Marine metabolites: metal binding and metal complexes of azole-based cyclic peptides of marine origin
Anna Bertram and Gerald Pattenden*
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 16th January 2007
Abstract
Azole-based cyclic peptides found in ascidians (‘sea squirts’) of the genus Lissoclinum have a high propensity to chelate metal ions. This Highlight summarises the current evidence for marine cyclic peptide–metal congruence, and the structural and stereochemical features in cyclic peptides which seem necessary to facilitate metal complexation. The biological relevance of the metal ions in these associations, including their possible role in the assembly of cyclic peptides in the marine milieu, is also briefly considered. Finally, the synthesis of natural, and some novel non-natural, azole-based cyclic peptides from the cyclooligomerisation and assembly of azole-based amino acids, including in the presence of metal ions, is presented.
 Anna Bertram Anna Bertram | Anna Bertram graduated, BSc Hons in Chemistry, from the University of Nottingham in 1997. She then went on to study for a PhD at Nottingham under the supervision of Professor Gerry Pattenden, investigating the synthesis of a range of biologically important natural cyclic peptides via a novel self-assembly approach, also involving metal-templation. Anna stayed in Nottingham as a Postdoctoral Fellow for a further two years, where she researched in the areas of synthesis of steroids and polyoxazole-based telomerase inhibitors. In 2002 Anna was appointed to a Fixed Term Lectureship in Organic Chemistry and she moved to her current position, as Manager of Undergraduate Teaching Laboratories, in the School of Chemistry at Nottingham, in 2005. |
 Gerald Pattenden Gerald Pattenden | ‘Gerry’ Pattenden has been Professor of Organic Chemistry at the University of Nottingham since 1980. His research interests are in designing new synthetic methods in organic chemistry and in target natural product total synthesis, often drawing inspiration from nature’s biosynthetic pathways and assembly methods. His scientific contributions have been recognised by a number of awards from learned societies including, recently, the Robert Robinson Lectureship and the Award for Natural Products Chemistry of the RSC. Professor Pattenden was the first Chairman of the Editorial Board of NPR, when it was established in 1984, and held the position until 1991. |
1 Introduction†
The marine environment is a rich source of novel and structurally diverse bioactive natural products. In 1993 we published a review with the somewhat provocative title “Marine Metabolites and Metal Ion Chelation. The Facts and the Fantasies”, which drew attention to the potential for marine organisms to sequester and transport metal ions.1 In the review we pondered the following questions: How widespread is the evidence for natural product–metal congruence in the marine milieu? What structural and stereochemical features in marine metabolites are necessary to facilitate metal complexation and transport? What is the biological role of the metal ions in these associations? Could metal ions be involved in the assembly of marine natural products in vivo? Finally, could answers to the aforementioned questions be exploited in synthetic studies with marine natural products?Ascidians (‘sea squirts’) of the genus Lissoclinum are a prolific source of unusual cyclic peptides which