Actively moving polymers
Marc
Behl
and
Andreas
Lendlein
*
Center for Biomaterial Development, Institute of Polymer Research, GKSS Research Center Geesthacht, Kantstr. 55, D-14513 Teltow, Germany. E-mail: andreas.lendlein@gkss.de
First published on 14th November 2006
Abstract
The ability of polymers to move actively in response to an external stimulus such as heat or light is of high scientific and technological significance. In any instance stimuli-responsive effects on the molecular level are converted into macroscopic movement, whereby generally two different moving behaviors have to be differentiated for polymer-based materials: the shape-memory effect and the shape-changing capability. Basic concepts for the molecular design of suitable polymer architectures for shape-memory polymers as well as tailored programming processes are presented. The thermally-induced shape-memory effect of polymers is described as well as the extension of this concept to other stimuli than heat. Indirect actuation of the thermally-induced effect by IR-irradiation, electric current, humidity or alternating magnetic fields are outlined as well as recent work on light-induced shape-memory polymers. For shape-changing polymers, two basic concepts are presented: shape changes occurring during phase orientation of liquid crystal elastomers (LCE) and the photomechanical effect based on photoisomerization of moieties, such as azo-groups incorporated in suitable polymer systems.
 Marc Behl | Marc Behl received his diploma in Chemistry from the University of Wuppertal and his doctor’s degree from Johannes Gutenberg University, Mainz. Currently, he holds a post-doctoral position at the Institute of Polymer Research at the GKSS Research Centre. His primary research interests are design, synthesis and characterization of shape-memory polymers. |
 Andreas Lendlein | Andreas Lendlein is Director of the Institute of Polymer Research at GKSS Research Center (member of the Helmholtz Association) in Teltow and Professor for Materials in Life Sciences at the University of Potsdam since 2002. In addition since 2006 he is Vice Director of the Berlin-Brandenburg Center for Regenerative Therapies (BCRT). He is internationally known for his work on polymer science and biomaterial research particularly for Regenerative Medicine. He did his postdoctoral lecture qualification in Macromolecular Chemistry 2002 at the University of Technology (RWTH) Aachen and received his PhD from the Swiss Federal Institute of Technology (ETH) in Zurich, Department for Material Sciences with Prof. Dr U. W. Suter. His current research interests are focussed on biomaterial interactions with physiological ambience, polymer technologies, and preclinical application development. |
1. Introduction
Stimuli-sensitive materials are able to respond to specific changes in their environment by adopting certain macroscopic properties such as shape, color or refractive index. In gels, reviewed in ref. 1, sensitivity to heat, light, magnetic fields, ion strength or control of the pH-value has been achieved. In non-swollen polymers active movement is stimulated by exposition to heat or light. Two types of active moving behaviors are visualized in Fig. 1, which are both temperature controlled. A shape-memory polymer (Fig. 1A) can be deformed and fixed in a second, temporary shape—e.g. a straight bar (upper picture). When this programmed material is heated to 60 °C, its original shape, a corkscrew like spiral, is recovered (lower pictures). A shape-changing polymer (Fig. 1B) contracts as long as exposed to the stimulus but returns into its initial shape when the stimulation is terminated. Here heat is used as stimulus. Both polymer concepts are based on elastic polymer networks, which are equipped with molecular switches or exhibit stimuli-sensitive domains. On activation the switches are triggered, resulting in the stimulated movement. Molecular architectures and the processes occurring during stimulation are discussed for both types of actively moving polymers. | ||
| Fig. 1 Shape-memory effect (A) and shape-changing capability (B). (A) The photoseries shows from top to bottom the thermally-induced transition from the temporary form of a stretched bar, to the permanent shape, a spiral for a thermoplastic shape-memory polymer. After heating to 60 °C the recovery process takes 35 s. (B) A strip of nematic liquid crystal elastomers contracts when heated and extends when cooled. Fig. 1B taken from the chapter: A Bird’s Eye View of Liquid Crystal Elastomers, Fig. 1.3, p. 3 from Liquid Crystal Elastomers by Warner and Terentjev.82 By permission of Oxford University Press; http://www.oup.com. | ||
2. Shape-memory polymers
2.1 General concept of shape-memory polymers
Shape-memory polymers are materials which can be deformed and fixed in a temporary shape, from which they recover their original shape only when exposed to an appropriate stimulus. This change of shape is predefined and determined by the mechanical deformation leading to the temporary shape. Heat or light have been used as stimuli in shape-memory polymers reported so far. Indirect actuation of the thermally-induced shape-memory effect is possible by irradiation with IR-light, application of electrical current, exposure to alternating magnetic fields or immersion in water.The shape-memory effect is not an intrinsic material property but a functionalization of a material. This functionalization results from the combination of the polymer's molecular architecture with a tailored processing and programming technology. In a first step the shape-memory polymer is formed into its initial shape by conventional processing methods. Afterwards, the polymer sample is deformed and fixed in an individual temporary shape. This process is called programming. The cycle of programming and recovery can be repeated several times whereby the temporary shapes can be varied from one to the next cycle. The shape-memory effect only relies on the polymer architecture; it is not limited to specific repeating units. This allows the adjustment of intrinsic material properties e.g. mechanic properties by variation of molecular parameters such as type of monomer or copolymer ratio to the needs of specific applications.
Generally shape-memory polymers are polymer networks equipped with suitable molecular switches (Fig. 2), which are sensitive to an external stimulus. Polymer networks consist of chain segments and netpoints. The netpoints crosslink the chain segments and determine the permanent shape of the polymer. The crosslinks can be either of a chemical nature (covalent bonds) or of a physical nature (intermolecular interactions). Physical crosslinking is obtained in a polymer, whose morphology consists of at least two segregated domains as found e.g. in block copolymers. Here the phase with the highest thermal transition (Tperm) acts as netpoint. In thermally-induced shape-memory polymers the chain segments associated to the domain with the second highest thermal transition Ttrans are called switching segments. Switching segments are flexible if the working temperature is higher than Ttrans. As a consequence the polymer networks show entropy elastic behavior above Ttrans: if a sample is deformed by application of an external stress, it snaps back to its original shape once the external stress is released.
