phenomenex (Torrance, CA, USA). Before their use for HPLC analysis, the buffer solutions were filtered through cellulose nitrate membrane filters (0.45 µm) purchased from Phenomenex. Dialysis and concentration of enzymatic solution was carried out using an Amicon concentrator (30 mL, Millipore, Billerica, MA) and a centrifuge (Eppendorff Instruments, Enfield, USA).Buffers
The purification and storage buffer was: triethanolamine (100.0 mM, pH 7.5), containing 1.0 mM β-mercaptoethanol, 1.0 mM ethylenediaminetetraacetic acid (EDTA), 30.0 mM sodium arsenate heptahydrate (NaHAsO·7H2O), 1.0 mM phenylmethylsulfonyl fluoride (PMSF), 50.0 mM NAD+, 1.0 mM pepstatin, 1.0 mM leupeptin, 0.5 M KCl, and glycerol 10%.Buffer 1: triethanolamine (100.0 mM, pH 7.5), containing 1.0 mM β-mercaptoethanol, 1.0 mM EDTA, 30.0 mM NaHAsO·7H2O, 1.0 mM PMSF, and 0.5 M KCl.
Buffer 2: phosphate buffer (50.0 mM, pH 7.0).
Buffer 3: 20.0 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 8.2.
Buffer 4: 20.0 mM ammonium acetate, pH 8.0.
Buffer 5: 20.0 mM sodium borate, pH 8.6.
Buffer 6: triethylamine (TEA) (1% in water v/v acidified with AcOH, pH = 6.0).
Buffer 7: triethanolamine (100.0 mM, pH 7.5), containing 1.0 mM EDTA, 1.0 mM PMSF, 1.0 mM β-mercaptoethanol.
Buffer 8: Tris-HCl (50.0 mM, pH 8.6), containing 1.0 mM β-mercaptoethanol, 30.0 mM NaHAsO·7H2O, and 1.0 mM EDTA.
T. cruzi GAPDH
T. cruzi GAPDH was over-expressed and purified as previously reported.9Chromatographic systems
The immobilization of the enzyme was carried out using a syringe-pump 341B (Sage Instruments, Boston, USA).Two modular HPLC systems were setup in order to carry out the on-line studies and the systems were connected as depicted in Fig. 1. The chromatographic experiments were carried out using a Shimadzu HPLC system (Shimadzu, Kyoto, Japan), which consisted of the two LC 10 AD VP pumps with one of the pumps having a valve FCV-10AL for selecting solvent, a UV-Vis detector (SPD-M10AV VP), an autosampler equipment with a 500 µL loop (SIL 10 AD VP). The column containing the immobilized GAPDH enzyme (GAPDH-IMER) was coupled on-line to an octyl column (Luna-Phenomenex®, 100 Å, 10 µm, 10 cm × 0.46 mm I.D.). A six-way switching sample-valve, Valco Nitronic 7000 EA (Supelco, St. Louis, MO, USA), was used to connect the two columns. Data acquisition was done on a Shimadzu SCL 10 AVP system interface with a computer equipped with Shimadzu-LCsolution (LCsolution 2.1) software (Shimadzu, Kyoto, Japan).
 | ||
| Fig. 1 Schematic diagram of multidimensional chromatographic system. | ||
Free GAPDH storage conditions
Conditions of storage were evaluated for the free soluble GAPDH. After purification, the enzyme was stored at low temperature (–80 °C) in the purification buffer. The enzyme activity and stability were evaluated by weekly measurements of the activity of the stored enzyme. The procedure for measuring the activity is described in the section ‘Free GAPDH activity assays’.Optimization of the immobilization conditions for GAPDH on fused silica capillary
Previously reported immobilization procedure was evaluated and then modified11 as follows: three different immobilization buffers were tested: buffer 3, buffer 4, and buffer 5.The fused capillary cleaning procedure was changed from NaOH 2.0 mol L–1 to HCl 2.0 mol L–1 and the effect on the silanization procedure was investigated by passing 3-aminopropyltriethoxysilane (APTS) solution, once or twice, thorough the capillary.
