Polymer–surfactant complexation in polyelectrolyte multilayer assemblies
Malkiat S.
Johal
*a and
Peter A.
Chiarelli†
b
aDepartment of Chemistry, Pomona College, 645 North College Avenue, Seaver North, Claremont, CA 91711-6338, USA. Web: http://pages.pomona.edu/%E2%88%BCmsj04747/E-mail: malkiat.johal@pomona.edu; Fax: +1 (909) 607-7726
bFaculty of Clinical Medicine, Oxford University, Oxford, OX3 9DU, UK
First published on 18th October 2006
Abstract
Layer-by-layer self-assembly can be used to incorporate amphiphilic molecules into multilayered polyelectrolyte architectures. This review examines equilibrium LbL assemblies constructed by direct adsorption from aqueous solution. LbL systems have not only provided fundamental insight into the nature of polyion–surfactant complexation, but have also yielded functional materials with useful surface, optical, and electronic properties.
 Malkiat Johal | Malkiat Johal received his Ph.D. from the University of Cambridge and then was a post-doctoral research associate at Los Alamos National Laboratory. His research activities focus on investigating electrostatic self-assembly processes in soft composite polyelectrolyte materials, and their interaction with biological macromolecules and ionic surfactants. He is currently an Associate Professor of Physical Chemistry at Pomona College. |
 Peter Chiarelli | Peter Chiarelli obtained his B.A. at Pomona College, and subsequently attended the University of Oxford on a Rhodes Scholarship, where he obtained his D.Phil. His research has focused on the directed self- assembly of amphiphilic, polymeric, and biological systems. Peter is currently enrolled in the Health Sciences and Technology division of Harvard Medical School. |
Introduction
Polyelectrolyte–surfactant aggregation has been studied for decades, both in bulk aqueous systems and at interfacial regions.1–3 The increased availability of water-soluble synthetic polyelectrolytes and biopolymers has led to a great number of studies focusing on polymer–surfactant complexation at aqueous interfaces. Such complexed materials possess growing commercial relevance as materials for separation-membranes, solubilization, and compatibilization.1–5 This review focuses on recent work performed using the layer-by-layer (LbL) electrostatic self-assembly (ESA) method to incorporate amphiphilic molecules into multilayered polyelectrolyte architectures. We examine equilibrium LbL assemblies constructed by direct adsorption from aqueous solution, as well as non-equilibrium complexes formed by spin-assembly. LbL systems have not only provided fundamental insight into the nature of polyion–surfactant complexation, but have also yielded functional materials with useful surface, optical, and electronic properties. More exhaustive accounts of polymer–surfactant interactions in the bulk phase are captured in a number of excellent review articles.3,6,7–9Owing to the displacement of small counterions, polyelectrolyte–surfactant association can be both entropically and electrostatically driven, with a modest contribution from hydrophobic interactions.3,10–14 Due to the interplay between these interactions, polymer–surfactant hybrid structures often possess properties different from those of the corresponding pure materials. The expulsion of small counterions into the solvent during ion-pair complexation is the key driving force for many ionic aggregation processes involving macromolecules. Electrostatic interactions dominate non-cooperative association between a polyion and an oppositely charged surfactant. The hydrophobic moiety of the surfactant may favor aggregation by cooperative binding to the polyion.
The ESA method of LbL assembly was developed in 1992 by Gero Decher, using oppositely charged water-soluble polyelectrolytes.15,16 The technique has since been used to construct highly ordered three-dimensional, multifunctional, reactive thin films. Polyelectrolyte systems fabricated by ESA are currently being developed for use as lens coatings, drug-delivery systems, biosensors, and nonlinear optical devices.17–22 ESA is a process that involves the alternating deposition of a polycation and a polyanion, to yield a charge-alternating set of interlaced multilayers. Charge overcompensation and entropic gain due to counterion expulsion are generally cited as the driving mechanism behind successful layer formation.23Fig. 1(a) illustrates the general procedure for the sequential adsorption of polyelectrolytes using ESA. The densely charged surface allows for ionic surfactant immobilization, and subsequent hydrophobic association between hydrocarbon chains. ESA provides a means for conveniently anchoring complete surfactant monolayers that are physically robust and not susceptible to desorption.
 | ||
| Fig. 1 LbL methods of constructing polyelectrolyte multilayer films on solid substrates. (a) ESA is accomplished by sequentially adsorbing equilibrium amounts of a polycation and a polyanion from their aqueous solutions. (b) In spin-assembly, the aqueous polyelectrolyte solutions are directly dropped onto the surface of the rotating solid substrate. Typical rotation speeds are 1000 to 3000 revolutions per minute. In both methods, the process can be repeated to construct a pre-determined number of polyelectrolyte layers. The solid substrate is typically a glass slide, rendered anionic by treatment with a strong oxidizing agent (e.g. chemical etching using H2O2–H2SO4). | ||
The spin-assembly process of LbL assembly provides an alternative to ESA, in which the polyanion and polycation are sequentially dropped onto a rotating substrate. In spin-assembly, film formation is attributed to mechanical as well as electrostatic interactions (Fig. 1(b)).24,25 An important difference between ESA and spin-assembly is the liquid–substrate contact time. In ESA, the substrate is placed in the polyelectrolyte solution for a relatively long time (∼5 min) until equilibrium has been reached. In contrast, the contact time for spin-assembly depends largely on the rotation speed of the substrate and is often fractions of a second. This leads to large differences in multilayer composition, conformation, and coverage.26 Spin-assembly has made possible a number of new layer structures previously unattainable with ESA, including stable like-charged multilayers and trilayer-repeat films.27,28
A brief synopsis of bulk and interfacial complexation
Much of our understanding of polymer–surfactant solutions comes from the outstanding contributions by Jones and others.29 Polymer-assisted surfactant micellization in the bulk aqueous phase occurs above a threshold surfactant concentration – referred to as the critical aggregation concentration (cac). In solutions containing a polyion and an oppositely charged surfactant, the cac typically exists a few orders of magnitude below the critical micelle concentration (cmc), at which unassisted micelle formation occurs in pure surfactant solutions. Polymer-assisted micellization is driven, in part, by an ion-exchange reaction between polyion chain segments and the surfactant ion. Most of the evidence for such complexation is derived from binding isotherms by methods such as potentiometry, tensiometry, or surfactant self-diffusion studies.1,3Polymer–surfactant interactions can be weak (e.g. between non-ionic surfactant and polyions) or strong (e.g. between oppositely charged surfactants and polyelectrolytes). Non-ionic surfactants are also known to interact with polymers mainly through a combination of ion-dipole, electrostatic, and hydrophobic forces.1,30 The process of polymer-assisted surfactant micellization, in the presence of a polyelectrolyte, can be monitored by measuring the surface tension of the corresponding aqueous solution. For example, a recent study by Deo et al. investigated the interaction between a hydrophobically modified anionic polyelectrolyte (PMAOVE) and the non-ionic surfactant pentaethyleneglycol mono-n-dodecyl ether (C12EO5) in aqueous solution (Fig. 2).31 The group used surface tension measurements to study the micellization of C12EO5 with and without the hydrophobic polyanion. As shown in Fig. 2, a break in the surface tension versus concentration plot for pure aqueous solutions of C12EO5 demonstrates that the aqueous-phase cmc of the surfactant is 0.06 mM. The addition of the polymer induces a reduction of the surface tension at low surfactant concentrations, since the polymer enhances surfactant adsorption at the air–water interface where polymer–surfactant complexation takes place. Such interfacial complexation typically minimizes the repulsive interaction between charged or dipolar surfactant headgroups at the surface.
 | ||
| Fig. 2 Polymer–surfactant complexation between poly(maleic acid–octyl vinyl ether) (PMAOVE) and pentaethyleneglycol mono-n-dodecyl ether (C12EO5). Surface tension of the aqueous surfactant solutions in the presence and absence of 0.1% (wt/wt) PMAOVE is shown. The upper portion of the schematic depicts how polymer-assisted micellization causes changes in observed surface tension. Surface tension data reprinted with permission from ref. 31, copyright American Chemical Society. | ||
A minimum in the surface tension plot is reached at a C12EO5 concentration of 0.0075 mM, representing the onset of polyelectrolyte-assisted micelle formation (the cac). The increase in surface tension beyond this concentration indicates a reduced surfactant packing density at the air–water interface, caused by favorable formation of stable bulk phase polymer–surfactant aggregates. Finally, as the surfactant concentration becomes increasingly large, the added surfactant remains in monomer form and begins to re-populate the air–water interface until the monomer concentration is high enough to begin unassisted micelle formation (at 0.06 mM in this example). At this point (above the cmc), the solution resembles a polyelectrolyte-free solution.