 | ||
| Fig. 2 Molecular mechanism of the thermally-induced shape-memory effect; Ttrans = thermal transition temperature related to the switching phase (taken from ref. 3). Reprinted by permission from Angewandte Chemie – International Edition, 2002, 114, 2138–2162, Copyright 2002 Wiley-VCH, Germany. | ||
The thermally-induced shape-memory functionality is enabled, if the material can be temporarily fixed in a deformed state under environmental conditions relevant for the particular application. This requires the reversible prevention of the recoiling of the chain segments oriented under external stress and is achieved by the introduction of additional reversible netpoints as molecular switches. These additional netpoints may be formed by covalent bonds or physical interactions. Physical crosslinking is obtained by vitrification or crystallization of domains formed by side chains or the chain segments themselves. Chemical crosslinking is reached by the attachment of functional groups to the chain segments. These functional groups must be able to reversibly form covalent bonds by reaction with each other. The reversible chemical reaction is controlled by an external stimulus. The introduction of photoreversible functional groups extended the shape-memory technology to light as a stimulus.2 Other stimuli like electrical current or alternating magnetic fields can be applied for indirect heating of the material and in this way enable indirect actuation of the thermally-induced shape-memory effect.
Shape-memory properties are quantified in cyclic, thermo- or photomechanical tests.3–5 The data obtained from these tests allow the determination of the strain fixity rate Rf, the strain recovery rate Rr and the switching temperature Tswitch. Rf quantifies the ability to fix a mechanical deformation εm applied in the programming process. Rr quantifies the ability of the material to memorize its permanent shape after programming using a certain mechanical deformation εm.
During programming as well as recovery the thermal transition of the switching phase does not only result in its solidification or softening. Depending on the character of the transition, changes of the sample volume are also possible, the extent of which may depend on the degree of orientation of polymer chains in the sample. For the prediction of shape-memory properties several models for the mathematical description of the thermally-induced shape-memory effect have been described for polymers.6–9 All models describe uniaxial deformation processes but use either a mechanic modeling approach8,9 or a thermodynamic approach.6,7 They allow the prediction of irrecoverable strain,7,9 temperature dependent stress and strain6 or stress and strain8 for polymers under deformation.
2.2 Thermally-induced shape-memory polymers
The thermally-induced shape-memory effect has been achieved for thermoplastic as well as covalently crosslinked polymers. Linear block copolymers as an example for thermoplasts with Ttrans = Tm include polyesterurethanes, polyetherester and ABA triblock copolymers.3 ABA triblock copolymers with shape-memory were prepared with polytetrahydrofuran as the B-block and poly(2-methyl-2-oxazoline) as the A-block in which poly(2-methyl-2-oxazoline) acts as the hard segment and polytetrahydrofuran as the switching segment (Tm = 20–40 °C). In polyetherester poly(ethyleneterephthalate) segments form the phase related to Tperm while crystallizable poly(ethylene glycol) acts as the switching segment (Tm = 40–60 °C).10–12 In polyesterurethanes poly(ε-caprolactone) segments form the switching phase (Tm = 44–55 °C) while polyurethane segments act as the hard segment.13–15 In polyurethanes containing mesogenic groups the switching segment is based on poly(ε-caprolactone) (Tm = 38–59 °C) while mesogenic diols and 4,4-methylene bis(phenyl isocyanate) form the segments related to Tperm.16,17In a block copolymer with trans-poly(isoprene) switching segments and polyurethane hard segments, assembling of the polyurethane segments into spherical domains has been observed.18 Here the polyurethane segments form the physical crosslinks, while the trans-polyisoprene segments associated with a phase with a melting transition around 60 °C act as the switching segment. Block copolymers with 70 wt% switching segment content showed an Rr of about 85% and an Rf close to 100%.
In block copolymers Ttrans can also rely on a glass transition temperature (Ttrans = Tg). Examples are polyetherurethanes with a hard segment of methylenediisocyanate and butanediol with polytetrahydrofuran or poly(ethylene adipate) as the second segment.3 In most cases these polymers form a mixed domain acting as the switching phase. In polytetrahydrofuran segments containing polyetherurethane segments the molecular weight of the polytetrahydrofurandiols used as the educt determines the quality of phase separation between the polytetrahydrofuran and the polyurethane segments. Also the use of two polytetrahydrofurandiols differing in their molecular weight (Mn = 1000 and 1800 g mol−1) and 4,4-methylene bis(phenyl isocyanate) was investigated.19 Additionally, the sequence structure of the two polytetrahydrofuran blocks in the block copolymers has been varied between a more random and a blocky sequence structure. While in a random arrangement a single Tg has been observed, a blocky arrangement results in two Tgs. Both types of polyetherurethane showed higher stresses and strains at break than polyetherurethanes with only one kind of polytetrahydrofuran or blends of polytetrahydrofuran and polyurethane. In polymers with a blocky sequence structure Rr and Rf values of more than 90% have been determined.
Polymer analogous partial reduction of polyketones with NaBH4–THF results in poly(ketone-co-alcohol)s with shape-memory functionality.20 Polyketones were synthesized by late transition metal complex catalyzed polymerization of propene, hex-1-ene or a mixture of propene and ethene with carbon monoxide. The degree of reduction of the poly(ketone-co-alcohol) was adjusted by the amount of NaBH4–THF and was shown to influence Tg, polarity and mechanical properties of the polymer. The most promising material was a partly reduced poly(ethylene-co-propene-co-carbonoxide) with a phase-separated morphology. It consisted of hard microcrystalline ethylene/CO rich segments within a softer amorphous polyketone ethylene-propene/CO matrix. The microcrystalline domains crosslink the material physically. The switching temperature is a glass transition temperature (Ttrans = Tg) and enables the shape-memory effect. The Tg of the material could be controlled between below room temperature and 75 °C by partial reduction. Rr values between 90% and 95% were determined.
Covalently crosslinked polymer networks can be obtained by crosslinking linear or branched polymers as well as by (co)polymerization–poly(co)condensation of one or several monomers, whereby at least one has to be at least tri-functional. Crosslinking of polymers has been successfully achieved by irradiating polyethylene21 and its copolymers22,23 with ionizing radiation (γ-radiation, neutrons). Another method involves dehydrochlorination of poly(vinyl chloride) under vacuum at increased temperature and subsequent crosslinking in an HCl atmosphere.24 Dicumylperoxide is applied as a thermally-induced radical initiator for the crosslinking of poly(ethylene-co-vinylacetate).25 Semi-crystalline polycyclooctene obtained by ring-opening metathesis polymerization could be crosslinked with the same radical initiator.26 Here, the shape-memory effect relies on the melting of the crystallites, which depends on the trans-vinylene content. The crystallinity of the material is controlled by the radical initiator content and decreases with increasing crosslinking density. For pure polycyclooctene with 81 wt% trans-vinylene content, a melting temperature of 60 °C was determined. Shape recovery of these materials occurred within 0.7 s at 70 °C.