The modified immobilization procedures for GAPDH
Using a syringe-pump with a flow rate of 130 µL min–1, the fused silica capillary tube (100 µm I.D. × 0.375 mm × 50 cm) was cleaned by washing with 2.0 mL of a 2.0 mol L–1 HCl solution, followed by 1.0 mL of distilled water. After rinsing, the capillary was dried in an oven at 95 °C for at least 1 h, and then 1.0 mL of a solution of 3-aminopropyltriethoxysilane [10% (v/v)] in water was passed through the capillary and subsequently placed in an oven at 95 °C for 30 min. The capillary was stored overnight at room temperature.The enzymatic solution was exhaustively dialyzed from the storage buffer to buffer 3 and concentrated, using an Amicon concentrator, to a final concentration of the 1.0 mg mL–1 used in the immobilization step.
A glutaraldehyde solution 1% (v/v), in buffer 2 (2.0 mL), was passed through the aminopropylsilica (APS) capillary by syringe-pump at 130 µL min–1 flow rate. In order to remove free glutaraldehyde and thus avoid polymerization, the capillary tubing was rinsed with buffer 2 (0.5 mL at 130 µL min–1). After this, the capillary tube was rinsed with buffer 3 (0.5 mL), immediately followed by 0.5 mL of GAPDH enzyme solution (1.0 mg mL–1) in buffer 3. The enzyme solution was passed through the capillary a second time, and then the capillary tube was rinsed with 1.0 mL of buffer 1. When not in use the GAPDH-IMER was kept at 4 °C with the two ends of the capillary tubing immersed in buffer 1.
GAPDH-IMER storage conditions and mobile phase
Two different buffers – buffer 1 and buffer 8 – were evaluated in order to select the best working buffer and optimal conditions of storage for the GAPDH-IMERs. The pH and temperature effects on the enzyme stability were evaluated with the free enzyme using both buffers. This was done daily in order to estimate the decrease in activity at each day. The experimental details are specified in the section ‘Free GAPDH activity assays’.Chromatographic conditions
The analytical columns were packed by the ascending slurry method, using methanol for the preparation of the slurry (50.0 mL) and also for the packing. The packing was carried out at a pressure of 7500 p.s.i. (1 p.s.i. = 6894.76 Pa).The analytical columns listed below were evaluated in different chromatographic conditions and temperatures: Column A: diol-silica Spherex®-OH (100 Å, 10.0 cm × 0.46 mm I.D., 10 µm); Column B: octyl silica Luna® (100 Å, 10 µm, 10.0 cm × 0.46 mm I.D.). Mobile phases evaluated: (a) KH2PO4 10 mM, pH 6.0, flow rate 0.8 mL min–1; (b) TEA (1% in water v/v, pH 6.0)–MeOH (98 : 2; 91 : 9; 90 : 10, 97 : 3 v/v) flow rate 0.8, 0.6 mL min–1; (c) ammonium acetate 10.0 mM, pH 6.0, flow rate 0.8 mL min–1; (d) HEPES 10.0 mM, pH 6.0, flow rate 0.8 mL min–1; (e) TEA 10.0 mM, pH 6.0, flow rate 0.8 mL min–1; (f) TEA (1% in water v/v, pH 6.0)–MeCN (98 : 2; 97 : 3; 96 : 4, 96.5 : 3.5, 91 : 9, 90 : 10, v/v) flow rate 0.8, 0.6 mL min–1. Temperatures evaluated: (a) 22 °C, (b) 25 °C, (c) 28 °C, and (d) 35 °C.
The flow rate used in the GAPDH-IMER and time-width, to transfer the enzyme reaction products from the GAPDH-IMER to the analytical columns, was evaluated by injecting duplicate 15 µL aliquots of a solution containing NAD+ (20 mM) and NADH (2.0 mM).
The chromatographic separations between NAD+ and NADH were achieved by a multidimensional chromatography system, in which the GAPDH-IMER was used in the first dimension; at room temperature, coupled to the analytical octyl silica column (100 Å, 10 µm, 10 cm × 4.6 cm I.D.) using a switching six way valve (Fig. 1). The chromatographic conditions are specified on Table 1.