The aforementioned description of polymer assisted micellization is characteristic of strongly interacting polyelectrolytes and ionic surfactants. The surface tension behavior observed by Deo et al. is very similar, despite the fact that the surfactant is uncharged. However, Deo et al. proposed that C12EO5 gets incorporated into hydrophobic nanodomains of PMAOVE at 0.0075 mM (they refer to this concentration as the critical complexation concentration). Incorporation of the surfactant into these hydrophobic domains has the effect of depleting C12EO5 at the air–water interface and thus increasing the surface tension.
Polymeric charge density plays an important role in polyelectrolyte–surfactant complex formation. One of the most thoroughly studied water-soluble polycations is PEI (poly(ethylenimine)), in both the linear and branched forms. At low pH, PEI has the highest known charge density of all polyelectrolytes and has a strong tendency to form complexes with anionic surfactants.7,32,33 PEI–surfactant complexes have been found to form well-ordered mesomorphic complexes with the anionic surfactant sodium dodecylsulfate (SDS) exhibiting low surface energies.34,35 Recent neutron scattering studies indicated that at large SDS concentrations, the polymer–surfactant aggregate may become negatively charged, implying that the excess SDS is located at the surface of the aggregate.36 Mészáros and co-workers recently used dialysis measurements to determine the amount of SDS bound to PEI as a function of equilibrium surfactant concentration. They suggested that SDS binds to PEI as monomers, not micelles.37 Using amine–headgroup interactions, as indicated by pH, to determine bonding character, they concluded that the PEI–SDS system lacks a cac. At low pH, PEI is a strong electrolyte, while at high pH it behaves more like a neutral polymer.38 Because Mészáros' studies were conducted at high pH, it is possible that the distance between charged binding sites on PEI was too large for a cac to emerge at the concentrations studied.
The tendency for amphiphiles to accumulate at high energy interfaces implies a likewise aggregation of polyelectrolyte–surfactant complexes at such interfaces. In many cases, binding of surfactants to polyions at such interfaces is cooperative, leading to stabilized micelles within the polyelectrolyte. At the other extreme, complexation without a cac has also been observed.39–41 Strong intermolecular hydrophobic interactions drive cooperative binding of surfactants onto charged sites on the polyion. Davies and co-workers have recently used sum-frequency generation (SFG) spectroscopy to study PEI–SDS complexes at the solid–aqueous interface.42 The solid surface was strongly hydrophobic, composed of alkylthiols chemisorbed on a gold substrate. SFG spectra of SDS at the interface suggested that the structure and conformation of the surfactant monolayer was significantly affected by the presence of the polycation. They identified two types of regions on the PEI chain: one of high SDS density, and one of little or no SDS density. Davies and co-workers suggested that electrostatically bound SDS attracts additional SDS monomers to nearby sites to maximize inter-alkyl chain interactions.
Thomas, Penfold and co-workers have extensively used neutron reflectivity to provide evidence of the roles of the electrostatic interactions between the polyelectrolyte and surfactant on the adsorption behavior.38,43–46 Their work has explored the use of polyelectrolytes to manipulate surfactant adsorption at hydrophilic surfaces. For example, the cationic polyelectrolyte PDMDAAC (poly(dimethyldiallylammonium chloride)), can be used to manipulate the adsorption of SDS and mixtures of SDS and nonionic C12E6 (monododecyl hexaethylene glycol) onto the surface of hydrophilic silica.44 They have also shown that the pattern of SDS and PEI adsorption is indicative of a strong polymer–surfactant interaction at low pH, which decreases with increasing pH.38 They show that SDS adsorption at the interface is unexpectedly most pronounced when the pH is high. Under these conditions, the polymer is essentially a neutral polymer. Enhanced adsorption also occurs when the polymer architecture is branched rather than linear.45 Likewise, the adsorption of the anionic polyelectrolyte sodium poly(styrene sulfonate), (PSS) onto a polycation-coated surface results in a change to an anionic surface, which then modifies the relative affinities of cationic and anionic surfactants for the surface.46
This group has also explored the role of hydrophobicity of the surfactant in complexation at interfaces. For example, the adsorption of sodium poly(acrylic acid) (PAA), and dodecyltrimethylammonium bromide (C12TAB) at the air–water interface was studied using a combination of neutron reflectivity and surface tension.43 They showed that at pH 4.2 and 9.2 the addition of the polyanion results in a substantial reduction in the adsorption of polymer–surfactant complexes at the interface. At pH 4.2 a mixed polymer–surfactant monolayer (∼20 Å thick) is adsorbed at the interface over the entire surfactant concentration range measured. At high pH, where the polyelectrolyte is highly charged, their results imply that at the lower surfactant concentrations the adsorption is dominated by a hydrophobic interaction, whereas at higher surfactant concentrations it is dominated by the electrostatic interaction.
Fluorescence spectroscopy has yielded structural information on polymer–surfactant complexes.47–49 Fluorescent dyes can be added directly to polymer–surfactant mixtures, allowing determination of the cac, surfactant aggregation number, and the micro-environment of the complexes. Fluorophores have also been covalently attached to polyelectrolytes, serving as a polymer-bound label. These studies have revealed large differences in aggregation numbers of surfactants containing a polyelectrolyte compared to polymer-free solutions.47,48,50 Furthermore, surfactant complexation has dramatic effects on monomer and excimer emission, and some studies have shown how surfactant aggregation on the polyelectrolyte can favor excimer formation.51,52 For instance, Winnik et al. used pyrene-labeled PEI to investigate SDS complexation using protonated and neutral forms of PEI.53 By measuring the monomer and excimer emission of pyrene, they showed that surfactant aggregates of SDS form with the PEI. These aggregates solubilize the pyrene groups and effectively shield them from being quenched by the protonated amine groups of PEI.