Another synthesis route to obtain polymer networks is the copolymerization of monofunctional monomers with low molecular weight or oligomeric bifunctional crosslinkers. An example is copolymers of stearyl acrylate, methacrylate and N,N′-methylenebisacrylamide as the crosslinker.27 Here crystalline domains of stearyl side chains form the switching phase. Radical copolymerization of poly(octadecyl vinylether)diacrylates or -dimethylacrylates with butyl acrylate results in multiphase copolymer networks.28,29 In both cases the crystalline domains of the octadecyl side chains are the switching segments.
Reaction of liquid crystalline moieties with star-shaped reactive precursors results in the formation of liquid crystalline polymer networks.30 The shape-memory effect is triggered by the thermal transition of the liquid crystalline domains. Main chain smectic-C elastomers were formed by coupling tetrafunctional silanes, working as netpoints, with oligomeric silanes, working as spacers, to which two distinct benzoate-based mesogenic groups had been attached. This crosslinking process defines the permanent shape. For programming the elastomer is heated to the isotropic state of the liquid crystalline domains, stretched or twisted and finally cooled below the clearing transition (I–SmC) of the smectic-C domains. When the sample is reheated the permanent shape is recovered.
Models for the prediction of stress and strain behavior of thermally-induced shape-memory polymers have been developed for physically6–8 and covalently crosslinked materials.8,9 The models for the covalently crosslinked materials follow a mechanics approach, in which the physical description of expansion and contraction is adjusted to fit the conditions of a shape-memory polymer8 or the shape-memory material is described as a combination of spring or dashpot units.9 These models allow calculation of stress and strain for crystallizable shape-memory materials depending on crystal size,8 or prediction of irreversible strain at large deformations.9 The first model was used for simulating a uniaxial and a circular-shear deformation under constant stress or constant strain. The uniaxial deformation experiments are in exceptional congruence with experimental data as the model differentiates the four different polymer phases during the programming cycle (rubbery phase, crystallization process, semi-crystalline phase and melting process). In the inhomogeneous case of circular-shear deformation, the predictions of the model could not be compared with experimental data, as these data were not available. The mechanics model,9 which is in good congruence with radiation crosslinked polyethylene, contains two damping units with different viscosities. If these viscosities are known or good estimations are available, shape recovery rates can be predicted.
A thermodynamic approach, based on a model assuming active and frozen phases, represents the multiphase character of thermoplastic shape-memory polymers. This model allows prediction of stress and strain in dependence of temperature for small unidirectional deformations (±10%).6 It has been further developed to a combination of the micromechanics with the standard viscoelastic model and is capable of predicting the remaining strain during stress release for large three-dimensional deformations.7
2.3 Indirect actuation of thermally-induced shape-memory effect
Two different strategies have been developed for the indirect actuation of the thermally-induced shape-memory effect. One method involves indirect heating, e.g. warming by irradiation. The other strategy aims at lowering Ttrans by diffusion of a low molecular weight molecule into the polymer bulk acting as a plasticizer allowing triggering of the thermally-induced shape-memory effect, while the sample-temperature remains constant.Indirect actuation by illumination with infrared light was demonstrated in a laser-activated device from polyurethane.31,32 Responsiveness of the shape-memory polymers upon exposure to heat-transferring fluids or infrared light sources is limited by its heat capacity and thermal conductivity.33 The heat transfer can be enhanced by incorporation of conductive fillers such as conductive ceramics, carbon black and carbon nanotubes.33–35 At the same time, compounding shape-memory polymer with particles influences the mechanical properties. While incorporation of microscale particles into thermoset epoxy resins results in increased stiffness and recoverable strain levels,36,37 nano-scale particles increased modulus and strength even more.38,39 Besides particle size, the molecular structure of the particles has to be considered to reach an enhanced photothermal effect. The reinforcement of polyesterurethanes with carbon black and carbon-nanotubes of similar size increases stress and fixity in both cases.40 While carbon black reinforced materials showed Rr values of only 25–30%, in carbon-nanotube reinforced materials Rr values of almost 100% were determined. This increased shape recovery capability has been assigned to a synergism between the anisotropic carbon-nanotubes and the crystallizing switching segments of the polyurethane. Incorporation of carbon-nanotubes into shape-memory polyurethane also lead to a certain level of electrical conductivity.41 When an electrical current is applied, the sample temperature increases by acting as an ohmic resistance. This effect can be used to trigger the shape-memory effect. Potential applications for such composite materials are electroactive actuators in microaerial vehicles41 or deployable space structures.36
Remote actuation of the thermally-induced shape-memory effect in an alternating magnetic field is realized in composites of shape-memory thermoplasts and magnetic nanoparticles.42 Here inductive heating of the nanoparticles in an alternating magnetic field (f = 258 kHz, H = 30 kA m−1) increased the sample temperature. The thermoplastic material consisted either of a biodegradable multiblock copolymer (PDC), with poly(p-dioxanone) as the hard segment and poly(ε-caprolactone) as the switching segment, or an aliphatic polyetherurethane (TFX) from methylene bis(p-cyclohexyl isocyanate), butanediol and polytetrahydrofuran. Magnetic nanoparticles of iron(III)oxide cores in a silica matrix were incorporated in both polymers. While TFX has an amorphous switching phase, PDC has a crystallizable switching segment. The shape-memory effect could be triggered in both samples by exposure to an alternating magnetic field. In Fig. 3 the magnetically-induced uncoiling of a corkscrew-like spiral of a composite from TFX and 10 wt% magnetic nanoparticles is shown exemplarily. The Rr values of indirectly heated samples were comparable to samples where the environmental temperature has been increased. Magnetic field driven shape-memory composites have potential applications in areas where mechanical adjustments in a non-contact mode are required, e.g. smart implants or instruments.
 | ||
| Fig. 3 Magnetically-induced shape-memory effect of a thermoplastic shape-memory composite from nano-particles consisting of iron(III)oxide particles in a silica matrix and a polyetherurethane. (Taken from ref. 42). Copyright 2006 National Academy of Sciences, USA. | ||
An example of shape-memory polymers whose Ttrans can be lowered by diffusion of water into the polymer are polyurethanes43,44 and their composites with carbon nanotubes.45 The temporary shape of the samples is programmed by conventional methods for thermally-induced shape-memory polymers. When immersed into water, the sample recovers into its permanent shape, as Tg is lowered from 39 °C to a temperature below ambient temperature. Two types of adsorbed water have to be considered in the polymer, bound and free water. While the effect of free water in the polymer is negligible, bound water acts as a plasticizer and lowers Tg significantly.