| Pump (eluent)a | Time/min | Event | Valve position |
|---|---|---|---|
| a Pump 1: flow rate: 0.05 mL min–1, eluent A, buffer 1. Pump 2: flow rate: 0.8 mL min–1, eluent B, buffer 6: MeCN (96.5 : 3.5, v/v). λ = 340 nm. | |||
| 1 (A) | 0.00–2.00 | Elution of reagents though the GAPDH-IMER | 1 |
| 2 (B) | 0.00–2.00 | Conditioning of the analytical column | 1 |
| 1 (A) | 2.01–8.50 | Transfer of the analytes from the IMER to analytical column | 2 |
| 2 (B) | 2.01–8.50 | Conditioning analytical column | 2 |
| 1 (A) | 8.51–20.00 | GAPDH-IMER conditioning | 1 |
| 2 (B) | 8.51–20.00 | Separation of the analytes at the analytical column | 1 |
Method validation
The NADH calibration curve was obtained using the appropriate standard solutions of NADH. Sample solutions were prepared in triplicate at the following concentrations: 5, 10, 20, 40, 80, 160, 280, and 320 µmol L–1. To prepare these solutions, aliquots (60 µL) of the appropriate standard solution of NADH were added to 40 µL of buffer 1. The solutions were vortex-mixed for 10 s and aliquots of 90 µL were transferred to an autoinjector vial. Samples of 15 µL were injected to the GAPDH-IMER at the HPLC system. NADH calibration curves were constructed by plotting the peak area against the concentration of NADH.The intra- and inter-day precision and accuracy of the method were evaluated by analyzing quality control samples at three different concentrations: 12.0, 240.0 and 300.0 µmol L–1. Five samples of each concentration were prepared and analyzed on three non-consecutive days. The acceptance criteria for the limit of quantification were that the precision of three samples was under 20% of variability, while the limit of detection was calculated taking a signal-to-noise ratio of 3. Chromatograms of blank buffer were analyzed to evaluate the selectivity of the method.
Free GAPDH activity assays
The enzymatic activity of soluble GAPDH was evaluated by measuring the formation of NADH by the following modified spectrophotometric method.9 In a quartz cuvette (500 µL) the reaction mixture was composed as follows (final concentration): to 5 µL of enzyme solution (20.0 nmol L–1) was added 385 µL of buffer 7, 15 µL of NaHAsO·7H2O (30.0 mM), 30 µL of D,L-GA3P (800 µM, final concentration of D-GA3P), and 5 µL NAD+ (800 µM). The reaction was initiated with the addition of D,L-GA3P solution. The extent of the enzymatic conversion was monitored by following the increase in NADH at λ = 340 nm. Enzymatic activity was calculated from the initial slope of the curve obtained during 30 s of reaction. A sample prepared with buffer 7, NaHAsO·7H2O, and NAD+ was used as the reference sample.Spectrometric determinations were performed using a Shimadzu (Shimadzu, Kyoto, Japan) UV-1650 PC spectrophotometer, with a computer equipped with a UV Probe (Kinetics) software version 1.10 for data collection.
Kinetic parameters were determined using the Sigma Plot software version 7.0.
Kinetics studies of the immobilized enzyme
The enzymatic activity of GAPDH in the IMER format was evaluated by using the multidimensional chromatographic system. Kinetic studies were performed in order to determine saturating conditions for NAD+ and D-GA3P. Solutions with NAD+ concentrations ranging from 0.10 to 12.5 mM and D-GA3P concentrations between 0.10 and 12.5 mM were injected in duplicate. The parameters for NAD+ were determined under saturating conditions of D-GA3P (7.5 mM) while the parameters for D-GA3P were determined at saturating concentrations of NAD+ (10.0 mM). Samples were injected in duplicate (15 µL) and the chromatographic conditions are the ones specified in Table 1.Non-linear regression analysis using the Sigma Plot software version 7.0 was used to determine the Michaelis–Menten values (Km) for the studied systems. GAPDH-IMER stability was determined every day by injecting 15 µL of saturating concentrations of NAD+ (10.0 mM) and D-GA3P (7.5 mM).
Results and discussion
Free T. cruzi GAPDH storage conditions
Once the T. cruzi GAPDH was expressed and purified a crucial point was the long-term storage of this soluble enzyme with preservation of activity. The stability of GAPDH in the storage buffer at –80 °C was evaluated weekly for an eight month period. The results demonstrated that 99% of the enzymatic activity was retained under the storage conditions, which made it possible to use the same batch of purified enzymes in the preparation of the T. cruzi GAPDH-IMERs used in this study.Mobile phase and conditions of storage for GAPDH-IMERs
The stability and enzymatic activity are influenced by a number of factors including temperature, pH, and buffer composition. These variables were investigated using free T. cruzi GAPDH in order to determine the optimum immobilization and working buffers, temperature and storage conditions for the T. cruzi GAPDH-IMER. Two buffers were evaluated as working buffers: buffer 1 at pH 7.5 and buffer 8 at pH 8.6, at two different temperatures.Aliquots of free enzyme, kept under the storage conditions, were exhaustively dialyzed against buffer 1 and 8 respectively, and concentrated to 1.0 mg mL–1 before use. Following this preparation, one set of enzyme solutions was maintained at room temperature and a second at 4 °C. The enzymatic activity was evaluated daily at saturating concentrations of the substrate and cofactor.