Surfactants on polyion surfaces: wettability studies
The surface wettability of sequentially adsorbed polyelectrolyte multilayers is determined primarily by the electrostatic and hydrophobic properties of the terminal surface layer.54,55 Experimentally, solution pH and ionic strength can directly influence the polyelectrolyte charge density, layer thickness, packing density, and surface wettability of multilayered systems. Rubner and co-workers have measured water contact angles for various polyelectrolyte multilayers assembled from the polycation poly(allylaminehydrochloride) (PAH) and the polyanion poly(acrylic acid) (PAA). They measured the contact angles both as a function of ionic strength (Fig. 3a) and as a function of pH (Fig. 3b).56Fig. 3 also highlights the difference in wettability between the polycation and the polyanion. There is almost a 30° difference in the contact angle between the two different polyelectrolyte surfaces. The addition of salt in the PAH solution causes an elevation in the contact angle of the PAH surfaces. | ||
| Fig. 3 (a) Contact angle measurements of a PAH–PAA film as a function of layer number. Even numbers represent films with PAA as the outermost layer. There is almost a 30° difference in the contact angle between the two different polyelectrolyte surfaces. The addition of salt in the PAH solution causes an elevation in the contact angle of the PAH surfaces. (b) The effect of pH on wettability. Even numbers represent films with PAA as the outermost layer. Reprinted with permission from ref. 56, copyright American Chemical Society. | ||
As mentioned previously, the presence of an oppositely charged polyelectrolyte reduces the surfactant concentration needed to induce micellization to a few orders of magnitude below the cmc. Similar complexation and aggregation patterns may be expected in multilayered LbL polyelectrolyte–surfactant systems, and the hydrophobic moieties in surfactants can be used to control the wettability of polyelectrolyte films. Generally, such a system can be described as [polycation–anionic surfactant]n, where n is the number of ion-pair bilayers. The relationship between bulk phase cac behavior and LbL surfactant–polyelectrolyte aggregation has only recently been investigated. For instance, one study has explored the wetting properties of PEI surfaces produced by ESA with increasing amounts of adsorbed SDS.57 The multilayered system [PEI–PAZO–PEI–SDS] was assembled on a glass slide, with polyelectrolytes adsorbed from 1 mM aqueous solutions, and the SDS adsorbed from solutions of variable concentration (Fig. 4). The polyanion poly[1-[4-(3-carboxy-4-hydroxyphenylazo)benzenesulfonamido]-1,2-ethanediyl, sodium salt] (PAZO) was used as a spacer between two cationic PEI layers to increase the distance between the terminal PEI cationic layer and the underlying glass substrate, thus mitigating a potential substrate-related influence on the film properties. These studies showed that the dynamic advancing contact angle of a pure PEI surface bound to glass is ∼30°. This contact angle increases with SDS adsorption. Saturated adsorption of SDS from the aqueous phase onto the PEI surface requires approximately 100 s (Fig. 5a). The contact angle reaches a limiting value of ∼80° when the surfactant is adsorbed from a concentration, ∼0.01 mM, corresponding to the maximum coverage of the surfactant on PEI (Fig. 5b). The observed increase in contact angle suggests that the anionic SDS headgroups are buried in the PEI layer, while the hydrophobic tail groups are exposed at the surface. This work also showed that the initial PEI–PAZO–PEI surface films consist mainly of interpenetrated layers of PEI and PAZO. The outer PEI layer, electrostatically driving the SDS adsorption, is mainly interpenetrated into the PAZO layer due to electrostatic interactions. On a 38 Å terminal PEI layer, only 6 ± 2 Å consist of pure PEI.21 This value can be obtained by measuring both the wettability and the thickness changes upon SDS adsorption (Fig. 5c).57 The thickness of the adsorbed terminal PEI layer needs to be sufficiently large to ensure that there is a small layer of pure PEI to drive SDS or PAZO adsorption. The strong affinity of SDS to PEI surfaces has also been recently observed using neutron reflectivity.46
 | ||
| Fig. 4 Schematic of the PEI–PAZO–PEI trilayer film, containing adsorbed SDS. The thickness of the interpenetrated region was estimated from contact angle and ellipsometric thickness measurements (see ref. 57). Structures of both PEI and PAZO are also shown. Reprinted with permission from ref. 57, copyright American Chemical Society. | ||
 | ||
| Fig. 5 (a) Advancing contact angles of the PEI–PAZO–PEI–SDS surface as a function of adsorption time of SDS. (b) Advancing contact angles of the PEI–PAZO–PEI–SDS surface as a function of concentration used to adsorb SDS. The concentration used to adsorb both the PEI layer and PAZO layer in the PEI–PAZO–PEI trilayers was fixed at 1 mM. The concentration used to adsorb SDS in (a) was fixed at 0.001 mM. (c) Advancing contact angle of the PEI–PAZO–PEI surface as a function of concentration used to adsorb PEI (○). Ellipsometric thickness measurements as a function of concentration used to adsorb PEI (△). The concentration used to adsorb both the first (inner) PEI layer and the PAZO layer in the PEI–PAZO–PEI trilayers was fixed at 1 mM. Reprinted with permission from ref. 57, copyright American Chemical Society. | ||
The concentration of SDS required to attain saturated coverage on PEI is surprisingly low, three orders of magnitude below the cmc of SDS (∼8 mM).58 This observation raises the question whether the observed saturation concentration of SDS (0.01 mM) might correspond to the cac of SDS in the bulk phase with PEI. Despite the fact that complexation of PEI and SDS was suggested over 30 years ago,32,59 few studies have focused on the cac of PEI–SDS mixtures. Fig. 6 displays the surface tension behavior of the PEI–SDS system.60 The onset of polymer-assisted micellization is 3 orders of magnitude below the accepted cmc of pure aqueous SDS. The cac value (∼10−3 mM) is close to the point of saturation of SDS onto the PEI surface in the ESA system. The same electrostatic stabilizing effect of PEI observed for bulk phase micellization may also operate in the LbL system. Ultimately, the data show that the wettability of polyelectrolyte surfaces may be modulated or fine-tuned by surfactants in the vicinity of the cac.
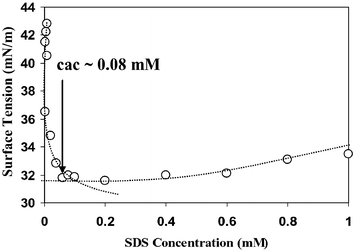 | ||
| Fig. 6 Surface tension of aqueous SDS as a function of concentration. PEI was present in all solutions at a concentration of 1 mM. The critical aggregation concentration of SDS in PEI is observed at ∼0.08 mM. | ||
In an interesting study, Thünemann et al. spin-cast multilayer complexes of PEI and a partially fluorinated C10 carboxylic acid surfactant onto silicon wafers.35,61 They measured the dynamic contact angles for the assemblies using multiple test liquids of varying surface tension. The Neumann and Li62 model was used to calculate the surface free energy,
 | (1) |
 | ||
| Fig. 7 (a) Zisman plots for complex-coated silicon wafers. The various test solutions were hexane, octane, decane, dodecane, and hexadecane. (b) The Girifalco–Good–Fowkes–Young plots for the determination of the dispersive surface free energy. (c) X-Ray reflectivity curve of the film on silicon. The inset shows a sketch of the film structure in the direction vertical to the substrate surface. Reprinted with permission from ref. 35, copyright American Chemical Society. | ||
The competitive adsorption of a similarly charged polyion and surfactant onto an oppositely charged polyion surface can lead to mixed surfactant–polyion surfaces with interesting wetting properties. For example, a recent study investigated the co-adsorpion of PAZO and SDS onto a PEI surface using both the ESA and spin-assembly methods.68 This generated a mixed surface containing polyanion and SDS. There exists a noticeable difference between the contact angle measurements of pure PEI and PAZO layers, demonstrating that the surface properties of the two materials are not the same. An increase in the contact angle of the PAZO surfaces was observed (from 64 ± 2° to 74 ± 2°) as the SDS concentration was increased from 0 to 10 mM, indicating that the surface was made more hydrophobic. This is in agreement with the conclusion that both SDS and PAZO adsorb to the substrate's surface, with both species competing for PEI binding sites. The increasing hydrophobicity of the PAZO surface upon SDS co-adsorption supported the conclusion that changes in PAZO adsorption kinetics are directly influenced by SDS on the surface of the film. The anionic headgroups of SDS bind to positive sites on the PEI layer, leaving the hydrophobic hydrocarbon tails exposed, which would increase the hydrophobicity of the PAZO–SDS composite surface.