2.4 Light-induced shape-memory polymers
Light-induced stimulation of shape-memory polymers has been realized in shape-memory polymers by incorporation of reversibly reacting molecular switches.2,5,46 In contrast to the above mentioned indirect actuation of thermally-induced shape-memory polymers, this molecular mechanism is independent of any temperature effects. The light-sensitive molecular switches consist of cinnamic acid (CA) or cinnamyliden acetic acid (CAA)2 moieties. These functional groups are able to form covalent bonds with each other upon irradiation of suitable wavelength. When irradiated with different suitable wavelengths the newly formed bonds are cleaved again.The permanent shape of the photoresponsive shape-memory polymers is determined by netpoints which are crosslinking the chain segments of an amorphous permanent polymer network. For programming, the polymer network is stretched resulting in an orientation of the coiled polymer segments between two netpoints. Afterwards, new covalent netpoints are created by irradiation with UV light, λ > 260 nm. The release of the external stress results in a temporary shape. Upon irradiation with UV light of λ < 260 nm the permanent shape recovers, as the crosslinks are cleaved.
Two alternative strategies were described to introduce light-sensitivity on the molecular level: grafting of photosensitive moieties onto a permanent polymer network (Fig. 4) or interpenetrating a permanent network with oligomeric molecules having several photosensitive moieties reversibly forming a polymer network.
 | ||
| Fig. 4 Molecular mechanism of light-induced shape-memory effect of a grafted polymer network: the chromophores (open triangles) are covalently grafted onto the permanent polymer network (filled circles, permanent crosslinks), forming photoreversible crosslinks (filled diamonds); fixation and recovery of the temporary shape are realized by UV light irradiation of suitable wavelengths. (Taken from ref. 2). Reprinted by permission from Macmillian Publishers Ltd: Nature, 2005, 434, 879–882, copyright 2005. | ||
The polymer networks with grafted CA molecules were synthesized by copolymerization of n-butylacrylate, hydroxyethyl methacrylate and ethylenegylcol-1-acrylate-2-CA with poly(propylene glycol)-dimethacrylate (Mn = 560 g mol−1) as the crosslinker.2 The photosensitive interpenetrating network is obtained by loading a permanent polymer network from butylacrylate and 3 wt% poly(propylene glycol)-dimethacrylate (Mn = 1000 g mol−1) as the crosslinker with 20 wt% star-poly(ethylene glycol) end-capped with terminal CAA groups.2 The grafted polymer showed an Rf of max. 52% and an Rr of max. 95% in the fifth cycle, while in the third cycle an Rf of 33% and an Rr of 98% were determined for the interpenetrating network.
As light-induced shape-memory polymers are independent of any external heating and do not heat up while irradiated, they offer an alternative mode of actuation e.g. for medical applications where significant temperature changes may damage tissue.
3. Shape-changing polymers
3.1 General concept of shape-changing polymers
Shape-changing polymers are materials which change their shape, e.g. shrink or bend, as long as they are exposed to a suitable stimulus. Once stimulation is terminated, the original shape is recovered. The geometry, how a workpiece is moving, is determined by its original three dimensional shape. While the process of stimulated deformation with subsequent recovery can be repeated several times, the geometry of the shape changes cannot be varied. This is in contrast to shape-memory polymers, where the temporary shape can be varied by tailored programming from one cycle to another. Stimuli for shape-changing polymers reported so far are heat, light and electro-magnetic fields. The shape-changing effect is based on a phase transition in liquid crystalline elastomers. Light-stimulated shape changes can also be realized utilizing the photomechanical effect which transfers geometric changes during photoisomerization of azo-groups from the molecular to the macroscopic level (see section 3.2).In 1975 de Gennes47 predicted, that phase transitions of liquid crystalline materials could lead to mechanical stress or strain. While in liquid crystals induced stress leads to static forces, which are balanced by flow, in liquid crystal elastomers the free flow is prevented through polymer network formation. In these LCEs the liquid crystalline (LC) moieties are bound to a flexible, crosslinked polymer backbone. This polymer backbone allows a change of the orientation of the mesogens, but not a free flow. The change of orientation can be stimulated thermally or by application of an electromagnetic field. The induced stresses are transformed from the mesogen to the polymer backbone and result in mechanical work. The reversible strain actuation and soft elasticity of LCEs makes them interesting candidates for actuators and artificial muscles.48,49
In LCEs the switching rate, meaning the time between stimulation and actuation, depends on the type of actuation. While in ferroelectric LC materials response times up to ∼10 ms can be achieved, their strain is low (∼4%).50 Otherwise in thermally stimulated LCEs, the switching rate depends on the heat transfer of the material and therefore on sample thickness. In thermally-stimulated materials, larger strains can be achieved. Furthermore thermally-stimulated LCEs can be actuated remotely by radiative heating. Assuming a uniform absorption, this offers fast activation, while the backwards way is limited by thermal conduction.
The geometry of the shape change in LCEs is always related to the permanent shape. Therefore the macroscopic alteration of the shape into more complex structures other than shrinking or expanding requires an even more sophisticated synthesis strategy. Bending or curling can be realized either by welding two materials in a bi-morph elastic material51 or by the creation of a concentration gradient of the LC moieties within a workpiece.