With buffer 1, 99% of the enzymatic activity was retained after 24 h and 57% after 120 h, Fig. 2. Temperature had no significant effect. When buffer 8 was used, 55% of the enzymatic activity was retained after dialysis, and after 48 h, the calculated activity had fallen to 25% (after storage at 4 °C) and 7% (after storage at room temperature), Fig. 2. Thus, both buffer composition and temperature had a significant effect on the enzymatic activity, and buffer 1 was utilized for the studies of the activity of free and immobilized T. cruzi GAPDH.
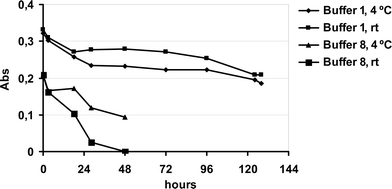 | ||
| Fig. 2 Effect of buffers 1 and 8 on the free T. cruzi GAPDH enzymatic activity kept at two different temperatures and measured as the absorbance of NADH produced. | ||
Multidimensional on-line GAPDH-IMER chromatographic system
In standard GAPDH assays, enzymatic activity is monitored by measuring the formation of NADH using UV-Vis detection. However, in the IMER format, a chromatographic separation of NAD+ from NADH is necessary. As previously observed,11 the GAPDH-IMER does not have sufficient chromatographic selectivity to achieve this separation and multidimensional chromatography is required. The column-switching system used in this study is illustrated in Fig. 1.In our previous work, the best resolution of NAD+ and NADH was achieved using a diol column.11 However, the use of buffer 1 with the T. cruzi GAPDH-IMER altered the chromatographic selectivity of the diol column and efficient resolution of NAD+ and NADH was not achieved. Therefore, a variety of analytical columns and chromatographic conditions was evaluated. The best chromatographic selectivity for NAD+ and NADH was obtained using an octyl column using buffer 6 as the mobile phase and a flow rate of 0.8 mL min–1.
In the optimized system, the retention factors (k) of NAD+ and NADH were 0.81 and 1.55 respectively, with a separation factor (α) of 5.2 and a resolution (Rs) of 12.1 (Fig. 3). The identity of the NADH peak was confirmed by injecting separated NADH and NAD+ standard solutions, at the same chromatographic conditions, and comparing retention times.
 | ||
Fig. 3 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) Representative chromatogram of the separation of NAD+ and NADH by an octyl analytical column coupled to the GAPDH-IMER. (—) Representative chromatogram of the on-line reduction of NAD+. D-GA3P (7.5 mM), NAD+ (10 mM). Separation obtained by an octyl analytical column coupled to the GAPDH-IMER, λmax = 340 nm. Experimental conditions as described in the text. ) Representative chromatogram of the separation of NAD+ and NADH by an octyl analytical column coupled to the GAPDH-IMER. (—) Representative chromatogram of the on-line reduction of NAD+. D-GA3P (7.5 mM), NAD+ (10 mM). Separation obtained by an octyl analytical column coupled to the GAPDH-IMER, λmax = 340 nm. Experimental conditions as described in the text. | ||
Method validation
NADH production by the T. cruzi GAPDH-IMER was evaluated using peak areas. A standard curve was constructed using NADH solutions ranging from 5 to 320 µM and a linear correlation was observed between injected concentration and peak area (y = 0.000287122x + 2.54127; r = 0.99987). Good precision and accuracy were obtained for triplicate analyses as the coefficient of variation (CV) ranged from 0.600 to 2.07% and the accuracy from 97 to 115%. The intra- and inter-day precision and accuracy of the method were determined by analyzing five replicates of three quality controls on three non-consecutive days. Precision was expressed as the CV and the accuracy was evaluated by back-calculation and expressed as the percentage deviation between the amount found and the amount prepared in the three concentrations examined. These results are shown in Table 2. The limit of quantification was of 5.0 µmol L–1 (CV = 2.07%, and accuracy 115%, n = 3) while the limit of detection was 2.0 µmol L–1.Immobilization conditions for T. cruzi GAPDH
Based on previous evaluation for the immobilization of human GAPDH enzyme,11 potassium phosphate buffer (50 mM, pH 8.6) was used as the initial immobilization buffer, but with poor results. These results reflected the stability of T. cruzi GAPDH in this buffer as only 50% of the initial enzymatic activity was retained after 24 h.Enzymatic stability is a key issue since, unlike human GAPDH enzyme, which is used as a lyophilized powder,11 T. cruzi GAPDH is stored in triethanolamine. Since glutaraldehyde is used in the immobilization step, the enzyme must be dialyzed into a buffer that is free of reactive amino moieties. Therefore, the enzyme needs to be stable in the buffer during the dialysis and immobilization procedures.