contain both D- and L-amino acids, many modified in the form of thiazole, oxazole, thiazoline or oxazoline rings. There is now a substantial literature which demonstrates that these marine metabolites, in particular, have a high propensity to chelate metal ions. Furthermore, this has led to speculation regarding the biological functions of cyclic peptide–metal conjugates in the marine milieu, and has suggested a role for metal ions in the assembly of novel and unusual cyclic peptides in vitro. In this Highlight we draw together this literature, published over the past decade, which has answered some of the questions posed in our earlier review.1,2
2 Azole-based cyclic peptides from sea squirts
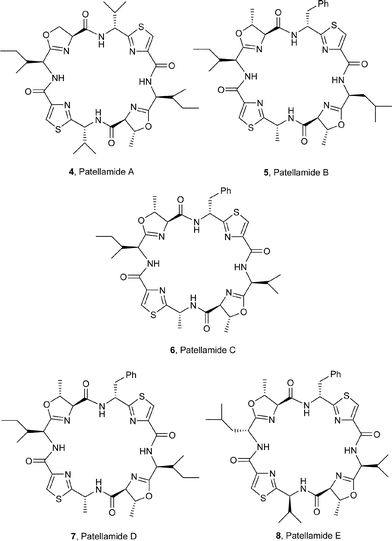
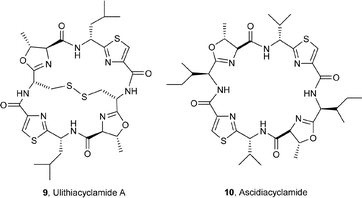
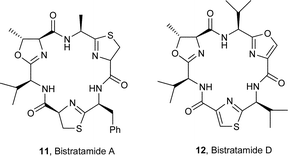
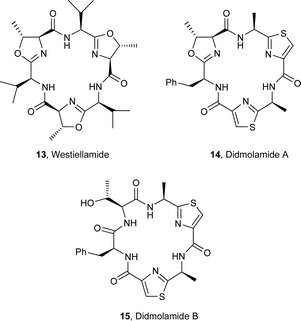
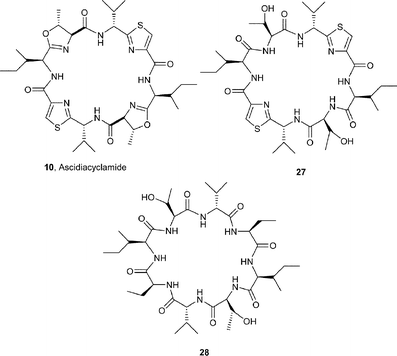
The sea squirt Lissoclinum patella is one of the most extensively investigated marine animals. It accumulates a particularly wide variety of oxazoline-, thiazole- and thiazoline-based cyclic hepta- and octapeptides, e.g. the lissoclinamides 1–3,3 the patellamides 4–8,4 ulithiacyclamide A (9)5 which incorporates an interesting disulfide bridge, and the C2-symmetric compound ascidiacyclamide 10.6 The ‘bistratamides’ e.g.11 and 12, by contrast, are oxazoline-based cyclic hexapeptides isolated from the Great Barrier Reef, Australia, and from the Philippine ascidian L. bistratum.7 These hexapeptides are related to the C3-symmetric tris-oxazoline westiellamide 13 found in L. bistratum,8 and to the bis-thiazole-based didmolamides A (14) and B (15), isolated from the ascidian Didemnum molle collected off the coast of Madagascar.9 Structures similar to the lissoclinamides, patellamides and the bistratamides have also been isolated from some terrestrial cyanobacteria. A special case in point is westiellamide 13, also known as ‘cycloxazoline’, which has been isolated from both L. bistratum and from the cyanobacterium Westiellopsis prolifica.8 The cyanobacterium Prochloron sp. has consistently been found in the tissues of L. patella, and this has led to considerable speculation that the patellamide family of cyclic peptides isolated from the ascidian are actually produced by the symbiotic cyanobacterium.10a More recently, Schmidt et al.10b have studied the genomic sequences of L. patella and its symbiont Prochloron didemni, and demonstrated that the latter has the necessary genes to produce patellamide cyclic peptides. However, despite some effort, it has not yet proved possible to culture P. didemni in the laboratory, independent of the ascidian, once it has been separated from the host.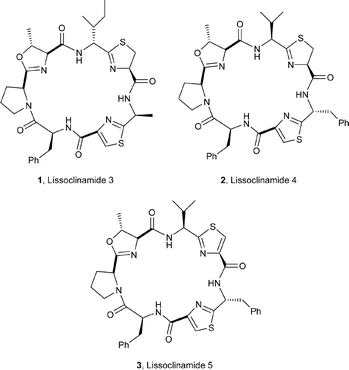
3 Conformational studies with cyclic peptides
Studies of the conformations of azole-based marine cyclic peptides, both in solution and in the solid state, have provided valuable insights into the factors which may affect the reactivities and the biology of these unique secondary metabolites. An early X-ray crystal structure determination of the C2-symmetric ascidiacyclamide 10, for example, showed that it has a ‘square’ (also sometimes referred to as ‘saddle-shaped’) conformation, i.e.16, with its thiazole and oxazoline rings occupying the corners of a rectangle.11 Patellamide A (4) has a similar rectangular shape in the solid state,12 but, by contrast, X-ray analysis of the