3.2 Thermally-stimulated shape-changing polymers
In LCEs as thermally-stimulated shape-changing polymers, the LC phase is macroscopically oriented in the network. Such networks can be prepared by orienting the LC moiety and subsequent polymerization or by a two step polymerization process combined with an orientation in the second step. In a first step the LCE is prepolymerized. In the second step it is formed into its permanent shape under load while the polymerization is completed. The load orders the LC moieties macroscopically. When the LC phase becomes isotropic it results in a change of the network dimensions, as the network shortens in the direction of the director axis. The magnitude of this shape-change is determined by the coupling of the nematic state of order and the conformation of the polymer main chains. In nematic side chain elastomers the shape-change is not translated to the polymer backbone. This explains the relative poor shape-changing behavior of nematic side chain LCEs. If the mesogens are incorporated in the main chain, large conformation changes occur. By the combination of nematic side chains with nematic main chains, materials with shape-changes between 350 and 400% could be created.52,53 In both cases a siloxane backbone has been coupled to a phenyl-benzoate based side chain mesogen with 1,4-alkenoxybenzene and a nematic main chain polymer of 1-biphenyl-2-biphenyl butane. A mechanical load was applied after gelation of the weakly crosslinked network to obtain a LCE with a macroscopically uniform orientation of the director. Afterwards the hydrosilylation reaction was completed under load. Like all nematic LCEs these materials change their shape within a narrow temperature interval at the nematic–isotropic transition. For a polymer network consisting of 57 mol% LC main chain polymer reversible deformation of 380% has been reported.Instead of a two step chemical crosslinking process macroscopic ordering can be achieved through processing by e.g. extrusion. It has been shown for the nematic main-chain mesogen 1-biphenyl-2-biphenyl butane modified with terphenyl moieties.54 Here, the macroscopic ordering is obtained during the extrusion process. The terphenyl moieties crosslink the thermoplastic elastomer physically throughout microphase separation of the nematic phase of the triblock copolymer. Fibers processed from these triblock copolymers display a shape-change (shrinkage) of 470%.
The thermally-stimulated shape-change effect has also been demonstrated on a nanometre scale in nanoparticles.55 For this purpose main-chain liquid crystalline polymer nanoparticles (MCLCP) based on the nematic main chain polyether 1-(4-hydroxy-4′-biphenyl)-2-(4-hydroxyphenyl)butane and other liquid crystalline main-chain moieties were synthesized. If the particle size in these polymer systems is below a critical size a shape change between ellipsoids and spheres can be observed. The shape-change is determined by the quasi-equilibrium between the intrinsic shape of the entangled MCLCP and the thermodynamically most stable form55 in the isotropic phase, the sphere. Fig. 5 shows SEM pictures of nanoparticles in a spherical shape, which were obtained by freezing the isotropic state with liquid nitrogen, and in an ellipsoidal shape, which is the intrinsic shape after slow crystallization.
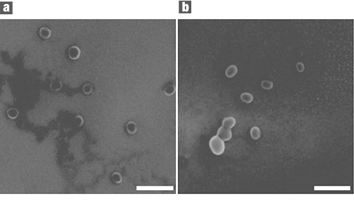 | ||
| Fig. 5 Scanning electron microscope images of main-chain polyether nanoparticles based on 1-(4-hydroxy-4′-biphenyl)-2-(4-hydroxyphenyl)butane. Particles are shown after being annealed at 101 °C for 40 s and (a) quickly quenched in liquid nitrogen; (b) slowly cooled to room temperature. The white bar represents 200 nm. (Taken from ref. 55). Reprinted by permission from Macmillian Publishers Ltd: Nature Materials, 2005, 4, 486–490, copyright 2005. | ||
Shape-changing has also been realized in smectic-C (SmC) elastomers.56 Although the coupling between elastic deformation and the SmC order has been theoretically described in the mid-nineties,57 the creation of monodomain samples was an experimental challenge. Monodomain samples with macroscopic C2 symmetry could be obtained by two successive deformation processes58,59 or by mechanical shear deformation.60 In contrast to nematic liquid crystalline elastomers SmC elastomers exhibit a biaxial shape-change effect:56 shrinkage (expansion) because of the smectic–isotropic (isotropic–smectic) transformation and shear deformation because of the tilting in the smectic phases, as shown in Fig. 6. Upon heating from 25 °C to 90 °C (Fig. 6 a to b) the SmC phase transforms into the SmA phase. This is accompanied by a reduction of the tilt angle from 23° to 11°. At further temperature increase from 90 to 130 °C (Fig. 6 b to c) the SmA phase transforms into the isotropic phase. As a result the sample shrinks from 8.5 to 6.3 mm. Fig. 6 c to e show the reverse cooling process.
 | ||
| Fig. 6 Photoseries of a monodomain SmC* elastomer in a heating and cooling process. The topside of the elastomer was fixed to a sample holder, while the lower end could freely move. The elastomer displays a rhomboid shape at room temperature (a), when heated to the temperature region of the SmA phase it transforms into a nearly rectangular shape (b). Further increase of temperature to the isotropic transition of the SmA phase results in shrinkage of the sample (c). On cooling from the isotropic to the smectic phase the sample elongates spontaneously into the rectangular-like film (d) and on further cooling into the rhombic shape (e). LE = sample length; θE = tilt angle of the elastomer film (taken from ref. 56). Reprinted with permission from Macromolecules, 2005, 7352–7357. Copyright 2005 American Chemical Society. | ||
Fig. 7 shows schematically the molecular realignment between the SmA and SmC phases in the shape-changing material. As the liquid crystalline moieties of each domain were macroscopically aligned and fixed, this molecular alignment results in a macroscopic shape-change of the material.
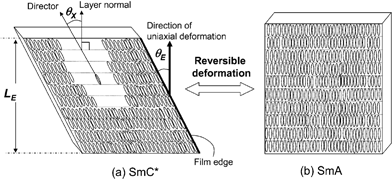 | ||
| Fig. 7 Schematic model explaining shape-changing behavior of elastomer film with molecular realignment. The elastomer film is based on a siloxane backbone with two different mesogenic moieties linked statistically to the monomer units. The macroscopic symmetry defined by the shape of the SmC* elastomer film corresponds to the microscopically local symmetry due to the molecular alignment of the liquid crystalline phase of SmC*. Fig. 7 (a) displays the SmC* phase with its sample length LE and the molecular tilt angle θx resulting in the elastomer film tilt angle θE. Upon heating the LC moieties reversibly rearrange into the SmA phase resulting in a lower θx and therefore a lower θE (b). LE = sample length; θx = molecular tilt angle; θE = tilt angle of the elastomer film (taken from ref. 56). Reprinted with permission from Macromolecules, 2005, 7352–7357. Copyright 2005 American Chemical Society. | ||
3.3 Light-stimulated shape-changing polymers
Responsiveness towards light can be introduced into shape-changing polymers by various functional groups,61–66 including azobenzene and triphenylmethane leuco derivatives. Light-stimulated shape-changes are based on photomechanical effects and light-stimulated phase transitions.Exposure of triphenylmethane leuco derivatives to UV light (λ > 270 nm) results in dissociation into ion pairs.67 During irradiation the intensely green colored triphenylmethyl cations are generated, the back reaction of the photo-generated cations with the counter anions occurs thermally in the dark. When triphenylmethane leuco derivatives are incorporated into polymers or gels, irradiation results in photo-generated charges and this variation of electrostatic repulsion leads to photo-induced expansion and shrinkage.68,69
Upon irradiation with UV light, azobenzene undergoes the reversible transformation from trans to cis, which is accompanied by a significant molecular length change from 9.0 Å in the trans form to 5.5 Å in the cis form. At the same time the dipole moment changes from 0.5 D to 3.1 D. Irradiation with light of wavelengths of 330–380 nm results in the trans → cis isomerization while irradiation with light with wavelengths of more than 420 nm or a temperature increase induces the backwards cis → trans isomerization reaction.
Different concepts have been evaluated in order to transfer the geometrical change caused by photoisomerization on the molecular level to macroscopic shape change. Azo-dye loaded nylon filament fabrics showed shrinkage of approximately 0.1% after irradiation under load.70 Azobenzene-containing crosslinkers enhanced this photomechanical effect in poly(ethyl acrylate) network films to 0.25%.71 Although the azobenzene moiety displays a contraction of around 60% on the molecular level the polymer sample does not show a shape change of this magnitude. In both cases the light-stimulated isomerization causes a conformational change of the adjacent polymer chain segments, but as the polymer chains have a random orientation, the conformational change of the polymers is equalized and does not result in a macroscopical change of the polymer sample. The photomechanical effect is significantly enhanced by aligning the azobenzene moieties. This is realized by incorporating azobenzene moieties into a mesogenic monomer and a crosslinker of a nematic liquid crystalline elastomer.72 The alignment of the chromophores is controlled by stretching the liquid crystalline elastomer in one direction.73 The photon absorption is limited to the surface region because of the strong absorption of the azobenzene moieties while the azo moieties in the bulk of the film remain in the trans form. Therefore the contraction of the liquid crystalline network takes place only at the surface. This results in a bending of the material towards the irradiation direction of the incident light.73,74 The bending direction can be precisely controlled by the use of linear polarized light,72 while the bending behavior can be controlled by the crosslink density.73,74
Incorporation of azobenzene moieties in nematic liquid crystalline polymer networks enables light-stimulated shape-changing.75–77 Here the UV light stimulated trans–cis isomerization triggers the phase transition of the liquid crystalline elastomer reducing the alignment order of the liquid crystalline moieties. While trans-azobenzene stabilizes the liquid crystalline alignment because of the rod like structure, the bent cis-azobenzene lowers the ordering. Therefore the trans–cis transformation results in an isotropic phase transition. The same phase transition can be reached by a temperature increase.77 Incorporation of azobenzene moieties into nematic elastomers, results in a contraction of about 20% upon irradiation with UV light having a wavelength of 365 nm for 60–90 minutes.75 Macroscopic alignment of the nematic elastomer is achieved by a two-step crosslinking process with an azobenzene-containing crosslinker, with the second step occurring under imposed extension. The time interval for the light-stimulated contraction process can be accelerated to less than one minute, when the azobenzene groups were incorporated as side-on part of the main chains of the liquid crystal elastomer.76 Here the light-stimulated trans → cis isomerization results in a complete loss of the nematic order. Light-induced contraction of up to 18% of the original length can be achieved. Full recovery of its original length requires heating to 100 °C in the dark for 30–60 minutes.
Conclusion and outlook
The field of actively moving polymers is developing rapidly. Fundamental research is presently focusing on new ways to let polymers actively move or to actuate polymers with other stimuli than heat. The first examples are the light-induced shape-memory polymers and light-stimulated shape-changing materials. In both cases stimuli depending alterations on the molecular level result in a variation of macroscopic shape. Another promising strategy to introduce new stimuli is heating of actively moving thermosensitive polymers e.g. by irradiation.The technology platform of available shape-memory polymers is progressing from laboratory demonstration objects to highly sophisticated applications. These applications span over various areas of everyday life and include fabrics, intelligent packaging, self repairing autobodies, switches and sensors. Other application areas can be seen in reusable composite tooling,78 aerospace industry or nano-actuators. An example for a mass market application was a choke used in automobile engines based on a shape-memory material. However, this product is obsolete today as engine technology developed to electronic fuel injection engines.
New shape-memory or shape-changing polymers in particular can be more expensive than established engineering plastics notably at the beginning of the commercialization phase. Reasons for this could be an additional process step for the functionalization, special monomers (e.g. for the molecular switches) or a high amount of synthesis steps. These cost factors could be reduced in the course of commercialization by integration of processes, up-scaling or optimization of synthesis.
Therefore, first applications for actively moving polymers can preferentially be found in areas, where only their functionality is really the key technology enabling a specific application. Aerospace and medicine industries seem to be two of these niches. Examples are self-deployable sun-sails for satellites or space structures based on shape-memory polymers.79 Here, the advantage arises from the fact, that no extra energy source for the one-time used sun-sail deployment is needed which therefore reduces weight.
Entering the mass market might be facilitated by prioritizing applications, which only require small amounts of actively moving polymers. An example is recycling. As recycling gets more and more important, solutions for economic disassembly have to be developed and can be realized by self-disassembling fasteners80 based on shape-memory polymers. The mobile phone chassis itself would still be made from conventional plastics.
Examples for applications requiring a larger amount of material are self repairing autobodies or wash and wear textiles. The implementation of stimuli such as alternating magnetic field or light will extend the application fields to non-contact operation. Here the area of active medical devices, such as implants81 or active prostheses is promising and the first potential applications were demonstrated.31,32 Besides demonstrating the application-relevant action of the shape-memory effect, an approval for each specific application has to be obtained, which often determines the time interval between a first demonstration object and begin of commercialization.
It has to be concluded, that only a few shape-memory polymers mainly having a thermally-induced effect, and even fewer shape-changing materials, have been investigated. Thus the research field of actively moving polymers is still at its beginning. Since research activities have strengthened over the last years, substantial advance can be expected in the near future.
References
- E. S. Gil and S. A. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- A. Lendlein, H. Y. Jiang, O. Jünger and R. Langer, Nature, 2005, 434, 879–882 CrossRef CAS.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- A. Lendlein, S. Kelch, J. Schulte and K. Kratz, Shape-Memory Polymers, in Encyclopedia of Materials: Science and Technology – Updates, ed. K. H. J. Buschow, Elsevier Science Ltd, New York, 2004, p. 5 Search PubMed.
- H. Y. Jiang, S. Kelch and A. Lendlein, Adv. Mater., 2006, 1471–1475 CrossRef CAS.
- Y. P. Liu, K. Gall, M. L. Dunn, A. R. Greenberg and J. Diani, Int. J. Plast., 2006, 22, 279–313 CrossRef CAS.
- J. Diani, Y. P. Liu and K. Gall, Polym. Eng. Sci., 2006, 46, 486–492 CrossRef CAS.
- G. Barot and I. Rao, ZAMP, 2006, 57, 652–681 Search PubMed.
- J. Morshedian, H. A. Khonakdar and S. Rasouli, Macromol. Theory Simul., 2005, 14, 428–434 CrossRef CAS.
- X. L. Luo, X. Y. Zhang, M. T. Wang, D. H. Ma, M. Xu and F. K. Li, J. Appl. Polym. Sci., 1997, 64, 2433–2440 CrossRef CAS.
- M. T. Wang, X. L. Luo, X. Y. Zhang and D. Z. Ma, Polym. Adv. Technol., 1997, 8, 136–139 CrossRef CAS.
- M. T. Wang and L. D. Zhang, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 101–112 CrossRef CAS.
- B. K. Kim, S. Y. Lee and M. Xu, Polymer, 1996, 37, 5781–5793 CrossRef CAS.
- F. K. Li, J. N. Hou, W. Zhu, X. Zhang, M. Xu, X. L. Luo, D. Z. Ma and B. K. Kim, J. Appl. Polym. Sci., 1996, 62, 631–638 CrossRef CAS.
- Z. L. Ma, W. G. Zhao, Y. F. Liu and J. R. Shi, J. Appl. Polym. Sci., 1997, 63, 1511–1515 CrossRef CAS.
- H. M. Jeong, B. K. Kim and Y. J. Choi, Polymer, 2000, 41, 1849–1855 CrossRef CAS.
- H. M. Jeong, J. B. Lee, S. Y. Lee and B. K. Kim, J. Mater. Sci., 2000, 35, 279–283 CrossRef CAS.
- X. Ni and X. Sun, J. Appl. Polym. Sci., 2006, 100, 879–885 CrossRef CAS.
- J. W. Cho, Y. C. Jung, Y. C. Chung and B. C. Chun, J. Appl. Polym. Sci., 2004, 93, 2410–2415 CrossRef CAS.
- D. Perez-Foullerat, S. Hild, A. Mucke and B. Rieger, Macromol. Chem. Phys., 2004, 205, 374–382 CrossRef CAS.
- A. Charlesby, Atomic Radiation and Polymers, Pergamon Press, Oxford, 1960, p. 198–257 Search PubMed.
- G. Kleinhans, W. Starkl and K. Nuffer, Kunststoffe, 1984, 74, 445–449 Search PubMed.
- G. Kleinhans and F. Heidenhain, Kunststoffe, 1986, 76, 1069–1073 Search PubMed.
- V. Skakalova, V. Lukes and M. Breza, Macromol. Chem. Phys., 1997, 198, 3161–3172 CrossRef CAS.
- F. K. Li, W. Zhu, X. Zhang, C. T. Zhao and M. Xu, J. Appl. Polym. Sci., 1999, 71, 1063–1070 CrossRef CAS.
- C. D. Liu, S. B. Chun, P. T. Mather, L. Zheng, E. H. Haley and E. B. Coughlin, Macromolecules, 2002, 35, 9868–9874 CrossRef CAS.
- Y. Kagami, J. P. Gong and Y. Osada, Macromol. Rapid Commun., 1996, 17, 539–543 CrossRef CAS.
- E. J. Goethals, W. Reyntjens and S. Lievens, Macromol. Symp., 1998, 132, 57–64 CAS.
- W. G. Reyntjens, F. E. Du Prez and E. J. Goethals, Macromol. Rapid Commun., 1999, 20, 251–255 CrossRef CAS.
- I. A. Rousseau and P. T. Mather, J. Am. Chem. Soc., 2003, 125, 15300–15301 CrossRef CAS.
- D. J. Maitland, M. F. Metzger, D. Schumann, A. Lee and T. S. Wilson, Lasers Surg. Med., 2002, 30, 1–11 CrossRef.
- W. Small, T. S. Wilson, W. J. Benett, J. M. Loge and D. J. Maitland, Opt. Express, 2005, 13, 8204–8213 CrossRef.
- C. D. Liu and P. T. Mather, ANTEC, 2003, 1962 Search PubMed.
- M. J. Biercuk, M. C. Llaguno, M. Radosavljevic, J. K. Hyun, A. T. Johnson and J. E. Fischer, Appl. Phys. Lett., 2002, 80, 2767–2769 CrossRef CAS.
- F. K. Li, L. Y. Qi, J. P. Yang, M. Xu, X. L. Luo and D. Z. Ma, J. Appl. Polym. Sci., 2000, 75, 68–77 CrossRef CAS.
- K. Gall, M. Mikulas, N. A. Munshi, F. Beavers and M. Tupper, J. Intell. Mat. Syst. Str., 2000, 11, 877–886 Search PubMed.
- C. Liang, C. A. Rogers and E. Malafeew, J. Intell. Mat. Syst. Str., 1997, 8, 380–386 Search PubMed.
- B. J. Ash, R. Stone, D. F. Rogers, L. S. Schadler, R. W. Siegel, B. C. Benicewicz and T. Apple, Investigation into the thermal mechanical behavior of PMMA/alumina nanocomposites, MRS Proceeding Volume 661, Symposium KK, ed. A. I. Nakatani, R. P. Hjelm, M. Gerspacher and R. Krishnammorti, Materials Research Society, Warrendale, PA, 2001 Search PubMed.
- S. K. Bhattacharya and R. R. Tummala, J. Electron. Packag., 2002, 124, 1–6 CrossRef CAS.
- H. Koerner, G. Price, N. A. Pearce, M. Alexander and R. A. Vaia, Nat. Mater., 2004, 3, 115–120 CrossRef CAS.
- J. W. Cho, J. W. Kim, Y. C. Jung and N. S. Goo, Macromol. Rapid Commun., 2005, 26, 412–416 CrossRef CAS.
- R. Mohr, K. Kratz, T. Weigel, M. Lucka-Gabor, M. Moneke and A. Lendlein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3540–3545 CrossRef CAS.
- W. M. Huang, B. Yang, L. An, C. Li and Y. S. Chan, Appl. Phys. Lett., 2005, 86.
- B. Yang, W. M. Huang, C. Li, C. M. Lee and L. Li, Smart Mater. Struct., 2004, 13, 191–195 CrossRef CAS.
- B. Yang, W. M. Huang, C. Li, L. Li and J. H. Chor, Scr. Mater., 2005, 53, 105–107 CrossRef CAS.
- Y. L. Yu and T. Ikeda, Macromol. Chem. Phys., 2005, 206, 1705–1708 CrossRef CAS.
- P. G. de Gennes, C. R. Seances Acad. Sci., Ser. B, 1975, 281, 101–103.
- Y. Bar-Cohen, J. Spacecr. Rockets, 2002, 39, 822–827 Search PubMed.
- J. D. W. Madden, N. A. Vandesteeg, P. A. Anquetil, P. G. A. Madden, A. Takshi, R. Z. Pytel, S. R. Lafontaine, P. A. Wieringa and I. W. Hunter, IEEE J. Oceanic Eng., 2004, 29, 706–728 CrossRef.
- W. Lehmann, H. Skupin, C. Tolksdorf, E. Gebhard, R. Zentel, P. Kruger, M. Losche and F. Kremer, Nature, 2001, 410, 447–450 CrossRef CAS.
- N. Assfalg and H. Finkelmann, Kautsch. Gummi, Kunstst., 1999, 52, 677–680 Search PubMed.
- H. Wermter and H. Finkelmann, e-Polym., 2001, 1–13 Search PubMed.
- A. R. Tajbakhsh and E. M. Terentjev, Eur. Phys. J. E, 2001, 6, 181–188 CrossRef CAS.
- S. V. Ahir, A. R. Tajbakhsh and E. M. Terentjev, Adv. Funct. Mater., 2006, 16, 556–560 CrossRef CAS.
- Z. Q. Yang, W. T. S. Huck, S. M. Clarke, A. R. Tajbakhsh and E. M. Terentjev, Nat. Mater., 2005, 4, 486–490 CrossRef CAS.
- K. Hiraoka, W. Sagano, T. Nose and H. Finkelmann, Macromolecules, 2005, 38, 7352–7357 CrossRef CAS.
- E. M. Terentjev and M. Warner, J. Phys. II, 1994, 4, 849–864 CrossRef CAS.
- K. Semmler and H. Finkelmann, Polym. Adv. Technol., 1994, 5, 231–235 CrossRef CAS.
- K. Semmler and H. Finkelmann, Macromol. Chem. Phys., 1995, 196, 3197–3208 CrossRef CAS.
- K. Hiraoka and H. Finkelmann, Macromol. Rapid Commun., 2001, 22, 456–460 CrossRef CAS.
- M. Irie, Adv. Polym. Sci., 1990, 94, 27–67 CAS.
- J. A. Delaire and K. Nakatani, Chem. Rev., 2000, 100, 1817–1845 CrossRef CAS.
- K. Ichimura, Chem. Rev., 2000, 100, 1847–1873 CrossRef CAS.
- N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875–1890 CrossRef CAS.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4175 CrossRef CAS.
- V. Shibaev, A. Bobrovsky and N. Boiko, Prog. Polym. Sci., 2003, 28, 729–836 CrossRef CAS.
- M. Irie and M. Hosoda, Makromol. Chem., Rapid Commun., 1985, 6, 533–536 CrossRef CAS.
- M. Irie and D. Kungwatchakun, Macromolecules, 1986, 19, 2476–2480 CrossRef CAS.
- A. Mamada, T. Tanaka, D. Kungwatchakun and M. Irie, Macromolecules, 1990, 23, 1517–1519 CrossRef CAS.
- E. Merian, Text. Res. J., 1966, 36, 612–618 Search PubMed.
- C. D. Eisenbach, Polymer, 1980, 21, 1175–1179 CrossRef CAS.
- Y. L. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145–145 CrossRef CAS.
- Y. L. Yu, M. Nakano and T. Ikeda, Pure Appl. Chem., 2004, 76, 1467–1477 CrossRef CAS.
- Y. L. Yu, M. Nakano, A. Shishido, T. Shiono and T. Ikeda, Chem. Mater., 2004, 16, 1637–1643 CrossRef CAS.
- H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 8701.
- M. H. Li, P. Keller, B. Li, X. G. Wang and M. Brunet, Adv. Mater., 2003, 15, 569–572 CrossRef CAS.
- M. Warner and E. Terentjev, Macromol. Symp., 2003, 200, 81–92 CrossRef CAS.
- M. C. Everhart and J. Stahl, High strain fiber reinforced reusable shape memory polymer mandrels, in International SAMPE Symposium and Exhibition (Proceedings), SAMPE (ISSEE), Covina, CA, 2005, vol. 50, pp. 955–965 Search PubMed.
- D. Campbell, M. S. Lake, M. R. Scherbarth, E. Nelson and R. W. Six, Elastic memory composite material: an enabling technology for future furable space structures, in 46th AIAA/ASME/ASCE/AHS/ASC Structures, Structural Dynamics and Materials Conference, AIAA, Reston, VA, 2005, article No AIAA 2005-2362 Search PubMed.
- H. Hussein and D. Harrison, Investigation into the use of engineering polymers as actuators to produce ‘automatic disassembly’ of electronic products, in Design and Manufacture for Sustainable Development 2004, ed. T. Bhamra and B. Hon, Professional Engineering Publishing, London, UK, 2004, pp. 36–49 Search PubMed.
- F. El Feninat, G. Laroche, M. Fiset and D. Mantovani, Adv. Eng. Mater., 2002, 4, 91–104 CrossRef CAS.
- M. Warner and E. Terentjev, Liquid Crystal Elastomers, International Series of Monographs on Physics, Clarendon Press, Oxford, 2003, vol. 120 Search PubMed.
| This journal is © The Royal Society of Chemistry 2007 |