In the selection of the immobilization buffer, two important characteristics were considered: (1) the buffer should not affect enzymatic activity during dialysis and; (2) the buffer should not react with glutaraldehyde during the immobilization process. Using these criteria, buffers 3, 4 and 5 were selected for study.
T. cruzi GAPDH in the storage buffer was dialyzed against each of the buffers followed by concentration to 1.0 mg mL–1. The resulting solutions were evaluated for residual enzymatic activity immediately after dialysis and after storage for 4 and 7 h at room temperature and 4 °C, Fig. 4.
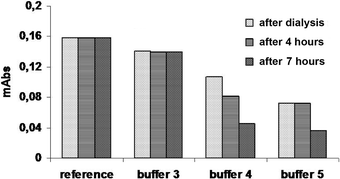
The results demonstrate that buffer 3 produced the best results and it was used in the immobilization studies. The stability of T. cruzi in buffer 3 was consistent with the use of this buffer in previous studies of this enzyme.12
Previous studies on the development of an IMER containing human GAPDH11 demonstrated that optimum activity and stability was obtained using a fused silica capillary. This approach was also used in this study. The initial step in the immobilization involves the cleaning and activation of the capillary's surface, which can be accomplished using NaOH or HCl.13,14
For the immobilization of GAPDH from T. cruzi, both conditions were investigated with the best results obtained when 2.0 mol L–1 HCl was used which was then selected to activate the capillaries.
The amount of T. cruzi GAPDH immobilized on the surface of the capillary was estimated by the difference in the absorbance at 210 and 280 nm of the solution of T. cruzi GAPDH passed through the activated capillary. The results indicate that between 160 and 180 µg (16–18% of the original protein content) was immobilized on a 50 cm capillary. The activity of the immobilized T. cruzi GAPDH was established by injecting the substrate, D-GA3P, and cofactor, NAD+, onto the T. cruzi GAPDH-IMER and measuring the resulting NADH, cf. Fig. 3. These results indicate that the immobilized T. cruzi GAPDH retained its enzymatic activity.
Stability and storage of GAPDH-IMERs
The activity of a T. cruzi GAPDH-IMER was assessed daily over a 35 day period. During the first 10 days, the IMER activity dropped to 10% of the original level and then remained stable for the remainder of the test period. The results indicated that in the IMER format, the stability of T. cruzi GAPDH was increased from hours to at least one month, when compared to the free enzyme. The high enzymatic activity and stability permitted the performance of several on-line kinetic studies.When the T. cruzi GAPDH-IMERs were not in use they were stored at 4 °C. The effect of storage on the enzymatic activity was examined using a second IMER that was used, then washed and stored for five days. This procedure was repeated during the same period of time that the first was in daily use. There were no significant differences in the time versus activity profiles of the two IMERs, indicating that the stability of the immobilized enzyme was independent of both use and storage. One possible explanation of these results is that only 10% of the estimated enzyme was actually covalently immobilized on the surface of the capillary and that the loss of activity reflects bleeding of the T. cruzi from the IMER.
Immobilization reproducibility
The reproducibility of the enzymatic activity in the T. cruzi GAPDH-IMER was investigated by simultaneously preparing six IMERs. The resulting IMERs exhibited nearly identical activities with production of NADH ranging from 360 to 270 µM.Using this new protocol a human GAPDH-IMER was also prepared, but no increase in activity was observed when compared to the previously reported method. Thus, for the immobilization of human GAPDH the previously reported11 conditions were maintained since the stability of the IMER was huge under the reported conditions.
Kinetics studies of the free and immobilized T. cruzi GAPDH
The Michaelis–Menten constants Km of the IMER format (immobilized) were determined for both substrate and cofactor following the experimental conditions and described in Table 1, respectively. The enzymatic activity of soluble GAPDH was evaluated by measuring the formation of NADH using the spectrophotometric method, as described in the Experimental section.Non-linear curve-fitting regression analysis was applied in order to determine the Km values from the collected experimental data. The results are presented in Table 3.