closely related patellamide D (7) shows that it assumes a ‘twisted figure of eight’ conformation, viz.17/18, stabilised by intramolecular hydrogen-bonds and by π-stacking between its thiazole rings (Fig. 1).13 Ishida et al.14 have suggested that the different conformations found in patellamides A (4) and D (7), in the solid state, are related to the degree of asymmetry in the latter. The authors argue that the ‘square’ conformation 16 in patellamide A (4), (and ascidiacyclamide 10), is able to resist, to a degree, the transition to the twisted figure-of-eight conformation 17/18 found in patellamide D, since this would lead to deformation of its preferred C2-symmetric structure. In agreement with this supposition, the solution conformations of the related non-C2-symmetric patellamides B and C, 5 and 6 respectively, have similar twisted figure-of-eight-like backbone conformations, viz.17, but without intramolecular hydrogen-bonding in chloroform solutions.14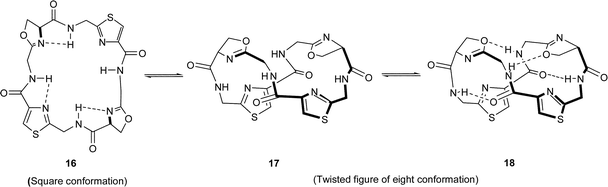 | ||
| Fig. 1 Conformations of ascidiacyclamide and patellamides A–D. | ||
As indicated earlier, the incorporation of both D- and L-amino acids and of azoles are common structural features found in cyclic peptides isolated from marine animals. In an interesting exercise, Fairlie et al.15 examined the possible effects of the presence of oxazolines and thiazoles on the conformation of cyclic peptides by studying the structures 19 and 20, which are likely biosynthetic precursors to the natural product ascidiacyclamide 10. Thus, molecular modelling studies indicated that the cyclic peptide 19 is very flexible and adopts several low-energy conformations. When the two cysteine residues in 19 are converted into their planar thiazole rings, 2D NMR analysis of the synthetic bis-thiazole-based cyclic peptide 20 showed that it had a pseudo-chair conformation restricted by the three intramolecular H-bonds (Fig. 2). In undergoing cyclodehydration of the two threonine units in 20, its conformation changes from chair to the constrained pseudo-boat (saddle-shaped), familiar to ascidiacyclamide 10. The comparison demonstrated that the presence of oxazolines and thiazoles in marine cyclic peptides restricts their conformations, thereby creating unusual shapes, which perhaps provide opportunities for hosting metal and other ions.
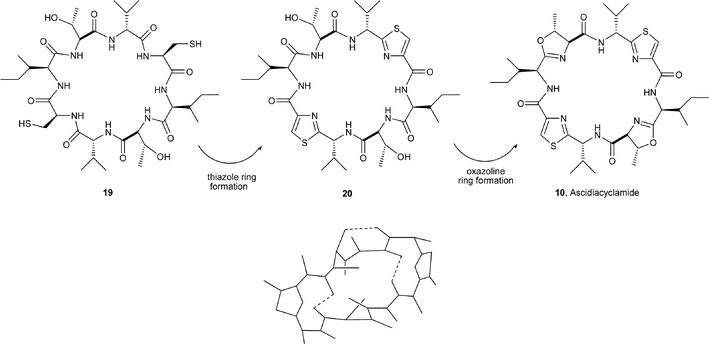 | ||
| Fig. 2 Intramolecular hydrogen bonding in the bis-thiazole 20. | ||
A study of the conformation of the 21-membered macrocycle cyclic heptapeptide lissoclinamide 7 (21) by Wipf et al.,16 both in the solid state and in solution, demonstrated that its conformation is dominated by a β-turn formed by the prolyloxazoline unit and by a β-loop around the thiazoline-phenylalanyl-thiazoline residue (Fig. 3). The cyclic heptapeptide is quite rigid conformationally and more closely resembles the twisted figure-of-eight conformation of patellamide D, i.e.18. The lissoclinamides, e.g.2, 3 and 21, are ostensibly derived from ‘non-natural’ D-amino acids, and Wipf et al.16 have postulated that the stereochemistries at C31 and C21 in lissoclinamide 7 (21) must be necessary to stabilise the overall conformation of the cyclic peptide. To examine this aspect, the authors synthesised the non-natural 31S-lissoclinamide 7 (22), and interestingly showed that it epimerised cleanly to the natural 31R epimer 21 on warming with pyridine in CDCl3 for a short time.