Polyelectrolyte–surfactant complexes often contain isolated polar (polyelectrolyte chain and surfactant headgroup) and apolar (hydrophobic tail) domains.64 Similar to the contact angle behavior for the PAZO outerlayer, the contact angle of PEI layers also increased when higher concentrations of SDS were used in the SDS–PAZO solution. However, for the PEI layers the contact angle increased from 57 ± 2° to 65 ± 2° as the concentration of SDS was increased from 0 mM to 10 mM. The smaller increase in contact angle for the PEI layer compared to the PAZO layer indicated that the hydrophobic alkyl chains of SDS are not as prevalent at the PEI surface. When PEI is added onto a coadsorbed SDS–PAZO layer, the alkyl chains of the SDS mixed with the PAZO likely interpenetrate through the new PEI layer, causing the slight increase in surface hydrophobicity.
LbL polyelectrolyte multilayer assemblies containing surfactants
Modified surfactants can be quickly and conveniently integrated into polyelectrolyte multilayered assemblies to generate materials with useful functionality. For example, azobenzene-derived surfactants have been studied in LbL assemblies as substitutes for vesicular membranes for control of membrane permeability65 and, more recently, for mechanosensitive protein channels.66 Another example involves a class of novel NLO-active surfactants incorporated into LbL assemblies for application in non-linear optics.67 Understanding the fundamental mechanism of polyelectrolyte–surfactant complexation in LbL systems should yield improved strategies for immobilizing functional amphiphiles within such assemblies.The internal structure of polyelectrolyte–surfactant complexes in ESA films is an important feature governing the overall layer-by-layer assembly in such films. In particular, the surfactant content and the degree of association between surfactant and polyelectrolyte will determine factors such as multilayer film thickness and packing density. The binding of surfactant to polyelectrolyte surfaces has been observed to proceed cooperatively in some systems, but establishing the presence of micelles within such assemblies is usually challenging. In a recent study, pre-adsorbed polyelectrolyte films were exposed to surfactant solutions to obtain information about long-range and adhesive forces in the polyelectrolyte layer.70,71 In these experiments, mica surfaces were coated with the polycation PCMA, poly((2-acryloxyethyl)trimethylammonium chloride) and then exposed to dilute SDS. Exposure to SDS concentrations of 0.01 to 0.02 times the cmc did not result in any change in the long-range interactions or desorption forces. At these low SDS concentrations, the adsorption of the surfactant was very limited. As the SDS concentration in solution was increased to 0.1 times the cmc, a long-range repulsive double-layer force could be observed. This force was overcome by an attraction at a separation of 110 Å, in which the surface is pulled inwards to a separation of 40 Å. This indicates a dense layer structure containing both polyelectrolyte and surfactant. The pull-off force of 25–30 mN m−1 is significantly lower than that observed for lower SDS concentrations (50–80 mN m−1). As the SDS concentration was increased further to 0.2 times the cmc, oscillations of the force curve were observed. These oscillations were further amplified when the SDS concentration was increased to the cmc, and had a periodicity of ∼40 Å. The repulsive double-layer force is the dominant force in these systems. These experiments demonstrate that the polyelectrolyte layer swells due to association with SDS, once 0.1 times the cmc concentration has been reached. At this point, some of the polyelectrolyte chain desorbs from the surface and the internal structure of the adsorbed layer changes to minimize the free energy of the system. The periodicity of the force profile is the same as that for bulk aggregates, suggesting that the internal structure of the adsorbed layer contains polyelectrolyte-stabilized rod-like micelles that are aligned parallel to the surface.
In addition to the formation of surfactant aggregates within polyelectrolyte films, pre-formed polyelectrolyte–surfactant complexes in the bulk have been found to directly adsorb on solid surfaces. For instance, the adsorption of polyelectrolyte–surfactant complexes onto a mica surface is initially rapid, resulting in a thick layer.70 The polyelectrolyte initially binds to the surface with multiple segments that have a large affinity for the surface, causing limited polyelectrolyte chain mobility, and the complexes take a long time to spread evenly on the surface. Equilibrium in these systems is reached slowly, sometimes after a period of several days. Due to the rate at which the complexes reach equilibrium on the surface, polyelectrolyte–surfactant aggregates are often trapped in non-equilibrium states. This phenomenon leads to an uneven distribution of material on the surface, compared to the more even adsorption of surfactant onto a pre-adsorbed polyelectrolyte layer.71
The effective formation of multilayers requires available binding sites on the terminal layer. In this respect, surfactant concentration will have an impact on multilayer growth. For instance, adsorption of a stoichiometric amount of SDS on a PEI terminal layer results in a lack of available cationic sites on PEI for further adsorption by ESA. However, SDS occupation of electrostatic binding sites does not preclude adsorption through hydrophobic or other interactions. This is demonstrated by our recent work with repeat trilayers of [PEI–SDS–PAZO]n (n = 1 to 10).60 In order to first understand film growth patterns without SDS, conventional multilayered films were constructed from pure PEI and PAZO using the LbL approach. Fig. 8 shows the UV-visible absorbance and ellipsometric thickness measurements of 10 bilayers of a PEI–PAZO film. The absorbance and thickness grow linearly, suggesting equivalent polymer deposition in each bilayer.
 | ||
| Fig. 8 Maximum absorbances at PAZO's λmax (= 360 nm) (△) and ellipsometric thicknesses (○) of 10 PEI–PAZO bilayers. | ||
Ten trilayers composed of PEI, SDS, and PAZO were constructed by the LbL approach. The UV-visible absorbance and the ellipsometric thickness were measured for SDS concentrations of 0 mM, 0.0001 mM, 0.001 mM, 0.01 mM and 0.1 mM. The concentrations used to adsorb PEI and PAZO were fixed at 1 mM (Fig. 9(a) and Fig 9(b)).60 Both the UV-absorbance and the ellipsometric thickness increase linearly as the number of bilayers increases. At very low SDS concentration, the amount of adsorbed PAZO decreases with increasing SDS concentration. This result was observed both for ionically self-assembled films and spin-assembled films. The amount of adsorbed PAZO reaches a minimum at an SDS concentration close to its cac (∼10−3 mM, as measured in the presence of 1 mM PEI). Beyond this concentration, increasing the concentration of SDS (up to 0.1 mM) results in films of relatively large thickness and absorbance.
 | ||
| Fig. 9 (a) Maximum absorbances at PAZO's λmax (= 360 nm) of 10 PEI–SDS–PAZO trilayers. Concentrations used to adsorb PEI and PAZO were fixed at 1 mM, while the ones used to adsorb SDS varied (○ 0 mM; □ 0.0001 mM; ▲ 0.001 mM; × 0.01 mM; ● 0.1 mM). (b) Ellipsometric thicknesses of 10 PEI–SDS–PAZO trilayers. Concentrations used to adsorb PEI and PAZO were fixed at 1 mM while the ones used to adsorb SDS varied (○ 0 mM; □ 0.0001 mM; ▲ 0.001 mM; × 0.01 mM; ● 0.1 mM). | ||
The observed decrease in PAZO adsorption up to the cac can be rationalized by considering the available binding sites in PEI. SDS molecules take up binding sites on the PEI surface, reducing the amount of PAZO adsorbed to the surface. The thickness and absorbance measurements suggest that the maximum number of amphiphilic molecules adsorb to the PEI surface when the concentration is at the cac. Above the cac (0.1 mM SDS), the thickness increases. Interestingly, the absorbance and thickness of the [PEI–SDS–PAZO]10 system, when [SDS] ∼0.1 mM, is the same as the [PEI–PAZO]10 system (thickness of about 350 Å and absorbance of 0.35), implying little or no SDS in the trilayer assemblies. Above the cac, adsorbed SDS would produce a strongly hydrophobic surface on the PEI. A strongly hydrophobic surface exposed to an aqueous phase is thermodynamically unstable. Rearrangement is likely to occur, and the film will be susceptible to desorption, especially in the presence of a similarly charged macromolecular species competing for adsorption. It is likely that near the cmc, PAZO displaces SDS into the bulk phase. Thus, films are formed with little or no SDS.
Taking a different approach, the SDS–polyelectrolyte multilayered system has also been investigated through the co-adsorption of SDS and PAZO from a common solution onto a PEI surface. In these studies, both SDS and PAZO were present in the bulk phase and SDS adsorption on PEI was examined over a concentration range spanning the surfactant's aqueous cmc.68 UV-visible absorbance (Fig. 10(a)) and ellipsometric thickness (Fig. 10(b)) measurements were taken for SDS concentrations of 0 mM, 0.1 mM, 1 mM, 3 mM, 5 mM, 8 mM and 10 mM with the PAZO concentration fixed at 1 mM. Co-adsorption of both SDS and PAZO was observed, which was attributed to strong electrostatic interactions. In this case, the desorption of SDS may have been limited by the presence of SDS in the bulk PAZO–SDS mixture. It was shown that increasing the concentration of SDS in the mixture resulted in films containing progressively less adsorbed PAZO, and no further PAZO reduction was observed above the SDS cmc (∼8 mM). This result is consistent with the maximum amount of amphiphilic molecules adsorbing to a surface when the concentration is at the cmc.69
 | ||
| Fig. 10 Co-adsorption of SDS and PAZO onto a pre-adsorbed PEI film. For ionically self-assembled films. (a) Absorption at λmax for the fourth bilayer as a function of SDS concentration. (b) Ellipsometric thickness of the fourth bilayer as a function of SDS concentration. Similar results were obtained for spin-assembled films. Reprinted with permission from ref. 68, copyright American Chemical Society. | ||
In these studies, co-adsorption of SDS onto PEI was shown to occur by a fast adsorption step (<100 s), followed by a slower adsorption of PAZO onto the remaining unoccupied cationic binding sites on PEI.68 The initial rate of PAZO adsorption on the PEI surface depended on the amount of SDS available to the surface. At low SDS concentrations the initial rate of PAZO adsorption was high, and this rate rapidly decreased as the SDS concentration was increased. The adsorption of PAZO on the PEI surface was found to saturate at around 5 minutes regardless of the SDS concentration. The effect of the SDS concentration on the PAZO adsorption kinetics indicates that SDS exists at the PEI surface prior to PAZO adsorption, caused largely by the high number of SDS molecules in solution and the high ionic mobility of SDS as compared with that of PAZO.
Although there are large differences in substrate–solution contact times, similar behavior in the co-adsorption of PAZO and SDS is observed in both spin-assembled and ESA systems. In particular, the presence of SDS in spin-assembled films reflects the rapid diffusion time for SDS and subsequent complex formation with PEI, even though these spin-assembled films may not be at equilibrium.24 Since the contact time in spin-assembly is fractions of a second, the SDS must adsorb quickly to the surface. At a 10 mM concentration of SDS, slightly more PAZO adsorbs in the spin-assembled than the ionically self-assembled system. In the spin-assembled film, polymer chain entanglement plays a greater role in deposition compared to the ESA system, which is driven by electrostatic interactions and entropic favorability. Therefore, at 10 mM SDS, more polymer is drawn to the surface when one chain entangles with the underlying PEI layer during spin-assembly. In electrostatic self-assembly, only an equilibrium amount of PAZO can adsorb.
Others have investigated the effect of surfactant concentration on the composition of co-adsorbed films. For example, in two studies by Dedinaite et al.,70,71 the polycation poly[2-(propionyloxy)ethyl]triethylammonium chloride (PMCA) and the negatively charged SDS were used to explore the adsorption of polyelectrolyte–surfactant mixtures. They compared the co-adsorbing system to the adsorption of surfactant onto a pure polyelectrolyte layer using XPS. Below a certain surfactant concentration (for the PMCA–SDS system this concentration was 0.01 times the cmc of SDS), there was no observable amount of surfactant detected in the layer. At higher SDS concentrations, a large amount of SDS was observed to be trapped in the adsorbed layer, and was found to be unaltered by dilution or transport through the air–solution interface.70
When the surface properties of a film fabricated by the adsorption of surfactant from a polyelectrolyte-free solution onto a pre-adsorbed polyelectrolyte layer were compared with those of a film fabricated by the co-adsorption of polyelectrolyte and surfactant from a bulk solution, several differences were observed. The first observed difference between the two films was the effect of surfactant concentration on the amount of SDS adsorbed to the surface. When the polyelectrolyte layer was pre-adsorbed, the adsorbed amount slowly decreased with SDS concentration, while a pronounced maximum was observed as the surfactant concentration was increased when the polyelectrolyte–surfactant complex was allowed to form in bulk.
Another difference between the two films was the surfactant concentration at which PCMA and SDS began to associate. The association between pre-adsorbed PCMA layers and SDS started at a surfactant concentration of about 0.1 times cmc. However, in bulk solution large aggregates are already formed at about 0.02 times cmc, and these aggregates adsorb to the surface to form thick, viscous layers. Finally, the association between pre-adsorbed PCMA layers and SDS at concentrations above 0.1 times the surfactant cmc resulted in an ordered adsorbed layer. No similar ordered arrangement was observed when polyelectrolyte–surfactant aggregates were adsorbed to the surface from a bulk solution.
LbL ESA assembly has demonstrated potential for optical materials and devices through the integration of photochromic species in mutilayered films. In particular, adding optical functionalities to amphiphilic molecules allows facile integration of these functionalities into polyelectrolyte films. Azo-benzene surfactants have been incorporated into bilayer and multilayer assemblies to exploit their photoisomerization, H–J aggregation, and nonlinear optical (NLO) properties.72–75 These NLO amphiphiles contained active chromophores at the alkyl chain terminus and are believed to form highly ordered structures similar to the SDS–PEI multilayered systems.
In a recent study, multilayered LBL assemblies of X-azo-O(CH2)10-SO3− and PEI were formed, where X was an electron-withdrawing group, either -NO2, -CN, or -COCH3 (Fig. 11). The electron-withdrawing group causes a change in the electron donor–π–electron acceptor properties of the azobenzene-derived surfactant.67 When the surfactants are incorporated into films with the SO3− headgroup anchored in the PEI layer, they show a prominent blue shift in the UV-visible spectrum from that of the bulk aqueous phase. These shifts are due to the presence of non-covalent interactions between the azobenzene chromophores in aggregated systems, normally absent in dilute solutions. The blue shift is attributed to H-type aggregation, in which the transition dipoles are arranged in a “head-to-head” arrangement. The J-type, or “tail-to-tail” arrangement would correspond to a red shift in the absorption spectrum.
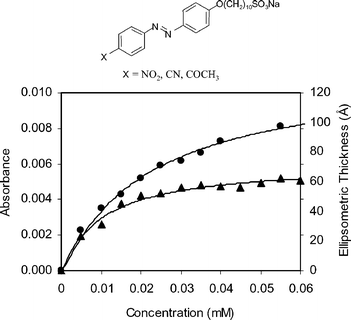 | ||
| Fig. 11 (a) The general structure of X-azo-(CH2)10-SO3−, where X is an electron-withdrawing group. (b) The absorbance at λmax (●) and ellipsometric thickness (▲) as a function of O2N-azo-(CH2)10-SO3− surfactant concentration. The solid line between the data points represents a Langmuir fit. Reprinted with permission from ref. 67, copyright American Chemical Society. | ||
In this work, adsorption kinetics of the azobenzene surfactants were monitored using dynamic force measurements and UV-visible spectroscopy. The dynamic force data indicated that an equilibrium amount of PEI is adsorbed in ∼150 s and an equilibrium amount of CN-azo-O(CH2)10-SO3− is adsorbed in ∼140 s. The UV-visible adsorption yielded a similar saturation adsorption value of the fifth layer of CN-azo-O(CH2)10-SO3−, and a shorter saturation adsorption time for the first surfactant layer (∼100 s). Coverage of the NO2- and CN-containing surfactants onto the PEI surface increases as a function of surfactant concentration in solution. As shown in Fig. 11, this adsorption pattern was found to follow Langmuir adsorption behavior.
Multilayered LbL [PEI–X-azo-O(CH2)10-SO3−] films were constructed by sequential adsorption of PEI and surfactant solutions on both glass and silicon substrates. Absorbance at λmax and the ellipsometric thickness for CN- and NO2-containing surfactants increased linearly with bilayer number (Fig. 12). The LbL system containing CH3CO-azo-O(CH2)10-SO3− also showed a linear increase of adsorbed surfactant with the layer number; however, the total amount of surfactant adsorbed was less than that of the other surfactants. This is likely to be due to the less polar nature of the acetyl headgroup compared to the CN and NO2 headgroups of the other surfactants, suggesting an interesting relationship between headgroup polarity and deposition amount. The adsorption studies yield further proof that film formation is a result of electrostatic physisorption of the surfactant, and is not due to aggregation.
 | ||
| Fig. 12 (a) Absorbance at λmax for films containing CN-azo-(CH2)10-SO3− (●) and O2N-azo-(CH2)10-SO3− (▲) (left axis) and CH3CO-azo-(CH2)10-SO3− (□) (right axis). (b) Ellipsometric thickness for films containing CN-azo-(CH2)10-SO3− (●) and O2N-azo-(CH2)10-SO3− (▲). Reprinted with permission from ref. 67, copyright American Chemical Society. | ||
The presence of an electron donor–π–electron acceptor system in the surfactant offers the potential for creating LbL nonlinear optical (NLO) materials. The challenge is to fabricate unidirectionally oriented layers of these surfactants within the polyelectrolyte assemblies. Second harmonic generation (SHG) is typically used to probe the NLO-properties of such materials. SHG measurements of the aforementioned films indicate that the bilayer containing X = -NO2 yields the largest signal, followed by X = -CN and X = -COCH3 (Fig. 13). The electron donor–π–electron acceptor strength is proportional to the degree of unidirectional alignment of the amphiphile in the film. When multilayered films containing X = -NO2 and X = -CN were studied, the first bilayer yields the largest SHG response. The response falls dramatically with increasing layer number, demonstrating that the first bilayer is the most asymmetrically ordered, with the -SO3− headgroup anchored into the PEI layer (forming ion pairs), and the hydrophobic interactions of the C10 chains helping to orient the NLO-active headgroups. The decrease in the SHG response is likely to be due to interpenetration of subsequent layers, yielding a film with no net asymmetric alignment of NLO-active headgroups. Highly interpenetrated films contain no discrete layer structure.
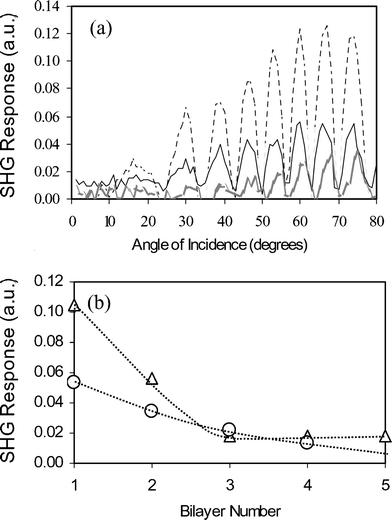 | ||
Fig. 13 (a) The SHG intensity as a function of the incidence angle for a single bilayer of PEI–O2N–azo–(CH2)10–SO3− (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ), PEI–CN-azo-(CH2)10–SO3− (—), and PEI–CH3CO-azo-(CH2)10–SO3− ( ), PEI–CN-azo-(CH2)10–SO3− (—), and PEI–CH3CO-azo-(CH2)10–SO3− (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ). (b) The SHG response at ∼60° angle of incidence (6th fringe) as a function of layer number for the O2N-azo-(CH2)10–SO3− (△) and CN-azo-(CH2)10-SO3− system (○). Reprinted with permission from ref. 67, copyright American Chemical Society. ). (b) The SHG response at ∼60° angle of incidence (6th fringe) as a function of layer number for the O2N-azo-(CH2)10–SO3− (△) and CN-azo-(CH2)10-SO3− system (○). Reprinted with permission from ref. 67, copyright American Chemical Society. | ||
The presence of an optical chromophore on the surfactant allows the determination of adsorbed amount within mixed films, which further allows conclusions to be drawn regarding cooperative and non-cooperative binding of surfactants to polyions. Adsorption behavior can often be described using very simple models. For example, the binding of CN-azo-O(CH2)10-SO3− to positive sites on PEI is found to be non-cooperative using the Hill equation. Adsorption of CN-azo-(CH2)10-SO3− on PEI can be described by the equilibrium PEI + AZO ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) PEI–AZO, characterized by the intrinsic equilibrium binding constant K = [PEI–AZO]/[PEI][AZO]. If the N binding sites are independent and identical, the intrinsic binding constant is related to the fraction (f) of positive sites on PEI occupied by CN-azo-O(CH2)10-SO3− as given by the Scatchard equation,
PEI–AZO, characterized by the intrinsic equilibrium binding constant K = [PEI–AZO]/[PEI][AZO]. If the N binding sites are independent and identical, the intrinsic binding constant is related to the fraction (f) of positive sites on PEI occupied by CN-azo-O(CH2)10-SO3− as given by the Scatchard equation,
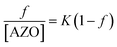 | (2) |
 | (3) |
 | (4) |
![(a) Hill plot for CN-azo-(CH2)10-SO3−. There are two distinct linear regions, with slopes of ∼1 and ∼3. The binding constants corresponding to these two regions can be obtained from the Scatchard plot of f/(1 −
f) versus [AZO]. (b) The non-cooperative binding has K = 74. However, after 0.03 M, K changes with concentration.](/image/article/2007/SM/b609045c/b609045c-f14.gif) | ||
| Fig. 14 (a) Hill plot for CN-azo-(CH2)10-SO3−. There are two distinct linear regions, with slopes of ∼1 and ∼3. The binding constants corresponding to these two regions can be obtained from the Scatchard plot of f/(1 − f) versus [AZO]. (b) The non-cooperative binding has K = 74. However, after 0.03 M, K changes with concentration. | ||
It is worth noting the effect of surfactants on the optical properties of polymers within polyelectrolyte assemblies. Polyelectrolyte–surfactant complexes have very different geometric conformations than the free polymer.76–79 Therefore, surfactants may be used to tune the optical and chemical properties by changing the conformation of conjugated polymers. As an example, we recently constructed LbL films (both by ESA and spin-assembly) of the cationic surfactant didodecyldimethylammonium bromide (DDAB) on poly[2-methoxy-5-(propyloxy sulfonate)phenylene vinylene] (MPS-PPV).60Fig. 15 demonstrates that increasing the amount of adsorbed surfactant changes the photoluminescence emission properties of MPS-PPV. The effect has also been observed in the bulk phase. For instance, Chen et al. have reported complexes between MPS-PPV and a series of cationic surfactants.80 They report a large increase in the fluorescence quantum yield of MPS-PPV by adding small amounts of the surfactant dodecyltrimethylammonium bromide (DTA) to the aqueous polymer solution. Fig. 16 shows how the fluorescence intensity and the quenching constant of aqueous solutions of MPS-PPV change as a function of added DTA. They show that non-radiative decay pathways in the polymer are essentially eliminated upon surfactant complexation.
 | ||
| Fig. 15 The effect of increasing DDAB content on the emission properties of an MPS-PPV layer. The data shown corresponds to an LbL film formed using ESA. (a) A much greater change in the emission wavelength occurs with increasing DDAB content compared to the absorption wavelength. (b) A minimum in the relative PLQE occurs near the surfactant cmc. | ||
 | ||
| Fig. 16 Fluorescence intensity of MPS-PPV as a function of DTA concentration (○). Reprinted with permission from ref. 80, copyright American Chemical Society. | ||
Conclusions
A complex interplay between various non-covalent interactions leads to polyelectrolyte–surfactant aggregation in electrostatically bound polyelectrolyte multilayered architectures and in aqueous solution. Such interactions can be exploited to provide polyelectrolyte surfaces of controlled wettabilty, or multilayered assemblies with remarkable optical and chemical properties. Modified surfactants containing useful functionalities can therefore be quickly and conveniently integrated into multilayered assemblies. Fluorinated surfactants are also becoming important in polyelectrolyte formulations. For example, Antonietti and co-workers have fabricated low energy coating formulations from polyelectrolyte–surfactant complexes with fluorinated surfactants.81 Despite the ease with which ESA can irreversibly immobilize amphiphiles, the complex interactions between polyelectrolytes and surfactants ultimately determine the surface structure, as highlighted in this review. These interactions are strongly influenced by factors such as macromolecular charge density, ionic strength of adsorbate solutions, the degree of polyelectrolyte interpenetration within multilayers, hydrophobicity of the surfactant and the temperature during film fabrication. The nature of these interactions is an important topic of current research. As our understanding of polyelectrolyte–surfactant interactions evolves, so too will our ability to construct complex materials for specific functions.Acknowledgements
We thank Miss Raea Hicks and Miss Talya Dayton for obtaining some of the unpublished data presented in this review. We also thank Miss Sarah Sherlock for her assistance.References
- E. D. Goddard and K. P. Ananthapadmanabhan, in Polymer–Surfactant Systems, ed. J. C. T. Kwak, Marcel Dekker, Inc., New York, 1998, pp. 22–64 Search PubMed.
- B.-H. Lee, S. D. Christian, E. E. Tucker and J. F. Scamehorn, Langmuir, 1991, 7, 1332 CrossRef CAS.
- Interactions of Surfactants with Polymers and Proteins, ed. E. D. Goddard and K. P. Ananthapadmanabhan, CRC Press, Boca Raton, 1993 Search PubMed.
- I. Iliopoulos, Curr. Opin. Colloid Interface Sci., 1998, 3, 149.
- W. J. MacKnight, E. A. Ponomarenko and D. A. Tirrell, Acc. Chem. Res., 1998, 31(12), 781–788 CrossRef CAS.
- K. Holmberg, B. Jönsson, B. Kronberg and B. Lindman, Surfactants and Polymers in Aqueous Solution, 2nd edn, John Wiley & Sons, New York, 2002 Search PubMed.
- J. C. T. Kwak, Polymer–Surfactant Systems (Surfactant Science Series), CRC Press, Boca Raton, 1998 Search PubMed.
- T. Sato, Stabilization of Colloidal Dispersions by Polymer Adsorption (Surfactant Science Series; vol. 9), Marcel Dekker, New York, 1980 Search PubMed.
- J. Goodwin, Colloids and Interfaces with Surfactants and Polymers, John Wiley & Sons, New York, 2004 Search PubMed.
- I. Satake and J. T. Yang, Biopolymers, 1976, 15, 226.
- T. K. Bronich, T. Cherry, S. V. Vinogradov, A. Eisenberg, V. A. Kabanov and A. V. Kabinov, Langmuir, 1998, 14, 6101 CrossRef CAS.
- A. F. Thünemann, Langmuir, 2000, 16, 824 CrossRef.
- T. Wallin and P. Linse, J. Phys. Chem., 1996, 100, 17873 CrossRef CAS.
- T. Wallin and P. Linse, Langmuir, 1996, 12, 305 CrossRef CAS.
- G. Decher, J.-D. Hong and J. Schmitt, Thin Solid Films, 1992, 210–211, 831–835 CrossRef.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- P. Hammond, Adv. Mater., 2004, 16, 1271 CrossRef CAS.
- M. Müller, J. Meier-Haack, S. Schwartz, H. M. Buchhammer, K.-L. Eichhorn, A. Janke, B. Kessler, J. Nagel, M. Oelmann, T. Reihs and K. Lunkwitz, J. Adhesion, 2004, 80, 521 Search PubMed.
- Multilayer Thin Films: Sequential Assembly of Nanocomposite Materials, ed. G. Decher and J. B. Schlenoff, John Wiley & Sons, New York, 2004 Search PubMed.
- M. S. Johal, M. Howland, J. M. Robinson, J. L. Casson and H.-L. Wang, Chem. Phys. Lett., 2004, 383(3–4), 276 CrossRef CAS.
- M. S. Johal, J. L. Casson, H.-L. Wang, J. M. Robinson and R. J. El-Khouri, Polym. Mater. Sci. Eng., 2003, 89, 841 Search PubMed.
- J. L. Casson, M. S. Johal, J. B. Roberts, H.-L. Wang and J. M. Robinson, J. Phys. Chem. B, 2000, 104, 11996–12001 CrossRef CAS.
- J. B. Schlenoff and S. T. Dubas, Macromolecules, 2001, 34(3), 592–598 CrossRef CAS.
- P. A. Chiarelli, M. S. Johal, J. L. Casson, J. B. Roberts, J. M. Robinson and H.-L. Wang, Adv. Mater., 2001, 13(15), 1167–1171 CrossRef CAS.
- P. A. Chiarelli, M. S. Johal, D. J. Holmes, J. L. Casson, J. M. Robinson and H.-L. Wang, Langmuir, 2002, 18, 168–173 CrossRef CAS.
- V. E. Campbell, P. A. Chiarelli, S. Kaur and M. S. Johal, Chem. Mater., 2005, 17, 186–190 CrossRef CAS.
- J. L. Casson, M. S. Johal, P. A. Chiarelli, D.-G. Liu, J. A. Shaw, J. M. Robinson and H.-L. Wang, Polym. Mater. Sci. Eng., 2003, 89, 33–34 Search PubMed.
- M. S. Johal, J. L. Casson, P. A. Chiarelli, D.-G. Lui, J. A. Shaw, J. M. Robinson and H.-L. Wang, Langmuir, 2003, 19(21), 8876 CrossRef CAS.
- N. M. Jones, J. Colloid Interface Sci., 1967, 23, 36 CAS.
- J. C. T. Kwak, Polymer–Surfactant Systems, Marcel Dekker, New York, 1998 Search PubMed.
- P. Deo and P. Somasundaran, Langmuir, 2005, 21, 3950–3956 CrossRef CAS.
- J. W. A. Van den Berg and A. J. Staverman, Recl. Trav. Chim. Pays-Bas, 1972, 91, 1151.
- H. Wang, Y. Wang, H. Yan, J. Zhang and R. K. Thomas, Langmuir, 2006, 22(4), 1526–1533 CrossRef CAS.
- A. F. Thünemann, Langmuir, 2000, 16, 824 CrossRef.
- A. F. Thünemann, S. Kubowicz and S. Pietsch, Langmuir, 2000, 16, 8562 CrossRef.
- P. M. Claesson, M. Bergström, A. Dedinaite, M. Kjellin, J.-F. Legrand and I. Grillo, J. Phys. Chem. B, 2000, 104, 11689 CrossRef CAS.
- R. Mészáros, L. Thompson, M. Bos, I. Varga and T. Gilányi, Langmuir, 2003, 19, 609 CrossRef CAS.
- J. Penfold, D. J. F. Taylor, R. K. Thomas, I. Tucker and L. J. Thompson, Langmuir, 2003, 19, 7740–7745 CrossRef CAS.
- C. Senan, J. Meadows, P. T. Shone and P. A. Williams, Langmuir, 1994, 10, 2471 CrossRef CAS.
- B. Mangy, I. Iliopoulos, R. Zana and R. Audebert, Langmuir, 1994, 10, 3180 CrossRef CAS.
- F. Guillemet and L. Piculell, J. Phys. Chem., 1995, 99, 9201 CrossRef CAS.
- R. Windsor, D. J. Neivandt and P. B. Davies, Langmuir, 2001, 17, 7306–7312 CrossRef CAS.
- J. Zhang, R. K. Thomas and J. Penfold, Soft Matter, 2005, 1(4), 310–318 RSC.
- J. Penfold, I. Tucker and R. K. Thomas, Langmuir, 2005, 21(25), 11757–11764 CrossRef CAS.
- J. Penfold, I. Tucker, R. K. Thomas and J. Zhang, Langmuir, 2005, 21(22), 10061–10073 CrossRef CAS.
- J. Penfold, I. Tucker, E. Staples and R. K. Thomas, Langmuir, 2004, 20(17), 7177–7182 CrossRef CAS.
- J. Fundin, P. Hansson, W. Brown and I. Lidggran, Macromolecules, 1997, 30, 1118 CrossRef CAS.
- K. Thalberg, J. van Stam, C. Lindblad, M. Almgren and B. Lindman, J. Phys. Chem., 1991, 95, 8975 CrossRef CAS.
- F. Guillemet and L. Piculell, J. Phys. Chem., 1995, 99, 9201 CrossRef CAS.
- P. Hansson and M. Almgren, J. Phys. Chem., 1995, 99, 16694 CrossRef CAS.
- F. Quina, E. Abuin and E. Lissi, Macromolecules, 1990, 23, 5173 CrossRef CAS.
- Y.-Z. Hu, C.-L. Zhao, M. A. Winnik and P. R. Sundararajan, Langmuir, 1991, 6, 880.
- M. A. Winnik, S. M. Bystryak and J. Siddiqui, Macromolecules, 1999, 32, 624–632 CrossRef CAS.
- D. Yoo and M. F. Rubner, SPE-Antec 95, Boston, 1995, p. 2568 Search PubMed.
- W. Chen and T. J. McCarthy, Macromolecules, 1997, 30, 78 CrossRef CAS.
- Y. D. Yoo, S. S. Shiratori and M. F. Rubner, Macromolecules, 1998, 31, 4309–4318 CrossRef CAS.
- R. J. El-Khouri and M. S. Johal, Langmuir, 2003, 19, 4880–4883 CrossRef CAS.
- C.-E. Lin, I.-J. Fang, Y.-J. Deng, W.-S. Liao, H.-T. Cheng and W.-P. Huang, J. Chromatogr., A, 2004, 1051(1–2), 85–94 CrossRef CAS.
- D. F. Evans and H. Wennerström, The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet, 2nd edn, John Wiley & Sons, New York, 1999 Search PubMed.
- These results are to be published.
- J. Israelachvili, Intermolecular and Surface Forces, 2nd edn, Academic Press, San Diego, 1992 Search PubMed.
- D. Li and A. W. Neumann, J. Colloid Interface Sci., 1992, 148, 190 CrossRef CAS.
- D. Y. Kwok, C. N. C. Lam, A. Li, A. Leung, R. Wu, E. Mok and A. W. Neumann, Colloids Surf., 1988, 142, 219.
- M. Antonietti, C. Burger and A. F. Thünemann, Trends Polym. Sci., 1997, 5(8), 262 CAS.
- T. Sata, Y. Shimokawa and K. Matsusaki, J. Membr. Sci., 2000, 171, 31 CrossRef CAS.
- J. M. Kuiper and J. B. F. N. Engberts, Langmuir, 2004, 20, 1152 CrossRef CAS.
- N. G. Caculitan, P. H. Scudder, A. Rodriguez, J. L. Casson, H.-L. Wang, J. M. Robinson and M. S. Johal, Langmuir, 2004, 20, 8735–8739 CrossRef CAS.
- M. S. Johal, B. H. Ozer, J. L. Casson, A. St. John, J. M. Robinson and H.-L. Wang, Langmuir, 2004, 20, 2792–2796 CrossRef CAS.
- M. J. Rosen, Surfactant and Interfacial Phenomena, 2nd edn, John Wiley & Sons, New York, 1989 Search PubMed.
- A. Dedinaite and P. Claesson, Langmuir, 2000, 16, 1951–1959 CrossRef CAS.
- A. Dedinaite, P. Claesson and M. Bergström, Langmuir, 2000, 16, 5257–5266 CrossRef CAS.
- M. S. Johal, A. N. Parikh, Y. Lee, J. L. Casson, L. Foster, B. I. Swanson, D. W. McBranch, D. Q. Li and J. M. Robinson, Langmuir, 1999, 15, 1275 CrossRef CAS.
- J. M. Pedrosa, M. T. M. Romero, L. Camcho and D. Möbius, J. Phys. Chem. B, 2002, 106, 2583 CrossRef CAS.
- D. G. Whitten, L. Chem, H. C. Geiger, J. Perlstein and X. Song, J. Phys. Chem., 1998, 102, 10098 Search PubMed.
- T. Kinoshita, J. Photochem. Photobiol., B, 1998, 42, 12 CrossRef CAS.
- W. J. MacKnight, E. A. Ponomarenko and D. A. Tirrell, Acc. Chem. Res., 1998, 31(12), 781–788 CrossRef CAS.
- A. V. Kabanov, T. K. Bronich, V. A. Kabinov, K. Yu and A. Eisenberg, J. Am. Chem. Soc., 1998, 120(38), 9941–9942 CrossRef CAS.
- A. F. Thünemann, Adv. Mater., 1999, 11(2), 127–130 CrossRef.
- K. Faiid and M. Leclerc, J. Am. Chem. Soc., 1998, 120, 3274.
- L. Chen, S. Xu and D. McBranch, J. Am. Chem. Soc., 2000, 122(38), 9302–9303 CrossRef CAS.
- K. H. Lochhaas, A. F. Thünemann and M. Antonietti, Surf. Coat. Int., 1999, 82(9), 451–455 Search PubMed.
Footnote |
| † Current address: Harvard Medical School, Vanderbilt Hall, 107 Avenue Louis Pasteur, Boston, MA 02115-5750, USA. |
| This journal is © The Royal Society of Chemistry 2007 |