The data demonstrate that the immobilization did not affect the affinity of the immobilized GAPDH relative to the free enzyme in solution. In the case of NAD+, the Km value for the immobilized enzyme was about two-fold higher than that measured for the enzyme in solution. However, when GA3P was used as the substrate, the Km values were almost the same for the immobilized and the free enzyme. This indicates that the immobilization process had a slightly more pronounced effect on the binding of the cofactor.
These results differ from the previous ones employing the human GAPDH-IMER system.11 Probably, because of the restriction in flexibility of structural components involved in the catalytic mechanism, the immobilization of human GAPDH reduced the binding-affinity for the substrate and the cofactor.
It is important to emphasize that the structural requirements for the binding of small molecules (inhibitor candidates of small molecular mass) to the target protein in the GAPDH-IMER are conserved as proved by the experiments carried out in this work. The covalent immobilization of the GAPDH enzyme not only retained the enzymatic activity, but also increased the enzyme stability. These are important achievements, allowing the biological screening of inhibitor candidates with improved accuracy and reproducibility. The differences in the Km values for the free GAPDH and GAPDH-IMER are a consequence of the conformational changes caused by the immobilization of the receptor target. However, the GAPDH-IMER system has retained the structural requirements for the search of competitive inhibitors at both ligand sites (NAD+ and GA3P), as indicated by the kinetic studies on the free GAPDH and immobilized enzyme.
The comparison of the immobilization effect, for human and T. Cruzi GAPDH-IMERs will be discussed elsewhere.
Conclusions
In the present work, the process of enzyme immobilization, which resulted in the GAPDH-IMER, is of substantial interest in drug discovery. Considering the high costs and difficulties regarding the purification of enzymes (GAPDH and others, from human or other organisms), this technique represents a useful way to preserve enzyme activity for a high number of enzyme assays. We consider this an important advance for the screening of synthetic and natural products in the search for new bioactive substances. This approach can also be useful for other target enzymes, considering that the structural requirements for the binding of substrates and small-molecule modulators could be preserved at both molecular and protein levels.Acknowledgements
This work was funded by grants of the São Paulo State Research Foundation (FAPESP). Q. B. C and C. L. C acknowledge CNPq and FAPESP for research and post-doctoral fellowship.References
- World Health Organization Statistical Information System Web-site, at http://www.who.ch/whosis/whosis.htm (accessed 25 September 2007).
- J. A. Urbina and R. Docampo, Trends Parasitol., 2003, 19, 495–501 Search PubMed.
- J. R. Coura and S. L. Castro, Mem. Inst. Oswaldo Cruz, 2002, 97, 3–24 Search PubMed.
- F. R. Operdoes, Annu. Rev. Microbiol., 1987, 41, 127–151 CrossRef.
- F. R. Opperdoes and P. A. M. Michels, Int. J. Parasitol., 2001, 31, 482–490 CrossRef CAS.
- B. M. Bakker, P. A. M. Michels, F. R. Opperdoes and H. V. Westerhoff, J. Biol. Chem., 1999, 274, 14551–14559 CrossRef CAS.
- J. J. Harris and M. Walters, in Enzymes, ed. P. D. Boyer, Academic Press, New York, 1976, vol. 13, p.1 Search PubMed.
- B. M. Bakker, H. V. Westerhoff, F. R. Opperdoes and P. A. M. Michels, Mol. Biochem. Parasitol., 2000, 106, 1–10 CrossRef CAS.
- D. H. F. Souza, R. C. Garratt, A. P. U. Arújo, B. G. Guimarães, W. D. P. Jesus, P. A. M. Michelis, V. Hannaert and G. Oliva, FEBS Lett., 1998, 424, 131–135 CrossRef CAS.
- A. M. Girelli and E. Mattei, J. Chromatogr., B, 2005, 819, 3–16 CrossRef CAS.
- C. L. Cardoso, V. V. Lima, A. Zottis, G. Oliva, A. D. Andricopulo, I. W. Wainer, R. Moaddel and Q. B. Cass, J. Chromatogr., A, 2006, 1120, 151–157 CrossRef CAS.
- E. C. Schirmer, J. Biol. Chem., 1998, 273, 15546–15552 CrossRef CAS.
- Y. Shi and S. R. Crouch, Anal. Chim. Acta, 1999, 381, 165–171 CrossRef CAS.
- Q. Yang, X.-Y. Liu, J. Miyake and H. Toyotama, Supramol. Sci., 1998, 5, 769–772 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |