Proton translocation by the cytochrome bc1 complexes of phototrophic bacteria: introducing the activated Q-cycle†
Armen Y.
Mulkidjanian
*ab
aA. N. Belozersky Institute of Physico-Chemical Biology, Moscow State University, 119899, Moscow, Russia
bSchool of Physics, University of Osnabrück, D-49069, Osnabrück, Germany. E-mail: amulkid@uos.de; Fax: +49-(541)-9692656; Tel: +49-(541)-9692680
First published on 7th December 2006
Abstract
The cytochrome bc1 complexes are proton-translocating, dimeric membrane ubiquinol:cytochrome c oxidoreductases that serve as “hubs” in the vast majority of electron transfer chains. After each ubiquinol molecule is oxidized in the catalytic center P at the positively charged membrane side, the two liberated electrons head out, according to the Mitchell's Q-cycle mechanism, to different acceptors. One is taken by the [2Fe-2S] iron–sulfur Rieske protein to be passed further to cytochrome c1. The other electron goes across the membrane, via the low- and high-potential hemes of cytochrome b, to another ubiquinone-binding site N at the opposite membrane side. It has been assumed that two ubiquinol molecules have to be oxidized by center P to yield first a semiquinone in center N and then to reduce this semiquinone to ubiquinol. This review is focused on the operation of cytochrome bc1 complexes in phototrophic purple bacteria. Their membranes provide a unique system where the generation of membrane voltage by light-driven, energy-converting enzymes can be traced via spectral shifts of native carotenoids and correlated with the electron and proton transfer reactions. An “activated Q-cycle” is proposed as a novel mechanism that is consistent with the available experimental data on the electron/proton coupling. Under physiological conditions, the dimeric cytochrome bc1 complex is suggested to be continually primed by prompt oxidation of membrane ubiquinol via center N yielding a bound semiquinone in this center and a reduced, high-potential heme b in the other monomer of the enzyme. Then the oxidation of each ubiquinol molecule in center P is followed by ubiquinol formation in center N, proton translocation and generation of membrane voltage.
 Armen Y. Mulkidjanian Armen Y. Mulkidjanian | Armen Y. Mulkidjanian received his academic degrees from the Moscow State University, Russia (PhD in 1984 and Dr. Sc. in 2006) and the University of Osnabrück, Germany (Dr. rer. nat. habil. in Biophysics, 2002). He is currently a Senior Research Scientist at the A. N. Belozerky Institute of Physico-Chemical Biology of the Moscow State University, an Adjunct Professor at the School of Bioengineering and Bioinformatics of the Moscow State University and a Visiting Lecturer at the University of Osnabrück. His research is focused on the molecular mechanisms of proton transfer by the membrane energy-converting enzymes. |
1. Introduction
Cytochrome bc1-complexes of animals and bacteria (hereafter bc1) are oligomeric membrane enzymes that function as quinol:cytochrome c oxidoreductases.1–7 They utilize the free energy of redox reaction to translocate protons across the energy-transducing membrane, from its negatively charged n-side to the positively charged p-side. Proton transfer leads to the formation of difference in electrochemical activity of protons across the membrane (Δ![[small mu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e0.gif) H+). The latter is contributed by chemical (ΔpH) and electrical (Δψ) components and reaches approx. 200–250 mV under physiological conditions.8 Thus, at steady state, bc1 translocates protons against remarkable backpressure.
H+). The latter is contributed by chemical (ΔpH) and electrical (Δψ) components and reaches approx. 200–250 mV under physiological conditions.8 Thus, at steady state, bc1 translocates protons against remarkable backpressure.
Recently bc1 has drawn attention as one of the major sources of reactive oxygen species (ROS), which are formed when oxygen molecules interact with semiquinone radicals that serve as intermediates in the catalytic cycle of bc1. The accumulation of the ROS-damaged cellular compounds is believed to cause aging; it has been reported that the worms Caenorhabditis elegans that carried mutations decreasing the activity of bc1 lived longer.9 Hence, the importance of clarifying the semiquinone chemistry inside the bc1 can be hardly overestimated.
The cytochrome bc1 complex (see Fig. 1) is an intertwined dimer both in crystals5–7,10–15 and in solution.16 The catalytic core of each bc1 monomer is formed by three subunits: the membrane-embedded cytochrome b and the membrane-anchored iron–sulfur Rieske protein and cytochrome c1. Each cytochrome b carries one low- and one high-potential heme (bl and bh, respectively). The number of subunits in cytochrome bc1-complexes varies from only 3 catalytic ones in some bacteria up to 11 in the mitochondrial bc1.
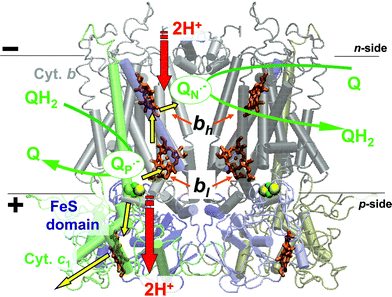 | ||
| Fig. 1 Overview of structure and function of the cytochrome bc1 complex. The scheme shows the Q-cycle scheme20 as plotted over the X-ray structure of a dimeric bc1 of Rb. capsulatus (PDB entry 1ZRT7). Colour code: grey, cytochrome b subunits; blue, the Rieske protein subunits; green, cytochromes c1; orange, hemes; yellow-green, the FeS clusters. Yellow arrows, ET steps; red arrows, proton transfer steps. The figure was produced with the help of the VMD software package.152 | ||
To explain the ability of the bc1-containing proteoliposomes to translocate two protons across the membrane per each oxidized quinol,17,18 Mitchell has suggested the Q-cycle mechanism.19,20 The mechanism invoked a bifurcation of electrons at the site of quinol oxidation, as originally suggested by Wikström and Berden,21 and, in addition, implied that electrons, after passing through cytochrome b, reduce a quinone molecule from the other side of the membrane, as shown in Fig. 1. According to the current, structure-based Q-cycle models,5,10–15,22–26 quinol molecules are oxidized at the interface between cytochrome b and the mobile [2Fe–2S] cluster-carrying domain of the Rieske protein (hereafter the FeS domain, see Fig. 1). This interface forms the catalytic center P of the enzyme (which corresponds to center o in the original Mitchell's notation). One electron is accepted by the FeS domain to be passed further, via cytochrome c1, to external, mobile c-type cytochromes.27 The other electron goes to heme bl and then moves across the membrane, via heme bh, to the further quinone-binding center N (center i in the Mitchell's notation). In center N, a ubiquinone molecule can be reduced first to a semiquinone anion QN˙− and then, after the oxidation of the next ubiquinol in center P, to a QNH2 ubiquinol. This ubiquinol molecule can be oxidized by bc1 as well, so that two charges are ultimately translocated across the membrane per each ubiquinol processed by bc1.
Depending on the presence of inhibitors and on crystallization conditions, the FeS domain was found in different positions, reflecting its rotation by approx. 60°, upon shuttling an electron from center P to cytochrome c1, from a position where the FeS cluster is docked to the heme bl (FeSb) into the position, where the cluster interacts with the cytochrome c1 heme (FeSc).5,7,10–14,28–31
The X-ray structures show that antimycin A (hereafter antimycin), which blocks the oxidation of heme bhvia center N,32,33 binds next to this heme.13,28,34 Confirming the earlier insightful suggestions,35,36 the crystal structures revealed two distinct, although partly overlapping inhibitor-binding sites in center P.5–7,11–15,28–30,37–39 Myxothiazol, methoxy-acrylate (MOA) stylbene-type inhibitors, and a non-oxidisable ubiquinol analogue 2,3,4-trimethoxy-5-decyl-6-methyl-phenol (TMDMP) occupy positions that are proximal to heme bl. Some other inhibitors bind distally, on the interface of cytochrome b and the docked FeS domain. These are, in particular, stigmatellin and 5-n-undecyl-6-hydroxy-4,7-dioxobenzothiazole (UHDBT)-type inhibitors. Stigmatellin, which seems to imitate either ubiquinol or anionic ubiquinone species,5,29,40 binds between His-161 of the Rieske protein and Glu-272 of cytochrome b (to conform with literature in the field, hereafter the avian numeration of amino acid residues is routinely used). Correspondingly, it has been argued that a ubiquinol molecule binds in the same way, and that His-161 and Glu-272 serve as acceptors of the first and second protons, respectively, upon ubiquinol oxidation.5,14,26,29,31,37,41
The Mitchell's idea of doubling the enzyme efficiency by internal electron cycling is generally accepted. The further details on enzyme operation remain, however, controversial, as discussed in several recent reviews.24–26,31,42 Under discussion are (i) the exact mechanism of ubiquinol oxidation and the role of the transiently formed QP˙− semiquinone in this reaction,24–26,31,42,43 (ii) the conformational cross-talk between the quinone-binding centers,22,26,44,45 and (iii) the possibility of electron exchange between the monomers via two bl hemes.10,22,46–49 One more debatable question is the mechanism of electron/proton coupling in bc1. This mechanism is routinely studied with chromatophores—the energosomes of purple phototrophic bacteria. Chromatophores can be obtained, as sealed inside-out vesicles of inner cellular membrane with diameter of 300–600 Å, by disruption of the cells of Rhodobacter sphaeroides and Rhodobacter capsulatus50–52 (see Fig. 2). With these vesicles, the Δψ generation by photosynthetic enzymes can be synchronously triggered by flashes of light, traced via electrochromic spectral changes of native carotenoids50 and correlated with the redox and pH changes measured either under the compatible conditions or even in the same samples.51–53 The flash-induced changes in the redox state of cytochromes and in pH can be monitored optically,50–56 whereas the generation of Δψ can be followed not only optically,50–58 but also by capacitive electrometry.58,59
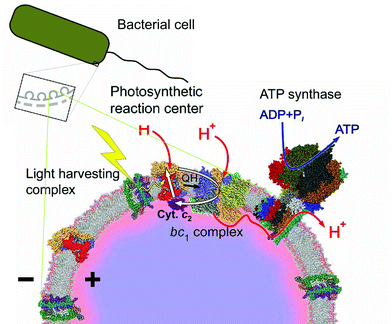 | ||
| Fig. 2 Schematic presentation of electron cycling in a chromatophore membrane (the figure is reproduced from ref. 26 with permission). White arrows depict electron cycling between the RC and the bc1. The red color of the interfacial water layer indicates its higher acidity at steady state, which can increase the protonic backpressure over the cytochrome bc1 complex.153 | ||
Besides providing the possibility to study bc1 in a pulsed mode and to follow the Δψ changes, chromatophores have several other advantages: (i) the cytochrome bc1 complexes of purple bacteria are simple and are made of only 3 or 4 subunits;7,60,61(ii) mutants with site-specific amino acid substitutions in the subunits of the bc1 are available;62 (iii) from an evolutionary perspective, purple bacteria are close to the bacterial ancestors of mitochondria;63 not surprisingly, the X-ray structure of a simple three-subunit bc1 from Rb. capsulatus overlaps with the three core subunits of the mitochondrial bc17 and the same specific inhibitors are efficient in both systems pointing to common catalytic mechanisms. The characteristic properties of the redox cofactors of Rhodobacter bc1 are summarized in Table 1.
| Redox cofactor | E 7m (apparent midpoint redox potential at pH 7.0 in mV) | λ max/nm | g x (EPR) |
|---|---|---|---|
| Heme bl | −90 ÷ −115 (heme bh reduced)51,60 | 558 and 56551,60 | |
| Approx. 0 (heme bh oxidized)128,149 | |||
| Heme bh | +50 ÷ +6051,115 | 560.551,115 | |
| Cytochrome c1 | +290 ÷ +34060,72 | 55260,72 | |
| [2Fe–2S] cluster | +270 (FeSc)60,145 ÷ +460 (FeSb)144 | — | 1.800 (ubiquinone present)150,151 |
| 1.774 (ubiquinone absent)150,151 |
In this review, only the flash-induced reactions in bc1 are surveyed. The data on generation of Δψ and ΔpH in response either to continuous illumination or to substrate addition are difficult to interpret in relation to molecular mechanisms of bc1. The reader has to consult earlier works52,64 for surveys on the steady operation of bc1 in photosynthetic membranes.
2. Electron cycling in chromatophore vesicles from purple phototrophic bacteria
As depicted in Fig. 2, an absorption of a light quantum leads to a transmembrane charge separation between the water-soluble cytochrome c2 and the secondary quinone QB of the photosynthetic reaction center (RC).51,52,55,56 The charge separation yields potential reductants and oxidants for the bc1, i.e. molecules of ubiquinol and of oxidized cytochrome c2, respectively. The bc1, in essence, catalyses the ET from ubiquinol back to cytochrome c2. The availability of specific inhibitors of center P, such as myxothiazol and stigmatellin, helps to discriminate the reactions in bc1 from other flash-induced events. In some cases, inhibitors of center N (e.g. antimycin) might be useful, in their presence electrons cannot pass heme bh, so that only one “half-turnover” of bc1 takes place.When the membrane ubiquinone pool is completely oxidized, the flash-induced turnover of bc1 is triggered by the arrival of a ubiquinol molecule that is formed in the RC. At neutral pH, the oxidation of this ubiquinol leads to partial reduction of heme bh at approx. 3 ms followed by slower (i) cytochrome c1 re-reduction by electrons coming from ubiquinol, (ii) Δψ generation, and (iii) proton release to the p-side of the membrane. These three reactions correlate with each other and take approx. 10–20 ms.22,55,56,58,65–70
When there is plenty of ubiquinol in the membrane (under reducing conditions that correspond to the physiological situation),71 the bc1 turnover is triggered by the migration of an electron vacancy from the flash-oxidized primary donor P870+, via cytochrome c1, to the FeS domain27,72 (see Fig. 1 and 2). The oxidation of QPH2 results in the re-reduction of cytochromes c at 2–5 ms. The Δψ generation proceeds with the same rate. No redox changes of cytochrome b can be resolved: apparently, under these conditions the hemes of cytochrome b are oxidized faster than are reduced.51,52,55,56,65–67
3. Correlations between Δψ generation, proton translocation, and ET reactions
The views on the mechanism of Δψ generation by bc1 have changed dramatically with time.‡ As the last summarising articles on this topic were published more than 15 years ago,58,73 a historical consideration of the subject might be appropriate.3.1 Electrogenic reactions in bc1: the pre-structural age
The major breakthrough was coupled with the introduction of center P inhibitors, UHDBT and myxothiazol in the first line.35,65,80,81 These inhibitors, when added over antimycin, helped to discriminate the reactions that accompanied the ubiquinol oxidation in center P. In the presence of antimycin, one ubiquinol molecule could be still oxidized in response to a flash and the concomitant reduction of heme bh, re-reduction of cytochrome c1 and Δψ generation proceeded with approx. same rate.82 For the bc1 of Rhodobacter, Crofts4,82 has suggested the modified Q-cycle scheme that incorporated some earlier ideas of Garland.83 According to this scheme, the center P turns over twice to produce a QNH2 ubiquinol. The oxidation of the first ubiquinol molecule in center P leads to the formation of a QN˙− semiquinone84 (in fact, the electron is shared between QN˙− and the reduced bh heme (bhred)). The redox equilibration in the low-potential branch of bc1 (after oxidation of QPH2 to QP˙− by the FeS cluster) can be written as:
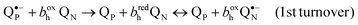 | (1) |
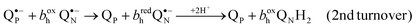 | (2) |
It is noteworthy that the Em value of heme bh in chromatophores of Rhodobacter, unlike that of the QN/QNH2 couple, is pH-independent at pH > 7.5,51 so that the Em of the ubiquinol/ubiquinone couple becomes lower than that of heme b under alkaline conditions. Therefore the heme bh could be reduced via center N by the RC-generated ubiquinol in the presence of myxothiazol at pH > 8.0.90,91 The reaction was accompanied by a blue carotenoid bandshift indicating a discharge of Δψ; this shift was not observed in the presence of antimycin.73,88 This negative electrogenesis was more pronounced in the presence of Δψ.73,88 It was suggested that the voltage generation by bc1 is a two-step ET reaction, where an electron goes first from heme bl, at the p-side of the membrane, to heme bh in the middle of the membrane, and then from heme bh to QN at the membrane n-side.73,88 The reversion of the latter reaction upon heme bh reduction via center N was suggested to account for the blue carotenoid bandshift. To explain why Δψ prevented the oxidation of heme bh but not its reduction, the transmembrane ET from heme bh to QN was considered to be the least favourable step of the cycle. The dielectric distance between heme bh and QN was estimated either as 40%73 or as 60%88 of the membrane dielectric. Accordingly, the proton transfer reactions were assumed to proceed at the membrane/water interfaces and to be electrically silent.73,88
Another considered possibility has been the involvement of transmembrane proton displacements in Δψ generation by bc1.20,58,59,68,69,92–96 In particular, the binding of the RC-generated ubiquinol to center N, as measured in the presence of myxothiazol by an electrometric technique, induced an antimycin-sensitive “negative” component of Δψ even at neutral pH when no reduction of heme bhvia center N could be expected. It was speculated that the binding of ubiquinol to center N could lead to the formation of an anion QH− and electrogenic proton release.58,59 A small myxothiazol-sensitive component of Δψ generation was resolved by capacitive electrometry in the mutants of Rb. sphaeroides lacking heme bh. It was suggested that proton release from center P might be electrogenic.96 In the resulting tentative picture of bc1, the centers P and N were protein-buried and connected with external aqueous phases by proton conducting channels.58 Proton transfer along these channels was assumed to be electrogenic.58,59
Furthermore, the above noted kinetic disparity between the faster reduction of heme bh, on one hand, and the slower Δψ generation and cytochrome c1 re-reduction, on the other hand, as seen under oxidizing conditions in the absence of antimycin,55,56,58,65,66,68,69,95 could indicate poor coupling between the transmembrane ET towards heme bh and the generation of Δψ. It was suggested that protons, which are released in center P upon ubiquinol oxidation, are not ejected out of bc1 but stay inside it for electrostatic compensation of the injected electron(s).68,69,95,97 It was hypothesized (i) that these protons are released after the negative charge at reduced cytochrome b is neutralized by proton binding in center N upon ubiquinol formation and (ii) that the coupled events of proton binding and release account for the major electrogenic reaction in bc1.68,69,95 It is worth mentioning that at the same time it was realised that the electrostatic neutralization of injected electrons by the trapped protons is an inherent feature of the cytochrome c oxidase.98
To measure the pH changes in the tiny internal space of chromatophores, they were soaked by dense solution of a hydrophilic pH dye phenol red that was then rinsed off. The proton release into the chromatophore “lumen” took about 20–30 ms at pH about 6.0.104 Saphon and Gräber measured the proton release into the interior of Rb. sphaeroides chromatophores via the fluorescence quenching of 9-aminoacridine.108 This technique enabled to estimate the relative extent of the flash-induced acidification as 0.1–0.3 pH units but was not fast enough to determine the time constant. Jackson and co-workers have measured the same proton release at the p-side with whole cells, spheroplasts and right-side-out vesicles of Rb. capsulatus; in all these cases, the p-side was facing the external phase.100–102 In whole cells and spheroplasts the proton release was distinctly slower than Δψ generation.100–102 In smaller right-side-out vesicles the proton release had the same rate as the voltage generation.102§ The relation between proton release from bc1, Δψ generation, and redox-reactions of cytochrome b was studied within chromatophores of Rb. capsulatus by using neutral red (NR) as an amphiphilic pH-dye.68 In the presence of NR, an additional flash-induced absorption rise with a time constant of approx. 10 ms was seen under oxidizing conditions. The transient could be abolished by the addition either of a penetrating pH buffer glycyl–glycin, or a K+/H+ exchanger nigericin, or myxothiazol.68 These absorption changes of NR were attributed to the acidification of chromatophore lumen by bc1. The onset of acidification correlated with Δψ generation by bc1 and was slower than the flash-induced reduction of heme bh at ∼3 ms.68,69
3.2 Electrogenic reactions in bc1: the post-structural era
The electron transfer between the bc1 monomers has been recently demonstrated.48,49 The support for the alternating sites mechanism has been provided by Trumpower and co-workers.42,44,45 The evidence in favour of the suggested, still controversial, mechanism of Δψ generation is surveyed in detail below.
Based on studies of Zn2+-treated preparations, the turnover of bc1 was suggested to proceed in two steps, at least26,53,113, as depicted in Fig. 3. During the first, apparently Zn2+-insensitive step (see Fig. 3A), ubiquinol is oxidized in center P, the FeS domain takes the first electron and first proton from ubiquinol, while the other electron is transferred via heme bl to heme bh across the membrane. The second proton remains in center P. During the second, Zn2+-sensitive step (see Fig. 3B), the FeS domain re-reduces cytochrome c1, protons are released into the chromatophore interior, heme bh is oxidized via center N and Δψ is generated.26,53,113
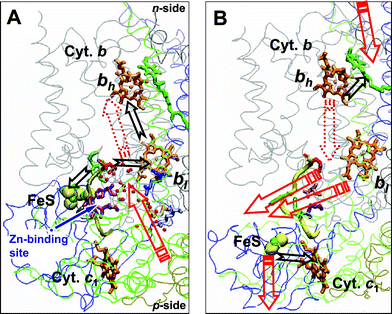 | ||
| Fig. 3 Tentative scheme of electron and proton transfer during the initial steps of bc1 turnover (the figure is reproduced from ref. 26 with permission). Black arrows, ET events; red arrows, proton transfer events; dark-red dotted arrows, dielectric relaxation of protein–water. The redox centers are colored as in Fig. 1. Below the yeast numbering of amino acid residues is given by straight letters, while that of Rb. capsulatus is given in italic letters. The Glu-272 (Glu-295) of cytochrome b is colored red. The segment of the ef loop that interferes with the movement of the FeS domain is shown as a thick yellow tube (cytochrome b residues from 260 to 270 (from 283 to 293 in Rb. capsulatus). The figure was produced with the help of the VMD software package.152 (A) Fast step of ubiquinol oxidation in center P. The picture is a compilation of two crystal structures of the yeast bc1 with the FeS domain docked to cytochrome b in the presence of stigmatellin: the water chains from the high resolution structure (PDB entry 1EZV14) are superimposed over the structure of a dimeric yeast bc1 co-crystallized with cytochrome c (PDB entry 1KYO15). The bound stigmatellin in center P was replaced by ubiquinol. Water molecules, which are found in the vicinity of center P are shown as red balls. The four amino acid residues, which correspond to the Zn2+-binding ligands of the chicken bc1, are depicted in violet. Thereby Ser-268 of yeast was replaced by histidine, as in Rb. capsulatus (His-291) and chicken; other residues are His-253 (His-276), Asp-255 (Asp-278) of cytochrome b and His-185 of cytochrome c1 (no evident counterpart in Rb. capsulatus). (B) Slower step of ubiquinone reduction in center N. The picture is based on the structure of the chicken bc1 with the FeS domain in the “cytochrome c1” position (PDB entry 1BCC11). Ubiquinol in center N is shown in the same position as it is found in the yeast bc1 (PDB entry 1EZV14). The four amino acid residues, which bind Zn2+ in the chicken bc1 are colored as follows: cytochrome b, Asp-253 (His-276), pink, Glu-255 (Asp-278), violet, His-268 (His-291), green, cytochrome c1, His-121 (no evident counterpart in Rb. capsulatus), light green. | ||
These studies of bc1 revealed several features that deserved explanation, namely:
(1) Only a half of the total heme bh content was reduced in response to a flash, even when the experimental conditions (pH, temperature, H2O/D2O) were varied.53,112–114 The reduction of only half of heme bh content was surprising. During the studies of Zn2+-treated samples, the ubiquinone pool was kept half-reduced by the succinate/fumarate redox couple; the redox poise of the sample corresponded to 80–100 mV at pH 7.5 of the measurements.53,112–114 The heme bh with E7.5m of ∼20 mV51 was expected to be oxidized in the dark and to be completely reduced in response to a flash. Such a complete reduction of the heme bh content could be indeed observed, but only after antimycin was added to the sample.53,112–114
(2) When weak exciting flash was used, so that ubiquinol was oxidized only by some bc1 monomers, the rate and relative extent of Δψ generation was comparable with that observed after a saturating flash.53,113,114 This observation was in variance with the behaviour of bc1 under oxidizing conditions when the Δψ generation slowed down and diminished dramatically under single-turnover settings.22,69,70
(3) The transmembrane ET from center P to heme bh took 1–2 ms and was not accompanied by notable Δψ generation.53,112–114 One would expect (i) that the transmembrane movement of a negative charge should contribute to Δψ generation and (ii) that the onsets of heme bh reduction and of voltage generation should match each other.
(4) Zn2+ ions concomitantly slowed down the Δψ generation, proton release into the lumen, re-reduction of cytochrome c1 and oxidation of heme bh.53,112–114 Such a kinetic match, as observed even under single-turnover conditions, deserves explanation as long as the oxidation of heme bh and the re-reduction of cytochrome c1 proceed at different sides of the membrane.
Below I attempt to justify these observations by considering the catalytic cycle of bc1 in some detail.
3.2.2.1. EPR-silent QN˙− semiquinone in a dimeric cytochrome bc1 complex. The observations (1) and (2) can be accounted for by invoking the data on potentiometric redox titrations of bc1. On one hand, these titrations have revealed a particular high-potential form of heme bh in the bc1 of Rhodobacter5,55 and of mitochondria.116–118 In chromatophores of Rhodobacter, the E7m of this “b155” was estimated as ∼150 mV.5,55 The “very high potential” cytochrome b was not detectable after the addition of antimycin; therefore it was suggested that b155 represented a special state of heme bh (hereafter denoted as bh,150) induced by its interaction with a quinone species in center N.5,90,118 On the other hand, Palmer and co-workers, while performing a parallel potentiometric titration of isolated yeast bc1 by optical and EPR spectroscopy,116,119 found that ∼50% of the oxidized heme bh was EPR silenced by its antiferromagnetic interaction with a semiubiquinone when the ubiquinone present was reduced by half. It was concluded that bc1 contains two populations of ubisemiquinone, namely a minor EPR-visible one (∼0.1 per bc1 monomer) and a major EPR-silent one (∼0.5 per bc1 monomer). Rich and co-workers came to the same conclusion upon analysing their data on potentiometric titration of bc1 in the beef hearth submitochondrial particles (SMP).118 By invoking the pioneering EPR spectroscopy data of De Vries and co-workers,46,47 Crofts argued that the bh,150 state resulted from “reversal of the second electron transfer of the normal forward reaction”.5,98 It was suggested that at Eh < 150 mV the oxidation of an ubiquinol molecule from the membrane pool via center N might yield a semiquinone and a reduced heme bh.5,90 The E7m value of the QN/QNH2 couple, as revealed from the potentiometric titration of the EPR-visible QN˙− signal, was indeed found to be ∼150 mV in the case of Rb. sphaeroides chromatophores.84
As long as a monomeric bc1 is considered, this explanation of the bh,150 phenomenon is rather paradoxical. It implies that an interaction of heme bh with a negatively charged QN˙− causes an increase in the Em value of the heme, conversely to expectations based on electrostatics. The paradox, however, can be solved by invoking the above noted electron exchange inside a bc1 dimer. Although such electron exchange, as explicitly suggested by some authors,10,22,24,46,47,120 has been recently experimentally demonstrated,48,49 it is not proved yet whether the exchange can proceed on the timescale of the bc1 turnover. However, in relation to the pulsed experiments, there is no thinkable way to prevent an electron equilibration between the bc1 monomers before the measurements, on a timescale of seconds. Such an equilibration is expected to proceed in the following way.
(1) Ubiquinol binding in center N would result in formation of a QN˙−bhred pair at redox poise corresponding to Eh < 150 mV, as suggested by Crofts and co-workers.5,90
(2) Provided that electron can migrate inside the bc1 dimer, the electrostatic repulsion between electrons at QN˙− and at bhred would favour the spillover of the electron from the bh heme next to QN˙− to the heme bh of the other bc1 monomer:
| QH2 + bhox | bhox → QN˙−bhred | bhox → QN˙−bhox | bhred | (3) |
(i) accounting for the data of Palmer and co-workers,116,119 one EPR silent semiquinone QN˙−bhox | bhred would be present in the majority of bc1 dimers (its estimated amount of 0.5 per one bc1 monomer116,118,119 corresponds to one semiquinone per one bc1 dimer).
(ii) in a minor fraction of enzymes, the QN˙− semiquinone could be EPR visible either because the electron has not got to the other monomer (QN˙−bhred | bhox), or because (one) heme bh was already pre-reduced at equilibrium (QN˙−bhred | bhred, note that the iron atom of bhred is diamagnetic116). At neutral pH the relative extent of EPR visible QN˙− seems to be ∼0.1 per bc1 monomer, at most.84,116,119,121
The EPR silence of the QN˙−bhox | bhred state is in agreement with the X-ray data that show QN only 5 Å away from heme bh14, i.e. close enough for antiferromagnetic quenching.
As already noted, the Em of heme bh in chromatophores of Rhodobacter, unlike that of the QN/QNH2 couple, is pH-independent at pH > 7.5.51 Then one should expect an increase in the relative amount of the EPR-visible QN˙−bhred | bhred state at alkaline pH owing to the increase in the amount of reduced heme bh next to QN˙−. This expectation is fulfilled: at alkaline pH, the relative extent of the EPR-visible QN˙− semiquinone increased up to 0.4 per bc1 monomer in chromatophores of Rb. sphaeroides.84 Supposedly because of the same reason, the relative amount of the EPR-visible QN˙− semiquinone increased upon alkalisation also in the cytochrome bc1 complexes from other sources: up to 0.26–0.5 in the SMP,84,122, and up to ∼0.5 in the bc1 of Paracoccus denitrificans.121 The pK value of 7.5, which is well-established for bh of Rhodobacter, is sufficient to explain the increase in the amount of the EPR-visible QN˙− at high pH values; there is no need to invoke additional functional pK of QN˙− in the neutral range.
Hence, when the ubiquinone pool is half-reduced, two electrons seem to be distributed over a bc1 dimer; they reside at heme bh and at QN˙− semiquinone of different monomers, as described by scheme (1). Under physiological conditions of a half-reduced ubiquinone pool,71 the state QN˙−bhox | bhred is the stable “ground” state of bc1; it is depicted as state A in Fig. 4 where a tentative catalytic cycle is schematically shown for a bc1 dimer.
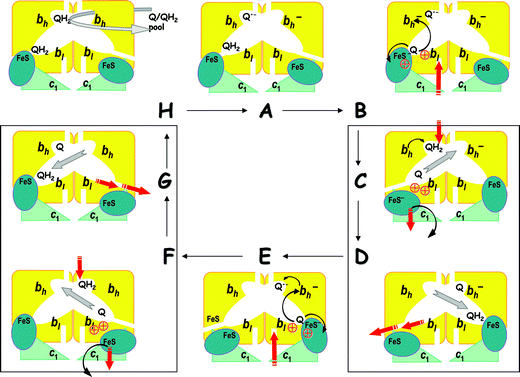 | ||
| Fig. 4 Activated Q-cycle in a dimeric cytochrome bc1 complex (the scheme has been first presented during the 13th EBEC Meeting).154 Thin black arrows, electron transfer steps; thick red arrows, proton transfer steps; thick gray arrows, quinone/quinol exchange reactions. Protons are depicted as red, circled crosses. The tentative mechanism of the first ubiquinol oxidation, as presented on panels from A to D, is based on the single-turnover data53,112–114 and on the comparative analysis of different bc1 structures.26,113,136 The following steps (panels from E to H), which describe the second turnover of center P, are hypothetical; they are based on symmetry considerations and on the need to complete the catalytic cycle. For the other explanations, see the text. (1) Panel A, initial activated state. When ubiquinol is present, the bc1 dimer is in an activated QN˙−bhox | bhred state. The docking of the oxidized FeS domain to cytochrome b leads to the formation of a quinol-oxidizing catalytic site in center P. (2) Panel B, the faster oxidation of QPH2 (see also Fig. 3A). The ubiquinol oxidation leads to the coupled electron and proton transfer to the FeS cluster yielding a QP˙− semiquinone. The reduced and protonated FeS domain remains docked to cytochrome b for the time being. The electron goes from QP˙− to heme bl; the released proton resides in center P. The electron then crosses the membrane going from heme bl to the only oxidized heme bh of the bc1 dimer, next to the QN˙− semiquinone. This transmembrane ET seems to be electrostatically compensated. (3) Panels C and D, the slower formation of QNH2 (see also Fig. 3B): The FeS domain undocks and moves towards cytochrome c1. This reaction seems to be coupled to the oxidation of heme bh by the QN˙− semiquinone, electrogenic binding of two protons from the n-side of the membrane, and formation of a QNH2 ubiquinol. The oxidation of the reduced and protonated FeS domain by cytochrome c1 is accompanied by the proton release into the water phase. In the same time, the protons, which have compensated the negative charge at cytochrome b hemes, get out via the now open proton exit. The transfer of all these protons across the membrane dielectric, as well as the re-orientation of the intra-membrane dipoles and charges, account for the observable electrogenic reaction. Only for illustrative purposes, the reactions accompanying this slower step of the catalytic cycle are depicted on two panels C and D; the panels are framed to emphasize that all these reactions are likely to be coupled to each other (this consideration relates also to the panels F and G). | ||
Then, in response to a flash of light:
(i) accounting for our observations53,112–114, not more than a half of the total bh content could be reduced—the other half is already pre-reduced in the dark as QN˙−bhox | bhred;
(ii) ubiquinol formation in center N would occur already in response to the first turnover of center P. Already the oxidation of the first QPH2 molecule would lead to the reduction of heme bh next to the QN˙− semiquinone followed by the oxidation of this heme and ubiquinol formation in center N (see Fig. 4):
| QP˙− + QN˙−bhox | bhred → QP + QN˙−bhred | bhred → QP + QNH2bhox | bhred | (4) |
Since the major electrogenic reaction in bc1 seems to be coupled with the formation of the QNH2 ubiquinol22 (see also section 3.2.2.3 below), the suggested rational explains why the flash-induced Δψ generation by bc1, as measured under reducing conditions in a single-turnover setup, was similar to that after a saturating flash.53,112–114
One could ask why the QN˙−bhox | bhred state is not oxidized by the membrane ubiquinone further to yield a QN˙−bhox | bhoxQN˙− state. The reason might be the inability of the bc1 dimer to stabilize two QN˙− semiquinones at once. The X-ray structure of crystals formed from bc1 dimers as crystallographic units has revealed one QN bound per bc1 dimer.15
According to the suggested scheme, already the first turnover of center P can yield QNH2 under reducing, physiological conditions. This feature is related to the long-lasting dilemma on the apparent incompleteness of a single Q-cycle turnover. Indeed, the oxidation of one QPH2 ubiquinol provides only one electron for center N, whereas two electrons are needed to yield a product—a QNH2 ubiquinol. Mitchell has been concerned by this problem; he has considered a possibility that while one electron comes to the quinone in center i (center N) from heme bh, another one is supplied by some other source. He hypothesized that this second electron is provided by the succinate dehydrogenase in mitochondria, by the RC in chromatophores of phototrophic bacteria, and by the photosystem II in plants.20 However, the bc1 operates with a H+/e− stoichiometry of 2 even alone, when incorporated into liposomes.17,18 Therefore, the common believe has been that a catalytic cycle of bc1 requires two turnovers of center P,4,82,83,123 as described by eqn (1) and (2) in section 3.1.1. The scheme (4) offers another solution: here an oxidation of only one ubiquinol molecule in center P by the “pre-activated” bc1 is sufficient to yield a QNH2 ubiquinol. In other words, the Q-cycle is complete in a single turnover of center P.
If reaction (3) is exothermic, then the QN˙−bhox | bhred state would be trapped once formed. Because of electrostatic attraction to the heme iron, the QN˙− semiquinone would be tightly bound (as it happens in the RC124). The bound QN˙− would be destined to wait for the second electron coming from center Pvia cytochrome b hemes. From the apparent Em of 150 mV both of bh,1505,55,115 and of the QN˙− semiquinone in Rb. sphaeroides,84 the ΔG value of reaction (3) can be estimated as ∼ −60 meV under conditions where the ubiquinone pool (E7m ∼ 90 mV) is half-reduced. This free energy is temporally borrowed from the ubiquinol pool to guarantee the steady presence of one QN˙− per one bc1 dimer; the loan is returned back when a new ubiquinol molecule is formed in center N. A continual priming of bc1 is secured by the promptness of ubiquinol binding in center N (<100 µs).59
The suggested ability of the QN˙−bhox | bhred state to exert control over the rate of bc1 turnover might solve an old controversy concerning the nature of the bound quinone in bc1. The earlier studies have revealed that the flash-induced electrogenesis and cytochrome c reduction in bc1 of Rhodobacter accelerated dramatically with an apparent E7m of ∼150 mV.55,65,67,77–79 Based on these observations, Dutton and co-workers have suggested a tightly bound ubiquinol (QZ) in center P with E7m of about 150 mV.55,77–79 This suggestion was supported by the observation that the prompt turnover of bc1 was unaffected by ubiquinone extraction until only 1–2 ubiquinones per bc1 remained.78 The X-ray structures of bc1 revealed neither ubiquinol nor ubiquinone in center P, but, instead, exposed ubiquinone molecule bound in center N.14,15,28 It looks like that the only binding site in bc1 with a notable affinity to ubiquinol/ubiquinone is the center N proper. Taken together, these data might indicate that Dutton and co-workers were correct when they suggested that the acceleration of the bc1 turnover is related to a bound quinone species with an apparent E7m of ∼150 mV. This quinone species, however, seems to be not the ubiquinol in center P, but a bound QN˙− semiquinone in center N. In fact, Dutton and co-workers have considered the possibility that the semiquinone in center N might control the kinetics of bc1.84 They, however, declined this possibility because the amount of the EPR-visible semiquinone was sub-stoichiometric at neutral pH.84 As argued above, one EPR-silent QN˙− semiquinone seems to be present per each bc1 dimer at Eh < 150 mV.116,118,119 Apparently, the formation of the EPR-silent QN˙−bhox | bhred state switches the cytochrome bc1 complex from the slower double-turnover mode of operation to the faster single-turnover one.
3.2.2.2. Electrostatic compensation of the transmembrane ET towards heme bh. As noted above, the faster reduction of heme bh, as compared to the Δψ generation, might indicate electrostatic compensation of the transmembrane electron transfer to heme bh.22,26,68,69,95 More widespread explanation is, however, that the genuine, slower kinetics of heme bh reduction is masked, in the absence of antimycin, by the oxidation of the heme, so that the observable prompt reduction of heme bh only seems to be faster than the voltage generation.125 To assess these two possibilities, it is useful to consider a simple kinetic scheme:
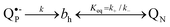 | (5) |
A partial flash-induced reduction of heme bh is expected if (i) k+ has the same order of magnitude as k (kinetic limitation) or (ii) if Keq is low (even when k+ ≫ k, thermodynamic limitation). Numerical simulations/analysis of the scheme (5) show that the oxidation of heme bh can cause apparent acceleration of its reduction rate only in case of kinetic limitation. In the case of thermodynamic limitation, the observable rate of heme bh reduction should be equal to the genuine rate of electron delivery to bh and independent of the observable extent of heme reduction. The extent of flash-induced heme bh reduction, as seen under oxidising conditions in the absence of antimycin, decreased in the presence of uncoupler55,126 and increased when the membrane potential was elevated by blocking the proton escape via ATP-ase.69 Thereby the time constant of heme bh reduction (∼3 ms) remained independent of the fraction of the heme staying reduced after the flash.69 As well, the distance of 5 Å between heme bh and QN14 is hardly compatible with a kinetically limited oxidation of heme bh. This evidence strongly supports the thermodynamic control over the oxidation of heme bh and, accordingly, suggests that the rate of the partial heme bh reduction in the absence of inhibitors reflects the genuine rate of electron delivery to the heme.
Still, when the membrane ubiquinol pool is oxidized and the substrate ubiquinol comes from the RC, it is difficult to exclude completely a prompt, non-electrogenic reduction of heme bhvia center N (A. R. Crofts, personal notion). Under reducing conditions, as maintained upon the studies of the Zn2+-treated bc1, this possibility could be ruled out. The heme bh was reduced at 1–2 ms by an electron that crossed the membrane coming from center P, and still no compatibly fast Δψ onset was observed.53,112–114
As argued in more detail elsewhere26,127, an electrostatic compensation is, in fact, a precondition of ET from heme bl (E7m ∼ 0 mV128) to heme bh (E7m ∼ 50 mV51,61) against the backpressure of Δψ (about 100 mV in chromatophores of Rb. capsulatus after a single saturating flash129). Such an electric silencing could be achieved, in particular, by proton transfer in the direction of ET,22,26,105e.g. along a water chain that connects the propionates of heme bl with the external p-phase14,130 While proton releasevia this path, as suggested by Crofts and co-workers41, would go against the transmembrane electric field, the same field would favour proton re-distribution in the opposite direction, towards heme bl. Additionally, electrostatic compensation might be achieved via re-orientation of water dipoles and of charged protein groups/domains, as discussed in more detail elsewhere26,53,113
3.2.2.3. Kinetic correlations in the cytochrome bc1 complex and the mechanism of Zn2+ binding. The mechanism of kinetic correlation between the re-reduction of cytochrome c1, Δψ generation, proton release into the chromatophore interior and the oxidation of heme bh53,112–114 can be tentatively elucidated if we consider the mode of Zn2+ binding by bc1.
Skulachev and co-workers were the first to show that Zn2+ ions increased the reduction level of cytochromes b and decreased that of cytochromes c in the mitochondrial bc1.110 Link and Jagow, who studied the inhibition of mitochondrial bc1 by Zn2+ as function of its concentration and pH, suggested that Zn2+ ions interfered with histidine residue(s) forming the proton outlet from center P.111 Relevantly to this conclusion, Zn2+ and some other divalent ions were found to block the proton inlet to QB in the RC of Rb. sphaeroides by binding to a cluster formed by His-126, His-128 and Asp-124 of the H subunit of the RC with an apparent pK of 6.8.131 As well, Zn2+ ions were found to block the proton paths in the cytochrome c oxidase.132,133 Incidentally or not, the affinity of negatively charged histidine-containing patches to Zn2+ ions seems to correlate with the proton trapping/proton conducting ability of these patches.134 In Rb. capsulatus, the Zn2+-due effects in bc1 have diminished with an apparent pK of approx. 7.0.114 This value is close to the pK values of Zn2+ binding to the mitochondrial bc1 (pK = 7.2)111 and to the Rb. sphaeroides RC (pK = 6.8)131 and points to histidine residue(s) as potential Zn2+ ligand(s) in Rb. capsulatus. Berry and co-workers have crystallized the avian bc1 in the presence of Zn2+. They found a binding site for Zn2+ close to center P that was formed by His-121 of cytochrome c1 and by Asp-253, Glu-255 and His-268 of cytochrome b135 (see Fig. 3A). All three Zn2+ ligands provided by the avian cytochrome b have counterparts in Rb. capsulatus; these are His-276, Asp-278 and His-291. The constellation of two histidine residues and a carboxyl strikingly resembles the Zn2+/proton binding cluster of the Rb. sphaeroides RC,131 so that this triad was suggested to bind Zn2+ in the bc1 of Rb. capsulatus.53,113,136 This suggestion has been recently confirmed by Venturoli and co-workers who, by using K-edge X-ray absorption spectroscopy, have found that two nitrogen and two oxygen atoms (at <2.20 Å) plus one oxygen (or nitrogen) atom serve as ligands for a single Zn2+ ion bound in the bc1 of Rb. capsulatus.137 The avian Asp-253 and Glu-255 (His 276 and Asp 278 of Rb. capsulatus) are close to Glu-272 (Glu-295 of Rb. capsulatus), the putative immediate acceptor of the second proton from ubiquinol.14,26,41 Thus, the path via the Zn2+-binding site seems to be the shortest potential proton exit from center P. The comparison of different bc1 structures shows that this exit is open when the FeS domain is not docked to cytochrome b (see Fig. 3B). Hence, it is thinkable that protons get the opportunity to escape from center P into the water phase only when the FeS domain relocates towards cytochrome c1. It is noteworthy that His-268 of cytochrome b belongs to its ef loop, which controls the motion of the FeS domain and serves as a slide-valve for the passing FeS domain.130,138,139 The participation of the ef loop in the binding of Zn2+ (via His-268 in avian bc1 and, most likely, via His-291 in the bc1 of Rb. capsulatus) might provide a mechanistic rationale for the slowing of the cytochrome c1 re-reduction by the FeS domain in the presence of Zn2+, as observed with chromatophores of Rb. capsulatus.53,112–114 (see Fig. 3). The binding of Zn2+ could constrain the mobility of the ef loop and thereby impede the motion of the FeS domain. This rational can account for the concurrent inhibition of cytochrome c1 re-reduction and Δψ generation by Zn2+: the binding of Zn2+ to the histidine patch can slow down (i) the movement of the FeS domain towards cytochrome c1 (via the above mentioned interaction with a histidine residue of the ef loop) and, accordingly, (ii) the electrogenic proton release from center P.
As it is discussed in more detail elsewhere,22,26,53,113,136 and as it is depicted in Fig. 3B and panels C and D of Fig. 4, the Δψ generation by bc1 seems to represent a chain of coupled electrogenic events. (i) The movement of the reduced and protonated FeS domain towards cytochrome c1 leads to the oxidation of the FeS cluster followed by proton release into the aqueous phase p; (ii) the formation/release of the QNH2 ubiquinol in center N is accompanied by electrogenic binding of two protons from the n-side of the membrane; (iii) the opening of the proton exit from center P lets proton(s), which do not compensate any negative charge now, to get out.¶ The transfer of all these protons across the membrane dielectric, together with the resetting of the intra-membrane charges and dipoles, generates Δψ. In agreement with experiment (i) the rate of Δψ generation correlates with the rate of cytochrome c1 re-reduction22,55,56,77,79,80 and (ii) both these reactions are sensitive to Zn.26,53,112–114
The Zn2+ treatment has also exposed a kinetic correlation between the re-reduction of cytochrome c1 (and Δψ generation), on one hand, and the oxidation of heme bh, on the other hand.53,112–114 It is noteworthy that such a kinetic correspondence has been observed not only with the bc1 of Rhodobacter55,65,66 but also upon pulsed studies of the mitochondrial cytochrome bc1 complex.140–142 Diverse earlier data on the steady operation of cytochrome bc1 complexes from different sources also showed correlation between the oxidation of cytochrome b and the re-reduction of cytochrome c1.32,55,56,143 Around 1981 Mitchell has mentioned in a conversation a possibility that “the electron might be getting hung up on the Rieske” (as noted recently by Crofts4). Independently, it has been hypothesized that the ET from the FeS cluster to cytochrome c1 might be mechanistically controlled by a redox reaction in center N.69 As argued elsewhere,26 the inability of the electron to leave the FeS cluster until the reaction in center N is completed helps to prevent the short-circuiting in bc1. The structural data provide some hints on how such a control can be achieved. As already noted, the relocation of the FeS domain between its two binding sites seems to be controlled by the cd and ef loops of cytochrome b.12,130,138,139,144,145 These loops connect, respectively, the C, D and E, F α-helices of cytochrome b. The quinone-binding site in center N, on the other hand, is formed with the participation of helices D and E of cytochrome b.14,28 The occupancy of center N might affect the mechanistic properties of the ef and cd loops and vice versa resulting in coupling between the relocation of the FeS domain and the oxidation of heme bh. Recently Cooley and coworkers, by increasing the spectral and spatial resolution obtainable with orientation-dependent EPR spectroscopic analysis of ordered membrane preparations, have indeed found correlations between the occupancy of center N and the EPR spectra of both cytochrome b hemes and the FeS cluster and suggested a hypothetical mechanism of coupling via helices of cytochrome b.146
The above considered correlation between the occupancy of center N and the mobility/position/EPR spectra of the FeS domain146 might be related to the “double occupancy” model of Dutton and co-workers (see ref. 3 and citations therein). These authors studied the EPR line shapes of the Fe–S cluster as function of (i) the amount of ubiquinone/ubiquinol present (varied by solvent extraction and reconstitution), (ii) the redox state of the ubiquinone pool, (iii) the presence of specific inhibitors and (iv) the site-specific mutations in cytochrome b. From data analysis, it has been suggested that the center P (QO site) can accommodate two ubiquinone molecules, namely the strongly (QOS) and the weakly (QOW) bound ones. After the crystal structures failed to reveal any bound quinone in center P, the double occupancy model has been apparently abandoned. The underlying experimental data, however, deserve explanation. These data can be accounted for, at least qualitatively, by suggestions that (i) QOS is the ubiquinone that occupies center N in the crystal structures,14,15,28 (ii) the extraction of this strongly bound ubiquinone affects the EPR spectra of the FeS cluster via the conformational coupling between centers N and P.
The oxidation of heme bhvia center N can lead either (i) to the QN˙− formation (if a quinone molecule pre-occupies center N, see eqn (1)) or (ii) to the formation of ubiquinol QNH2 (if a QN˙− semiquinone is present in the site, see eqn (2) and eqn (4)) . Which of these two reactions is coupled with the relocation of the FeS domain? This question can be tentatively answered by considering the single turnover data. When bc1 of Rb. capsulatus turned over only once under oxidizing conditions (case (i)), the kinetic correlation was absent: the re-reduction of cytochrome c1 took approx. 30–40 ms while the electron re-distribution between heme bh and the QN ubiquinone, according to eqn (1), proceeded at <3 ms.22,69,70 On the contrary, under reducing conditions (case (ii)) the re-reduction of cytochrome c1 correlated with the oxidation of heme bh even after weak flashes.53,112–114 Hence, it seems that the relocation of the FeS domain towards cytochrome c1 is coupled with the formation of the QNH2 ubiquinol and/or with its release from center N. After this point of “no return”, there is no danger of short-circuiting, so that the reduced FeS domain can move towards cytochrome c1 to be oxidized. Besides, the coupling between centers N and P could drive the unfavourable proton binding upon QNH2 formation by the thermodynamically gainful release of proton(s) from center P into the water phase.136
The idea of coupling between the “liberation” of the FeS domain and the formation/release of QNH2 gets further support from the already cited study of Cooley and co-workers who concluded that the bound antimycin, on one hand, imitates either the presence of QNH2 or the absence of quinone, and, on the other hand, increases the mobility of the reduced FeS domain maximally as if it were to facilitate its movement away from center P.146 This observation not only provides hints on how the reduced FeS domain can be “released” after QNH2 formation, but also might explain the difference in the behaviour of bc1 in the absence and in the presence of antimycin, respectively. In chromatophores, Δψ generation and proton release accelerated after the addition of antimycin, while dramatically diminishing in magnitude. Thereby these reactions proceeded approximately as fast as the reduction of heme bh.22,58,69,70,73,82,85,89,95 This kinetic match could not be due just to the oxidation of only one ubiquinol molecule in the presence of antimycin. As noted above, the kinetic discrepancy between the faster heme bh reduction, from one hand, and the slower cytochrome c1 re-reduction, Δψ generation and proton release, from the other hand, was still observed under single-turnover conditions, as created (in the absence of antimycin!) by ubiquinol shortage or weak flashes.22,53,69,70,112,114 Most likely, the turnover of center P proceeds differently in the absence of antimycin and in its presence. It is known that antimycin affects the mechanistic properties of bc1 and, in particular, the interaction of the Rieske protein with the cytochrome b (see ref. 32, 147 and citations therein). As argued elsewhere,26,53,69,70 it is thinkable that antimycin, by disrupting the conformational connection between centers N and P, enables the fast re-location of the reduced FeS domain towards cytochrome c1. The observation of Cooley and co-workers on the increased mobility of the reduced FeS domain in the presence of antimycin146 is compatible with this suggestion.
In this framework, it seems plausible that the electrogenic reaction, as observed in the presence of antimycin, is mostly due to the unconstrained and fast (due to the increased mobility of the FeS domain) proton release from center P, while the ET to heme bh is electrically silenced. This suggestion is supported by the relatively small extent of voltage generation in the presence of antimycin,58,73,85,88,89 by the ability of bc1 of Rb. sphaeroides to generate Δψ even in the absence of heme bh,96 and by the kinetic correspondence between proton release into the lumen of Rb. capsulatus and Δψ generation, when measured in the presence of antimycin.69
The anomalous absence of proton binding by bc1 under oxidizing conditions,56,105–107 as discussed in section 3.1.2, might be due to the pH-dependence of Em of heme bh.51 Because of this feature, the flash-induced turnover of bc1 should be accompanied by proton trapping from the n-side both in the absence of antimycin and in its presence. In the latter case, protons would be taken by ionizable groups next to heme bh. Hence, the −/+ antimycin difference might diminish to zero under certain conditions. The increase in proton binding after the addition of valinomycin56,105–107 might reflect the drop in the ΔH+ backpressure. When the flash-triggered proton release into the chromatophore lumen was measured by NR, the addition of valinomycin doubled the NR response.68 Hence, ΔH+, as generated in response to a single saturating flash (>100 mV129), was large enough to block the turnovers of bc1 beyond the first one. In view of these data, the amount of protons trapped in the absence of antimycin should approx. double after the valinomycin addition, while the proton binding by heme bh in the presence of antimycin should stay independent of valinomycin. As a result, the extent of −/+ antimycin proton binding should increase in the presence of valinomycin, in agreement with experimental observations.56,105–107 As well, a gradual elevation of the ubiquinol/ubiquinone ratio should increase the number of bc1 turnovers by raising the reaction driving force. Accordingly, the extent of the antimycin-sensitive proton binding should increase with lowering of Eh, also in agreement with experimental observations.56,105–107
Still, to adequately measure the proton trapping by bc1, one has to compare the intact enzyme with one that is “doubly killed” by the addition of antimycin and myxothiazol. These experiments have to be carried yet.
4. Activated Q-cycle as a consistent mechanism of the cytochrome bc1 complex
The different facets of proton translocation by the cytochrome bc1 complex are brought together in Fig. 4. This scheme can be denoted as activated Q-cycle where the cytochrome bc1 complex enters the catalytic cycle being in an “activated,” pre-reduced QN˙−bhox | bhred state, as shown on panel A. As long as ubiquinol is present in the membrane, the activated state is maintained by steady ubiquinol oxidation via center N, according to eqn (3), yielding a bound semiquinone in this center and a reduced high-potential heme b in the other monomer of the enzyme. The almost isoenergetic pre-activation of bc1 enables the formation of QNH2 upon each turnover of center P as described by eqn (4). Otherwise, if bc1 is not activated (that could happen under non-physiological, oxidizing conditions), the formation of ubiquinol in center N requires not one, but two turnovers of center P, according to the bicycle mechanism of Crofts et al.4,82 and Garland et al.,83 see eqn (1) and (2). For oxidizing conditions, a tentative scheme of a catalytic cycle in a dimeric bc1 has been presented elsewhere.22Upon designing the steps of quinone/quinol transfer inside bc1, as tentatively depicted in Fig. 4, the aim was to minimize the exchange reactions with the membrane quinone pool. Quite unexpectedly, a scheme was obtained where the external ubiquinol enters the catalytic cycle by binding in center N (upon the G → H transition), but not in center P, as commonly believed. Ubiquinol binding is followed by its oxidation and formation of the QN˙−bhox | bhred state (H → A transition in Fig. 4). The center P, in turn, is steadily supplied by ubiquinol molecules coming from center N, as shown in Fig. 4 (this possibility has been earlier considered by Rich148); it is not obligatory for external ubiquinol molecules to penetrate towards center P to be oxidized. This rather odd view on the Q-cycle mechanism is consistent with the higher accessibility of center N in the crystal structure10 and with otherwise peculiar observations that (i) ubiquinol binds in center N of Rhodobacter by order of magnitude tighter than ubiquinone84 although center N is believed to be the quinone-reducing one; (ii) external ubiquinol molecules interact with center N faster than with center P in chromatophores of Rb. sphaeroides;59,90,91 (iii) ubiquinone is seen bound in center N but not in center P in the crystal structures.5,14,15,28
At least in one point the presented model carries more resemblance with the original Q-cycle scheme of Mitchell20 than with its later modifications. Mitchell has considered the possibility of ubiquinol formation in center N already after the first turnover of center P.20 As already noted in section 3.2.2.1, he has hypothesised that one electron comes to QN from cytochrome b while the other one is donated by some different enzyme.20 The activated Q-cycle in Fig. 4 implies, similarly to the original Mitchell's scheme,20 that the oxidation of each ubiquinol in center P can be followed by ubiquinol production in center N. The mechanism, however, differs from that suggested by Peter Mitchell: according to the scheme in Fig. 4, the other electron(s) wait(s) inside the dimeric bc1 after its redox equilibration with the ubiquinol pool via center N.
Acknowledgements
The author is thankful to Cristiano Viappiani, Thomas Gensch, and Joachim Heberle for the opportunity to contribute to the special issue ‘Proton Transfer in Biological Systems’ in Photochemical & Photobiological Sciences.Stimulating discussions with Drs E. A. Berry, J. W. Cooley, W. A. Cramer, A. R. Crofts, F. Daldal, L. A. Drachev, R. B. Gennis, O. A. Gopta, C. Hunte, S. S. Klishin, A. A. Konstantinov, L. I. Krishtalik, C. R. D. Lancaster, M. D. Mamedov, P. Mitchell, W. Nitschke, P. Rich, V. P. Shinkarev, V. P. Skulachev, B. L. Trumpower, M. V. Verkhovsky, S. de Vries, M. Wikström, R. J. B. Williams, D. Xia and C.-A. Yu are gratefully appreciated. The author is especially thankful to Professor Wolfgang Junge and Dr Dmitry Cherepanov for their help with analysis of the kinetic scheme (5) and to Dr Natalia Voskoboynikova for reading the manuscript and valuable comments. The research of the author was supported by the Volkswagen Foundation, INTAS (2001–736), and by grants from the Deutsche Forschungsgemeinschaft (Mu-1285/1, SFB 431-P15, 436-RUS-113/210).
References
- D. B. Knaff, The cytochrome bc1 complexes of photosynthetic purple bacteria, Photosynth. Res., 1993, 35, 117–33 CrossRef CAS.
- B. L. Trumpower and R. B. Gennis, Energy transduction by cytochrome complexes in mitochondrial and bacterial respiration: the enzymology of coupling electron transfer reactions to transmembrane proton translocation, Annu. Rev. Biochem., 1994, 63, 675–716 CAS.
- R. E. Sharp, C. C. Moser, B. R. Gibney and P. L. Dutton, Primary steps in the energy conversion reaction of the cytochrome bc1 complex QO site, J. Bioenerg. Biomembr., 1999, 31, 225–233 CrossRef CAS.
- A. R. Crofts, The Q-cycle—a personal perspective, Photosynth. Res., 2004, 80, 223–43 CrossRef CAS.
- E. A. Berry, M. Guergova-Kuras, L. S. Huang and A. R. Crofts, Structure and function of cytochrome bc complexes, Annu. Rev. Biochem., 2000, 69, 1005–75 CrossRef CAS.
- J. L. Smith, H. Zhang, J. Yan, G. Kurisu and W. A. Cramer, Cytochrome bc complexes: a common core of structure and function surrounded by diversity in the outlying provinces, Curr. Opin. Struct. Biol., 2004, 14, 432–39 CrossRef CAS.
- E. A. Berry, L. S. Huang, L. K. Saechao, N. G. Pon, M. Valkova-Valchanova and F. Daldal, X-Ray structure of Rhodobacter capsulatus cytochrome bc1: comparison with its mitochondrial and chloroplast counterparts, Photosynt. Res., 2004, 81, 251–75 Search PubMed.
- W. A. Cramer and D. B. Knaff, Energy transduction in biological membranes: a textbook of bioenergetics, Springer-Verlag, New York, 1990 Search PubMed.
- S. Hekimi, S. and L. Guarente, Genetics and the specificity of the aging process, Science, 2003, 299, 1351–54 CrossRef CAS.
- D. Xia, C. A. Yu, H. Kim, J. Z. Xia, A. M. Kachurin, L. Zhang, L. Yu and J. Deisenhofer, Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria, Science, 1997, 277, 60–66 CrossRef CAS.
- Z. Zhang, L. Huang, V. M. Shulmeister, Y. I. Chi, K. K. Kim, L. W. Hung, A. R. Crofts, E. A. Berry and S. H. Kim, Electron transfer by domain movement in cytochrome bc1, Nature, 1998, 392, 677–84 CrossRef CAS.
- S. Iwata, J. W. Lee, K. Okada, J. K. Lee, M. Iwata, B. Rasmussen, T. A. Link, S. Ramaswamy and B. K. Jap, Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex, Science, 1998, 281, 64–71 CrossRef CAS.
- H. Kim, D. Xia, C. A. Yu, J. Z. Xia, A. M. Kachurin, L. Zhang, L. Yu and J. Deisenhofer, Inhibitor binding changes domain mobility in the iron–sulfur protein of the mitochondrial bc1 complex from bovine heart, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8026–33 CrossRef CAS.
- C. Hunte, J. Koepke, C. Lange, T. Rossmanith and H. Michel, Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment, Struct. Fold. Des., 2000, 8, 669–84 Search PubMed.
- C. Lange and C. Hunte, Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c, Proc. Acad. Sci. U. S. A., 2002, 99, 2800–05 Search PubMed.
- K. H. Xiao, A. Chandrasekaran, L. Yu and C. A. Yu, Evidence for the intertwined dimer of the cytochrome bc1 complex in solution, J. Biol. Chem., 2001, 276, 46125–31 CrossRef CAS.
- K. H. Leung and P. C. Hinkle, Reconstitution of ion transport and respiratory control in vesicles formed from reduced coenzyme Q-cytochrome c reductase and phospholipids, J. Biol. Chem., 1975, 250, 8467–71 CAS.
- E. C. Hurt, N. Gabellini, Y. Shahak, W. Lockau and G. Hauska, Extra proton translocation and membrane potential generation-universal properties of cytochrome bc1/b6f complexes reconstituted into liposomes, Arch. Biochem. Biophys., 1983, 225, 879–85 CrossRef CAS.
- P. Mitchell, Proton motive redox mechanism of the cytochrome b–c1 complex in the respiratory chain: Proton motive ubiquinone cycle, FEBS Lett., 1975, 56, 1–6 CrossRef CAS.
- P. Mitchell, Possible molecular mechanisms of the protonmotive function of cytochrome systems, J. Theor. Biol., 1976, 62, 327–67 CrossRef CAS.
- M. K. Wikstrom and J. A. Berden, Oxidoreduction of cytochrome b in the presence of antimycin, Biochim. Biophys. Acta, 1972, 283, 403–20 CAS.
- O. A. Gopta, B. A. Feniouk, W. Junge and A. Y. Mulkidjanian, The cytochrome bc1 complex of Rhodobacter capsulatus: ubiquinol oxidation in a dimeric Q-cycle?, FEBS Lett., 1998, 431, 291–96 CrossRef CAS.
- C. Hunte, H. Palsdottir and B. L. Trumpower, Protonmotive pathways and mechanisms in the cytochrome bc1 complex, FEBS Lett., 2003, 545, 39–46 CrossRef CAS.
- A. Osyczka, C. C. Moser, F. Daldal and P. L. Dutton, Reversible redox energy coupling in electron transfer chains, Nature, 2004, 427, 607–12 CrossRef CAS.
- P. R. Rich, The quinone chemistry of bc complexes, Biochim. Biophys. Acta, 2004, 1658, 165–171 CAS.
- A. Y. Mulkidjanian, Ubiquinol oxidation in the cytochrome bc complex: Reaction mechanism and prevention of short-circuiting, Biochim. Biophys. Acta, 2005, 1709, 5–34 CAS.
- H. Myllykallio, F. Drepper, P. Mathis and F. Daldal, Electron-transfer supercomplexes in photosynthesis and respiration, Trends Microbiol., 2000, 8, 493–94 CrossRef CAS.
- X. Gao, X. Wen, L. Esser, B. Quinn, L. Yu, C. A. Yu and D. Xia, Structural basis for the quinone reduction in the bc1 complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site, Biochemistry, 2003, 42, 9067–80 CrossRef CAS.
- H. Palsdottir, C. G. Lojero, B. L. Trumpower and C. Hunte, Structure of the yeast cytochrome bc1 complex with a hydroxyquinone anion Qo site inhibitor bound, J. Biol. Chem., 2003, 278, 31303–11 CrossRef CAS.
- L. Esser, B. Quinn, Y. F. Li, M. Zhang, M. Elberry, L. Yu, C. A. Yu and D. Xia, Crystallographic studies of quinol oxidation site inhibitors: a modified classification of inhibitors for the cytochrome bc1 complex, J. Mol. Biol., 2004, 341, 281–302 CrossRef CAS.
- A. R. Crofts, The cytochrome bc1 complex: function in the context of structure, Annu. Rev. Physiol., 2004, 66, 689–733 CrossRef CAS.
- M. K. Wikstrom, The different cytochrome b components in the respiratory chain of animal mitochondria and their role in electron transport and energy conservation, Biochim. Biophys. Acta, 1973, 301, 155–93 CAS.
- W. H. van den Berg, R. C. Prince, C. L. Bashford, K. I. Takamiya, W. D. Bonner, Jr. and P. L. Dutton, Electron and proton transport in the ubiquinone cytochrome b–c2 oxidoreductase of Rhodopseudomonas sphaeroides. Patterns of binding and inhibition by antimycin, J. Biol. Chem., 1979, 254, 8594–604 CAS.
- L. S. Huang, D. Cobessi, E. Y. Tung and E. A. Berry, Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen-bonding pattern, J. Mol. Biol., 2005, 351, 573–97 CrossRef CAS.
- S. W. Meinhardt and A. R. Crofts, The site and mechanism of action of myxothiazol as an inhibitor of electron transfer in Rhodopseudomonas sphaeroides, FEBS Lett., 1982, 149, 217–22 CrossRef.
- U. Brandt, U. Haase, H. Schagger and G. von Jagow, Significance of the “Rieske” iron–sulfur protein for formation and function of the ubiquinol-oxidation pocket of mitochondrial cytochrome c reductase (bc1 complex), J. Biol. Chem., 1991, 266, 19958–64 CAS.
- A. R. Crofts, B. Barquera, R. B. Gennis, R. Kuras, M. Guergova-Kuras and E. A. Berry, Mechanism of ubiquinol oxidation by the bc1 complex: Different domains of the quinol binding pocket and their role in the mechanism and binding of inhibitors, Biochemistry, 1999, 38, 15807–26 CrossRef CAS.
- L. Zhang, Z. Li, B. Quinn, L. Yu and C. A. Yu, Nonoxidizable ubiquinol derivatives that are suitable for the study of the ubiquinol oxidation site in the cytochrome bc1 complex, Biochim. Biophys. Acta, 2002, 1556, 226–32 CAS.
- L. Esser, X. Gong, S. Yang, L. Yu, C. A. Yu and D. Xia, Surface-modulated motion switch: capture and release of iron–sulfur protein in the cytochrome bc1 complex, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13045–50 CrossRef CAS.
- C. R. D. Lancaster and H. Michel, The coupling of light-induced electron transfer and proton uptake as derived from crystal structures of reaction centres from Rhodopseudomonas viridis modified at the binding site of the secondary quinone, QB, Structure, 1997, 5, 1339–59 CrossRef CAS.
- A. R. Crofts, S. Hong, N. Ugulava, B. Barquera, R. Gennis, M. Guergova-Kuras and E. A. Berry, Pathways for proton release during ubihydroquinone oxidation by the bc1 complex, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10021–26 CrossRef CAS.
- B. L. Trumpower, A concerted, alternating sites mechanism of ubiquinol oxidation by the dimeric cytochrome bc1 complex, Biochim. Biophys. Acta, 2002, 1555, 166–73 CAS.
- J. L. Cape, M. K. Bowman and D. M. Kramer, Reaction intermediates of quinol oxidation in a photoactivatable system that mimics electron transfer in the cytochrome bc1 complex, J. Am. Chem. Soc., 2005, 127, 4208–15 CrossRef CAS.
- E. B. Gutierrez-Cirlos and B. L. Trumpower, Inhibitory analogs of ubiquinol act anti-cooperatively on the yeast cytochrome bc1 complex. Evidence for an alternating, half-of-the-sites mechanism of ubiquinol oxidation, J. Biol. Chem., 2002, 277, 1195–202 CrossRef CAS.
- R. Covian and B. L. Trumpower, Regulatory interactions between ubiquinol oxidation and ubiquinone reduction sites in the dimeric cytochrome bc1 complex, J. Biol. Chem., 2006, 281, 30925–32 CrossRef CAS.
- S. de Vries, S. P. Albracht, J. A. Berden and E. C. Slater, The pathway of electrons through QH2:cytochrome c oxidoreductase studied by pre-steady-state kinetics, Biochim. Biophys. Acta, 1982, 681, 41–53 CAS.
- S. de Vries, S. P. Albracht, J. A. Berden, C. A. Marres and E. C. Slater, The effect of pH, ubiquinone depletion and myxothiazol on the reduction kinetics of the prosthetic groups of ubiquinol:cytochrome c oxidoreductase, Biochim. Biophys. Acta, 1983, 723, 91–103 CAS.
- R. Covian and B. L. Trumpower, Rapid electron transfer between monomers when the cytochrome bc1 complex dimer is reduced through center N, J. Biol. Chem., 2005, 280, 22732–40 CrossRef CAS.
- X. Gong, L. Yu, D. Xia and C. A. Yu, Evidence for electron equilibrium between the two hemes bL in the dimeric cytochrome bc1 complex, J. Biol. Chem., 2005, 280, 9251–57 CAS.
- W. Junge and J. B. Jackson, The development of electrochemical potential gradients across photosynthetic membranes, in Photosynthesis, ed. Govindjee, Academic Press, New York, 1982, vol. 1, pp. 589–646 Search PubMed.
- A. R. Crofts and C. A. Wraight, The electrochemical domain of photosynthesis, Biochim. Biophys. Acta, 1983, 726, 149–85 CrossRef CAS.
- J. B. Jackson, Bacterial photosynthesis, in Bacterial energy transduction, ed. C. Anthony, Academic Press, London, 1988, pp. 317–376 Search PubMed.
- S. S. Klishin, W. Junge and A. Y. Mulkidjanian, Flash-induced turnover of the cytochrome bc1 complex in chromatophores of Rhodobacter capsulatus: binding of Zn2+ decelerates likewise the oxidation of cytochrome b, the reduction of cytochrome c1 and the voltage generation, Biochim. Biophys. Acta, 2002, 1553, 177–82 CAS.
- W. W. Parson, Bacterial photosynthesis, Annu. Rev. Microbiol., 1974, 28, 41–59 CrossRef CAS.
- P. L. Dutton and R. C. Prince, Reaction center driven cytochrome interactions in electron and proton translocation and energy conservation, in The Photosynthetic Bacteria, ed. R. K. Clayton and W. R. Sistrom, Plenum Press, New York, 1978, pp. 525–570 Search PubMed.
- C. A. Wraight, R. J. Cogdell and B. Chance, Ion transport and electrochemical gradients in photosynthetic bacteria, in The Photosynthetic Bacteria, ed. R. K. Clayton and W. R. Sistrom, Plenum Press, New York, 1978, pp. 471–511 Search PubMed.
- J. B. Jackson and A. R. Crofts, The kinetics of light induced carotenoid changes in Rhodopseudomonas sphaeroides and their relation to electrical field generation across the chromatophore membrane, Eur. J. Biochem., 1971, 18, 120–30 CrossRef CAS.
- L. A. Drachev, M. D. Mamedov, A. Y. Mulkidjanian, A. Y. Semenov, V. P. Shinkarev, M. I. Verkhovsky, B. S. Kaurov and V. P. Skulachev, Flash-induced electrogenic events in the photosynthetic reaction center and cytochrome bc1-complex of Rhodobacter sphaeroides chromatophores, Biochim. Biophys. Acta, 1989, 973, 189–97 CAS.
- A. Y. Mulkidjanian, M. D. Mamedov, A. Y. Semenov, V. P. Shinkarev, M. I. Verkhovsky and L. A. Drachev, Partial reversion of electrogenic reaction in ubiquinol: cytochrome c2 oxidoreductase of Rhodobacter sphaeroides under the neutral and alkaline conditions, FEBS Lett., 1990, 277, 127–30 CrossRef.
- D. E. Robertson, H. Ding, P. R. Chelminski, C. Slaughter, J. Hsu, C. Moomaw, M. Tokito, F. Daldal and P. L. Dutton, Hydroubiquinone–cytochrome c2 oxidoreductase from Rhodobacter capsulatus: definition of a minimal, functional isolated preparation, Biochemistry, 1993, 32, 1310–17 CrossRef CAS.
- R. B. Gennis, B. Barquera, B. Hacker, S. R. Van Doren, S. Arnaud, A. R. Crofts, E. Davidson, K. A. Gray and F. Daldal, The bc1 complexes of Rhodobacter sphaeroides and Rhodobacter capsulatus, J. Bioenerg. Biomembr., 1993, 25, 195–209 CrossRef CAS.
- G. Brasseur, A. S. Saribas and F. Daldal, A compilation of mutations located in the cytochrome b subunit of the bacterial and mitochondrial bc1 complex, Biochim. Biophys. Acta, 1996, 1275, 61–69.
- S. G. E. Andersson, A. Zomorodipour, J. O. Andersson, T. Sicheritz-Ponten, U. C. M. Alsmark, R. M. Podowski, A. K. Naslund, A. S. Eriksson, H. H. Winkler and C. G. Kurland, The genome sequence of Rickettsia prowazekii and the origin of mitochondria, Nature, 1998, 396, 133–40 CrossRef CAS.
- The Photosynthetic Bacteria, ed. R. K. Clayton and W. R. Sistrom, Plenum Press, New York, 1978 Search PubMed.
- J. R. Bowyer and A. R. Crofts, On the mechanism of photosynthetic electron transfer in Rhodopseudomonas capsulata and Rhodopseudomonas sphaeroides, Biochim. Biophys. Acta, 1981, 636, 218–33 CAS.
- D. P. O'Keefe and P. L. Dutton, Cytochrome b oxidation and reduction reactions in the ubiquinone–cytochrome b/c2 oxidoreductase from Rhodopseudomonas sphaeroides, Biochim. Biophys. Acta, 1981, 635, 149–66 CAS.
- G. Venturoli, J. G. Fernandez-Velasco, A. R. Crofts and B. A. Melandri, Demonstration of a collisional interaction of ubiquinol with the ubiquinol–cytochrome c2 oxidoreductase complex in chromatophores from Rhodobacter sphaeroides, Biochim. Biophys. Acta, 1986, 851, 340–52 CAS.
- A. Y. Mulkidjanian and W. Junge, Calibration and time resolution of lumenal pH-transients in chromatophores of Rhodobacter capsulatus following a single turnover flash of light: proton release by the cytochrome bc1-complex is strongly electrogenic, FEBS Lett., 1994, 353, 189–93 CrossRef CAS.
- A. Y. Mulkidjanian and W. Junge, Electrogenic proton displacements in the cytochrome bc1 complex of Rhodobacter capsulatus, in Photosynthesis: from Light to Biosphere, ed. P. Mathis, Kluwer Academic Publishers, Dordrecht, 1995, pp. 547–550 Search PubMed.
- O. A. Gopta and A. Y. Mulkidjanian, The cytochrome bc1-complex of Rhodobacter capsulatus: does the reaction in the presence of antimycin A correspond to a single turnover of an untreated enzyme? in Photosynthesis: Mechanisms and Effects, ed. G. Garab, Kluwer Academic Publishers, Dordrecht, 1998, pp. 1533–1536 Search PubMed.
- K. I. Takamiya and P. L. Dutton, Ubiquinone in Rhodopseudomonas sphaeroides. Some thermodynamic properties, Biochim. Biophys. Acta, 1979, 546, 1–16 CAS.
- S. Meinhardt and A. R. Crofts, Kinetic and thermodynamic resolution of cytochrome c1 and cytochrome c2 from Rhodopseudomonas sphaeroides, FEBS Lett., 1982, 149, 223–27 CrossRef CAS.
- D. E. Robertson and P. L. Dutton, The nature and magnitude of the charge-separation reactions of ubiquinol cytochrome c2 oxidoreductase, Biochim. Biophys. Acta, 1988, 935, 273–91 CAS.
- J. B. Jackson and A. R. Crofts, High energy state in chromatophores of Rhodopseudomonas sphaeroides, FEBS Lett., 1969, 4, 185–89 CrossRef CAS.
- R. J. Cogdell, J. B. Jackson and A. R. Crofts, The effect of redox potential on the coupling between rapid hydrogen-ion binding and electron transport in chromatophores from Rhodopseudomonas spheroides, J. Bioenerg, 1973, 4, 211–27 Search PubMed.
- J. B. Jackson and P. L. Dutton, The kinetic and redox potentiometric resolution of the carotenoid shifts in Rhodopseudomonas sphaeroides chromatophores: their relationship to electric field alterations in electron transport and energy coupling, Biochim. Biophys. Acta, 1973, 325, 102–13 CAS.
- R. C. Prince and P. L. Dutton, Single and multiple turnover reactions in the ubiquinone-cytochrome b–c2 oxidoreductase of Rhodopseudomonas sphaeroides: the physical chemistry of the major electron donor to cytochrome c2, and its coupled reactions, Biochim. Biophys. Acta, 1977, 462, 731–47 CAS.
- K. Takamiya, R. C. Prince and P. L. Dutton, The recognition of a special ubiquinone functionally central in the ubiquinone–cytochrome b–c2 oxidoreductase, J. Biol. Chem., 1979, 254, 11307–11 CAS.
- C. L. Bashford, R. C. Prince, K. I. Takamiya and P. L. Dutton, Electrogenic events in the ubiquinone–cytochrome b/c2 oxidoreductase of Rhodopseudomonas sphaeroides, Biochim. Biophys. Acta, 1979, 545, 223–35 CAS.
- J. R. Bowyer, P. L. Dutton, R. C. Prince and A. R. Crofts, The role of the Rieske iron–sulfur center as the electron donor to ferricytochrome c2 in Rhodopseudomonas sphaeroides, Biochim. Biophys. Acta, 1980, 592, 445–60 CAS.
- W. A. Cramer and A. R. Crofts, Electron and proton transport, in Photosynthesis, ed. Govindjee, Academic Press, New York, 1982, vol. 1, pp. 387–467 Search PubMed.
- A. R. Crofts, S. W. Meinhardt, K. R. Jones and M. Snozzi, The role of the quinone pool in the cyclic electron transfer chain of Rhodopseudomonas sphaeroides, Biochim. Biophys. Acta, 1983, 723, 202–18 CAS.
- P. B. Garland, R. A. Clegg, D. Boxer, J. A. Downie and B. A. Haddock, Proton-translocating nitrate reductase of Escherichia coli, in Electron Transfer Chains and Oxidative Phosphorylation, ed. E. Quagliariello, S. Papa, F. Palmieri, E. C. Slater and N. Siliprandi, North-Holland Publishing, Amsterdam, 1975, pp. 351–358 Search PubMed.
- D. E. Robertson, R. C. Prince, J. R. Bowyer, K. Matsuura, P. L. Dutton and T. Ohnishi, Thermodynamic properties of the semiquinone and its binding site in the ubiquinol–cytochrome c (c2) oxidoreductase of respiratory and photosynthetic systems, J. Biol. Chem., 1984, 259, 1758–63 CAS.
- E. G. Glaser and A. R. Crofts, A new electrogenic step in the ubiquinol:cytochrome c2 oxidoreductase complex of Rhodopseudomonas sphaeroides, Biochim. Biophys. Acta, 1984, 766, 322–33 CAS.
- M. Saraste, Location of haem-binding sites in the mitochondrial cytochrome b, FEBS Lett., 1984, 166, 367–72 CrossRef CAS.
- A. R. Crofts, H. H. Robinson, K. Andrews, S. Van Doren and E. Berry, Catalytic sites for reduction and oxidation of quinones, in Cytochrome Systems: Molecular Biology and Bioenergetics, ed. S. Papa, B. Chance and L. Ernster, Plenum Publisher, New York, 1987, pp. 617–624 Search PubMed.
- E. Glaser and A. R. Crofts, Studies of the electrogenicity of the reduction of cytochrome b561 through the antimycin-sensitive site of the ubiquinol–cytochrome c2 complex of Rhodobacter sphaeroides, in Cytochrome systems: Molecular Biology and Bioenergetics, ed. S. Papa, B. Chance and L. Ernster, Plenum Press, New York, 1988, pp. 625–631 Search PubMed.
- V. P. Shinkarev, A. L. Drachev, M. D. Mamedov, A. Y. Mulkidjanian, A. Y. Semenov and M. I. Verkhovsky, Light-induced electron transfer and electrogenic reactions in the bc1-complex of photosynthetic purple bacterium Rhodobacter sphaeroides, in Molecular Biology of Membrane-Bound Complexes in Phototrophic Bacteria, ed. G. Drews and E. A. Dawes, Plenum Press, New York, 1990, pp. 393–400 Search PubMed.
- E. G. Glaser, S. W. Meinhardt and A. R. Crofts, Reduction of cytochrome b-561 through the antimycin-sensitive site of the ubiquinol–cytochrome c2 oxidoreductase complex of Rhodopseudomonas sphaeroides, FEBS Lett., 1984, 178, 336–42 CrossRef CAS.
- D. E. Robertson, K. M. Giangiacomo, S. de Vries, C. C. Moser and P. L. Dutton, Two distinct quinone-modulated modes of antimycin-sensitive cytochrome b reduction in the cytochrome bc1 complex, FEBS Lett., 1984, 178, 343–50 CrossRef CAS.
- A. A. Konstantinov and E. Popova, Topography and Protonmotive Mechanism of Mitochondrial Coupling Site 2, in Cytochrome systems: Molecular Biology and Bioenergetics, ed. S. Papa, B. Chance and L. Ernster, Plenum Press, New York, 1988, pp. 751–765 Search PubMed.
- A. A. Konstantinov, Vectorial electron and proton transfer in the cytochrome bc1 complex, Biochim. Biophys. Acta, 1990, 1018, 138–41 CAS.
- P. Mitchell, The classical mobile carrier function of lipophilic quinones in the osmochemistry of electron-driven proton translocation, in Highlights in Ubiquinone Research, ed. G. Lenaz, Taylor and Francis, London, 1990, pp. 77–82 Search PubMed.
- A. Y. Mulkidjanian, M. D. Mamedov and L. A. Drachev, Slow electrogenic events in the cytochrome bc1-complex of Rhodobacter sphaeroides: The electron transfer between cytochrome b hemes can be non-electrogenic, FEBS Lett., 1991, 284, 227–31 CrossRef.
- A. Y. Semenov, D. A. Bloch, A. R. Crofts, L. A. Drachev, R. B. Gennis, A. Y. Mulkidjanian, C.-H. Yun, V. P. Shinkarev, M. I. Verkhovsky, B. S. Kaurov and V. P. Skulachev, Electrogenic events in chromatophores from Rhodobacter sphaeroides lacking high-potential cytochrome b of the bc1-complex, Biochim. Biophys. Acta, 1992, 1101, 166–67 CAS.
- A. Y. Mulkidjanian, M. I. Verkhovsky, V. P. Shinkarev, V. D. Sled', N. P. Grishanova and B. S. Kaurov, Study of photosynthetic electron transfer reactions by redox probes: Electron transport in non-sulfur purple bacteria, Biokhimiya, 1985, 50, 1786–96 Search PubMed.
- R. Mitchell and P. R. Rich, Proton uptake by cytochrome c oxidase on reduction and on ligand binding, Biochim. Biophys. Acta, 1994, 1186, 19–26 CAS.
- B. Chance, A. R. Crofts, M. Nishimura and B. Price, Fast membrane H+ binding in the light-activated state of Chromatium chromatophores, Eur. J. Biochem., 1970, 13, 364–74 CrossRef CAS.
- M. A. Taylor and J. B. Jackson, Rapid proton release accompanying photosynthetic electron transport in intact cells of Rhodopseudomonas capsulata, FEBS Lett., 1985, 180, 145–49 CrossRef CAS.
- M. R. Jones and J. B. Jackson, Proton release by the quinol oxidase site of the cytochrome b/c1 complex following single turnover flash excitation of intact cells of Rhodobacter capsulatus, Biochim. Biophys. Acta, 1989, 975, 34–43 CAS.
- M. R. Jones and J. B. Jackson, Proton efflux from right-side-out membrane vesicles of Rhodobacter sphaeroides after short flashes, Biochim. Biophys. Acta, 1990, 1019, 51–58 CAS.
- K. M. Petty and P. L. Dutton, Properties of the flash-induced proton binding encountered in membranes of Rhodopseudomonas sphaeroides: a functional pK on the ubisemiquinone?, Arch. Biochem. Biophys., 1976, 172, 335–45 CrossRef CAS.
- K. M. Petty and P. L. Dutton, Ubiquinone–cytochrome b electron and proton transfer: a functional pK on cytochrome b50 in Rhodopseudomonas sphaeroides membranes, Arch. Biochem. Biophys., 1976, 172, 346–53 CrossRef CAS.
- K. M. Petty, J. B. Jackson and P. L. Dutton, Kinetics and stoichiometry of proton binding in Rhodopseudomonas sphaeroides chromatophores, FEBS Lett., 1977, 84, 299–303 CrossRef CAS.
- K. Petty, J. B. Jackson and P. L. Dutton, Factors controlling the binding of two protons per electron transferred through the ubiquinone and cytochrome b/c2 segment of Rhodopseudomonas sphaeroides chromatophores, Biochim. Biophys. Acta, 1979, 546, 17–42 CAS.
- K. Matsuura, D. P. O'Keefe and P. L. Dutton, A reevaluation of the events leading to the electrogenic reaction and proton translocation in the ubiquinol–cytochrome c oxidoreductase of Rhodopseudomonas sphaeroides, Biochim. Biophys. Acta, 1983, 722, 12–22 CAS.
- S. Saphon and P. Gräber, External proton uptake, internal proton release and internal pH changes in chromatophores from Rps. sphaeroides following single turnover flashes, Z. Naturforsch., C: Biosci., 1978, 33C, 715–22 CAS.
- C. C. Moser, J. M. Keske, K. Warncke, R. S. Farid and P. L. Dutton, Nature of biological electron transfer, Nature, 1992, 355, 796–802 CrossRef CAS.
- V. P. Skulachev, V. V. Chistyakov, A. A. Jasaitis and E. G. Smirnova, Inhibition of the respiratory chain by zinc ions, Biochem. Biophys. Res. Commun., 1967, 26, 1–6 CrossRef CAS.
- T. A. Link and G. von Jagow, Zinc ions inhibit the QP center of bovine heart mitochondrial bc1 complex by blocking a protonatable group, J. Biol. Chem., 1995, 270, 25001–06 CrossRef CAS.
- S. S. Klishin, D. A. Cherepanov and A. Y. Mulkidjanian, Resolution of proton and electron transfer events in the photosynthetic reaction center and the cytochrome-bc1 complex of phototrophic bacteria, in PS2001. 12th International Congress of Photosynthesis, Brisbane, 2001, pp. S12-005 Search PubMed.
- S. S. Klishin and A. Y. Mulkidjanian, Proton transfer paths at the quinol oxidizing site of the Rb. capsulatus cytochrome bc complex, in Photosynthesis: Fundamental aspects to global perspectives, ed. A. van der Est and D. Bruce, Montreal, vol. 1, 2005, pp. 260–262 Search PubMed.
- S. S. Klishin, Resolution of Partial Steps in the Catalytic Cycle of Rhodobacter capsulatus Cytochrome bc1 Complex with Help of Zinc Ions, PhD Thesis, Lomonosov University, Moscow, 2005 Search PubMed.
- P. L. Dutton and J. B. Jackson, Thermodynamic and kinetic characterization of electron transfer components in situ in Rhodopseudomonas sphaeroides and Rhodospirillum rubrum, Eur. J. Biochem., 1972, 30, 495–510 CrossRef CAS.
- J. N. Siedow, S. Power, F. F. de la Rosa and G. Palmer, The preparation and characterization of highly purified, enzymically active complex III from baker's yeast, J. Biol. Chem., 1978, 253, 2392–99 CAS.
- J. C. Salerno, Y. Xu, M. P. Osgood, C. H. Kim and T. E. King, Thermodynamic and spectroscopic characteristics of the cytochrome-bc1 complex—role of quinone in the behavior of cytochrome-b562, J. Biol. Chem., 1989, 264, 15398–403 CAS.
- P. R. Rich, A. E. Jeal, S. A. Madgwick and A. J. Moody, Inhibitor effects on redox-linked protonations of the b haems of the mitochondrial bc1 complex, Biochim. Biophys. Acta, 1990, 1018, 29–40 CAS.
- F. F. de la Rosa and G. Palmer, Reductive titration of CoQ-depleted Complex III from baker's yeast. Evidence for an exchange-coupled complex between QH˙ and low-spin ferricytochrome b, FEBS Lett., 1983, 163, 140–43 CrossRef CAS.
- S. de Vries, The pathway of electron transfer in the dimeric QH2: cytochrome c oxidoreductase, J. Bioenerg. Biomembr., 1986, 18, 195–224 CrossRef CAS.
- S. W. Meinhardt, X. H. Yang, B. L. Trumpower and T. Ohnishi, Identification of a stable ubisemiquinone and characterization of the effects of ubiquinone oxidation–reduction status on the Rieske iron–sulfur protein in the three-subunit ubiquinol–cytochrome c oxidoreductase complex of Paracoccus denitrificans, J. Biol. Chem., 1987, 262, 8702–06 CAS.
- S. de Vries, J. A. Berden and E. C. Slater, Properties of a semiquinone anion located in the QH2:cytochrome c oxidoreductase segment of the mitochondrial respiratory chain, FEBS Lett., 1980, 122, 143–48 CrossRef CAS.
- A. R. Crofts, The mechanism of the ubiquinol:cytochrome c oxidoreductases of mitochondria and of Rhodopseudomonas sphaeroides, in The Enzymes of Biological Membranes. Vol. 4. Bioenergetics of Electron and Proton Transport, ed. A. N. Martonosi, Plenum Press, New York and London, 1985, pp. 347–382 Search PubMed.
- A. Y. Mulkidjanian, V. P. Shinkarev, M. I. Verkhovsky and B. S. Kaurov, A study of kinetic properties of the stable semiquinone of the reaction center secondary acceptor in chromatophores of nonsulfur purple bacteria, Biochim. Biophys. Acta, 1986, 849, 150–61.
- A. R. Crofts, V. P. Shinkarev, D. R. Kolling and S. Hong, The modified Q-cycle explains the apparent mismatch between the kinetics of reduction of cytochrome c1 and bh in the bc1 complex, J. Biol. Chem., 2003, 278, 36191–201 CrossRef CAS.
- K. Takamiya and P. L. Dutton, The influence of transmembrane potentials of the redox equilibrium between cytochrome c2 and the reaction center in Rhodopseudomonas sphaeroides chromatophores, FEBS Lett., 1977, 80, 279–84 CrossRef CAS.
- D. A. Cherepanov, L. I. Krishtalik and A. Y. Mulkidjanian, Photosynthetic electron transfer controlled by protein relaxation: analysis by Langevin stochastic approach, Biophys. J., 2001, 80, 1033–49 CrossRef CAS.
- V. P. Shinkarev, A. R. Crofts and C. A. Wraight, The electric field generated by photosynthetic reaction center induces rapid reversed electron transfer in the bc1 complex, Biochemistry, 2001, 40, 12584–90 CrossRef CAS.
- B. A. Feniouk, M. A. Kozlova, D. A. Knorre, D. A. Cherepanov, A. Y. Mulkidjanian and W. Junge, The proton driven rotor of ATP synthase: Ohmic conductance (10 fS), and absence of voltage gating, Biophys. J., 2004, 86, 665–80 CrossRef CAS.
- S. Izrailev, A. R. Crofts, E. A. Berry and K. Schulten, Steered molecular dynamics simulation of the Rieske subunit motion in the cytochrome bc1 complex, Biophys. J., 1999, 77, 1753–68 CrossRef CAS.
- M. L. Paddock, L. Sagle, A. Tehrani, J. T. Beatty, G. Feher and M. Y. Okamura, Mechanism of proton transfer inhibition by Cd2+ binding to bacterial reaction centers: determination of the pKA of functionally important histidine residues, Biochemistry, 2003, 42, 9626–32 CrossRef CAS.
- D. A. Mills, B. Schmidt, C. Hiser, E. Westley and S. Ferguson-Miller, Membrane potential-controlled inhibition of cytochrome c oxidase by zinc, J. Biol. Chem., 2002, 277, 14894–901 CAS.
- A. Aagaard, A. Namslauer and P. Brzezinski, Inhibition of proton transfer in cytochrome c oxidase by zinc ions: delayed proton uptake during oxygen reduction, Biochim. Biophys. Acta, 2002, 1555, 133–39 CAS.
- T. E. DeCoursey, Voltage-gated proton channels and other proton transfer pathways, Physiol. Rev., 2003, 83, 475–579 Search PubMed.
- E. A. Berry, Z. Zhang, H. D. Bellamy and L. Huang, Crystallographic location of two Zn2+-binding sites in the avian cytochrome bc1 complex, Biochim. Biophys. Acta, 2000, 1459, 440–48 CAS.
- A. Y. Mulkidjanian, Proton in the well and through the desolvation barrier, Biochim. Biophys. Acta, 2006, 1757, 415–27 CAS.
- L. Giachini, F. Francia, D.-W. Lee, F. Daldal, L.-S. Huang, E. A. Berry, T. Cocco, S. Papa, F. Boscherini and G. Venturoli, X-Ray absorption studies of Zn2+ binding sites in bacterial, avian and bovine cytochrome bc1 complexes, Biochim. Biophys. Acta, 2006, 1757(Supplement 1, 14th EBEC Short Reports), 169–170.
- E. Darrouzet and F. Daldal, Movement of the iron–sulfur subunit beyond the ef loop of cytochrome b is required for multiple turnovers of the bc1 complex but not for single turnover QO site catalysis, J. Biol. Chem., 2002, 277, 3471–76 CrossRef CAS.
- E. Darrouzet and F. Daldal, Protein–protein interactions between cytochrome b and the Fe–S protein subunits during QH2 oxidation and large-scale domain movement in the bc1 complex, Biochemistry, 2003, 42, 1499–507 CrossRef CAS.
- T. E. King, C. A. Yu, L. Yu and Y. L. Chiang, An examination of the components, sequence, mechanisms and their uncertainties involved in mitochondrial electron transport from succinate to cytochrome c, in Electron Transfer Chain and Oxidative Phosphorylation, ed. E. Quagliariello, S. Papa, F. Palmieri, E. C. Slater and N. Siliprandi, North-Holland Publ. Co, Amsterdam, 1975, pp. 105–118 Search PubMed.
- C. H. Snyder and B. L. Trumpower, Ubiquinone at center N is responsible for triphasic reduction of cytochrome b in the cytochrome bc1 complex, J. Biol. Chem., 1999, 274, 31209–16 CrossRef CAS.
- K. C. Hansen, B. E. Schultz, G. Wang and S. I. Chan, Reaction of Escherichia coli cytochrome bo3 and mitochondrial cytochrome bc1 with a photoreleasable decylubiquinol, Biochim. Biophys. Acta, 2000, 1456, 121–37 CAS.
- M. Wikstrom, K. Krab and M. Saraste, Proton-translocating cytochrome complexes, Annu. Rev. Biochem., 1981, 50, 623–55 CrossRef CAS.
- E. Darrouzet, M. Valkova-Valchanova and F. Daldal, The [2Fe–2S] cluster Em as an indicator of the iron–sulfur subunit position in the ubihydroquinone oxidation site of the cytochrome bc1 complex, J. Biol. Chem., 2002, 277, 3464–70 CrossRef CAS.
- V. P. Shinkarev, D. R. J. Kolling, T. J. Miller and A. R. Crofts, Modulation of the midpoint potential of the [2Fe–2S] Rieske iron sulfur center by QO occupants in the bc1 complex, Biochemistry, 2002, 41, 14372–82 CrossRef CAS.
- J. W. Cooley, T. Ohnishi and F. Daldal, Binding dynamics at the quinone reduction (Qi) site influence the equilibrium interactions of the iron sulfur protein and hydroquinone oxidation (QO) site of the cytochrome bc1 complex, Biochemistry, 2005, 44, 10520–32 CrossRef CAS.
- M. Valkova-Valchanova, E. Darrouzet, C. R. Moomaw, C. A. Slaughter and F. Daldal, Proteolytic cleavage of the Fe–S subunit hinge region of Rhodobacter capsulatus bc1 complex: effects of inhibitors and mutations, Biochemistry, 2000, 39, 15484–92 CrossRef CAS.
- P. R. Rich, Electron and proton transfers through quinones and cytochrome bc complexes, Biochim. Biophys. Acta, 1984, 768, 53–79 CrossRef CAS.
- D. E. Robertson, E. Davidson, R. C. Prince, W. H. van den Berg, B. L. Marrs and P. L. Dutton, Discrete catalytic sites for quinone in the ubiquinol–cytochrome c2 oxidoreductase of Rhodopseudomonas capsulata. Evidence from a mutant defective in ubiquinol oxidation, J. Biol. Chem., 1986, 261, 584–91 CAS.
- K. Matsuura, J. R. Bowyer, T. Ohnishi and P. L. Dutton, Inhibition of electron transfer by 3-alkyl-2-hydroxy-1,4-naphthoquinones in the ubiquinol–cytochrome c oxidoreductases of Rhodopseudomonas sphaeroides and mammalian mitochondria. Interaction with a ubiquinone-binding site and the Rieske iron–sulfur cluster, J. Biol. Chem., 1983, 258, 1571–79 CAS.
- H. Ding, D. E. Robertson, F. Daldal and P. L. Dutton, Cytochrome bc1 complex [2Fe–2S] cluster and its interaction with ubiquinone and ubihydroquinone at the Qo site: a double-occupancy Qo site model, Biochemistry, 1992, 31, 3144–58 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef.
- A. Y. Mulkidjanian, J. Heberle and D. A. Cherepanov, Protons @ interfaces: Implications for biological energy conversion, Biochim. Biophys. Acta, 2006, 1757, 913–30 CAS.
- S. S. Klishin, S. S. and A. Y. Mulkidjanian, Breaking down the flash-induced turnover of the cytochrome bc1 complex into separate steps, Biochim. Biophys. Acta, 2004, 1658(Supplement 1, 13th EBEC Short Reports), 170.
- N. Gabellini and G. Hauska, Characterization of cytochrome b in the isolated ubiquinol–cytochrome c2 oxidoreductase from Rhodopseudomonas sphaeroides GA, FEBS Lett., 1983, 153, 146–50 CrossRef CAS.
- G. Hauska, E. Hurt, N. Gabellini and W. Lockau, Comparative aspects of quinol–cytochrome c/plastocyanin oxidoreductases, Biochim. Biophys. Acta, 1983, 726, 97–133 CrossRef CAS.
- D. A. Cherepanov, W. Junge and A. Y. Mulkidjanian, Proton transfer dynamics at the membrane/water interface: Dependence on the fixed and mobile pH buffers, the geometry of membrane particles, and the interfacial potential barrier, Biophys. J., 2004, 86, 665–80 CrossRef CAS.
- M. B. Partenskii and P. C. Jordan, Theoretical perspectives on ion-channel electrostatics: continuum and microscopic approach, Q. Rev. Biophys., 1992, 25, 477–510 CrossRef CAS.
Footnotes |
| † Submitted as part of the special issue ‘Proton Transfer in Biological Systems’, Photochem. Photobiol. Sci., 2006, 5(6). |
| ‡ It took some time until the bc1 of Rhodobacter was isolated and the relation of the cytochrome b component of chromatophores to the ubiquinol:cytochrome c oxidoreductase (complex III) of mitochondria became unambiguous.4,155,156 For simplicity, the notion bc1 is used hereafter even in relation to earlier studies where the cytochrome bc1 complex has not been identified as such yet. |
| § The slower acidification in larger cells and spheroplasts, as compared to the vesicles, was caused, most likely, by the dependence of the surface protonic equilibration rate on the size of cells/vesicles, as elucidated elsewhere.157 |
| ¶ The expulsion of non-compensating, redundant proton(s) out of center P, even via a short dielectric distance, can be highly electrogenic. By polarizing water molecules, a non-compensated charge can decrease, up to ∼5, the dielectric permittivity inside a water-filled cavity intruding in a hydrophobic medium.158 |
| This journal is © The Royal Society of Chemistry and Owner Societies 2007 |