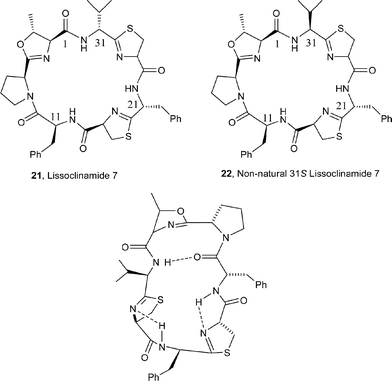 | ||
| Fig. 3 H-bonding network in the β-loop and β-turn regions of lissoclinamide 7 (21). | ||
4 Association and binding of metals in marine cyclic peptides
On the face of it, the similarity of azole-based cyclic peptides to macrocyclic ligands, such as porphyrins and aza-crown ethers, suggests that they are tailor-made for association and binding to metal ions in the marine milieu. However, before we published our review in 1993 there was only scarce mention of the possibility of cyclic peptide–metal congruence within cyclic peptides from sea squirts in the literature.17 Much has changed since this time, and we now have a much deeper understanding of, and evidence for, the association and binding of metals to azole-based marine cyclic peptides of the type shown by structures 1–15.Pioneering studies in this area were carried out by Gahan and his collaborators at the University of Queensland in Brisbane, Australia. In 1994, these researchers were the first to characterise a bis-copper(II) complex of ascidiacyclamide 10, isolated from L. patella by X-ray crystallography.18 This beautiful structure (Fig. 4) has two copper ions separated by a bridging carbonate anion embedded in the saddle-shaped ascidiacyclamide ligand. The copper ions in the complex are further coordinated to three nitrogen donors, and a deprotonated amide, and to two water molecules. The affinity of the ascidiacyclamide–copper complex for carbonate is especially interesting and has suggested that these metal cyclic peptide conjugates could be involved in the activation and transport of CO2in vivo for specific biochemical processes. Contemporaneously, the Queensland group of researchers studied the binding of Cu(II) to patellamide D (7), which has the twisted ‘figure-of-eight’ conformation 17/18 in the solid state, using a combination of electronic absorption, CD, EPR and MS, and determined a closely similar bis-copper complex for this cyclic peptide.19
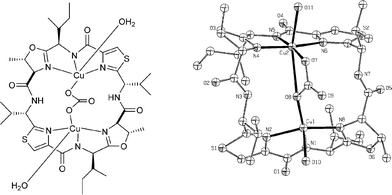 | ||
| Fig. 4 Bis-copper(II) complex of asciacyclamide (10). | ||
During the same period of time, in 1994, Wipf et al.20 described the synthesis and X-ray structure of a novel silver complex of the C3-symmetric cyclic peptide westiellamide 13 isolated from L. bistratum. This complex, shown in Fig. 5, has four silver atoms sandwiched between two cyclic peptide ligands. Interestingly, in the free ligand 13 each of the N-atoms point towards the centre of the flat planar macrocycle. Upon complexation with Ag+ however, the macrocycle turns inside out and the carbonyl groups all point towards the centre of the macrocycle with the oxazoline rings orientated almost orthogonal to the macrocycle. These studies, by Wipf et al. and by the University of Queensland researchers, provided the first clear evidence for the binding of Lissoclinum peptides to metal ions and, no doubt, provided the impetus for further investigations in this area.
 | ||
| Fig. 5 X-Ray crystal structure of a silver complex of westiellamide (13). | ||
A large number of analytical and spectroscopic methods have been used to study the metal complexation behaviour of Lissoclinum cyclic peptides. Prominent amongst the spectroscopic methods has been circular dichroism (CD),21 and our own research group used this method to evaluate the selectivity and level of binding of various metal ions to Lissoclinum cyclic peptides.22 Thus, it was established that although patellamides A (4),B (5) and E (8) could bind comfortably to Zn2+ and Cu2+, they showed no affinity for binding to such metals as Mg2+ and Ca2+. Analysis of CD spectra showed that the symmetrical patellamide A (4), like ascidiacyclamide 10, has predominantly a ‘square’ (‘saddle-shape’) conformation, cf. 16. The non-symmetric patellamides B (5) and E (8), however, assume largely the alternative twisted ‘figure-of-eight’ conformation, viz. 18. Variable-temperature CD spectra of the patellamides 4, 5 and 8 correlate with these extreme conformations and complemented the earlier NMR studies of Ishida14 and others.15 CD studies of methanol solutions of 4, 5, and 8, titrated with methanol solutions of Cu2+ and Zn2+ salts, demonstrated that the cyclic peptides can each bind two metal ions per molecule with binding constants in the range 2 × 104 to 2 × 105 for the first binding domain and 20–230 for the second binding site.22 Furthermore, the CD spectra of the patellamide–metal conjugates could all be correlated with the ‘square’ conformation of the cyclic peptide ligands, cf. the bis-Cu2+ complex (Fig. 4) of ascidiacylamide.
Later independent studies by Jaspars et al.,23 with patellamides A (4) and C (6), and ulithiacyclamide A (9), using combinations of CD and NMR spectroscopy, together with mass spectrometry, endorsed these general findings. Other studies by Fairlie et al.24 also revealed the relevance of the nature of the metal anion and the presence or absence of base, together with the importance of kinetics to the overall binding processes.
Jaspars et al.23 found that patellamide C (6) showed a greater affinity for copper ions than patellamide A (4), a feature which they attributed to the ability of patellamide C to adjust its conformation from the ‘figure-of-eight,’ cf. 17/18, to a ‘square’ form, cf. 16, to accommodate the metal. Complexation with a second copper ion then occurs without any further change in conformation, suggesting that the first-formed Cu(II) complex probably forms a preorganised binding site for the second Cu2+ ion. The greater specificity of patellamide C (6) for copper ions, compared to patellamide A (4), could also be attributed to the presence of a phenylalanine substituent in the former, which distorts its planar conformation sufficiently to provide the ideal binding site for Cu2+ ions.
The relative affinities of patellamides A and C for Cu2+ and Zn2+ ions was also studied by Jaspars et al.,23 using an interesting competition experiment and monitoring changes in CD spectra. The experiment demonstrated that in the presence of Zn2+ ions, patellamide C(6) remains uncomplexed and retains its closed ‘figure-of-eight’ conformation. When Cu2+ ions are added, however, the cyclic peptide switches its conformation to the ‘square’ form to host the Cu2+ in a selective manner, cf. Fig. 1. When the same experiment was carried out with patellamide A (4), no changes in CD spectra were observed in the presence of Cu2+ or Zn2+ ions, suggesting that this cyclic peptide has no preference for either metal. Jaspars et al. have suggested that this lack of selectivity could be associated with the availability of nitrogen binding sites for Zn2+ and Cu2+ ions in the ‘square’ conformation of patellamide A. Zinc prefers a tetrahedral arrangement of nitrogen centres for binding, and when three nitrogen atoms are orientated to complex to zinc, the patellamide backbone adopts a ‘square’ conformation. Copper, however, prefers a ‘square’ pyramidal arrangement of ligands for binding, which is only available when the patellamides adopt a ‘square’ conformation. Nevertheless, when the patellamide backbone is distorted to a figure-of-eight conformation, as in patellamide C (6), only two nitrogen centres are available for complexation to Zn2+ ions, and necessarily the cyclic peptide shows selectivity for Cu2+ ions. There is no doubt that copper is the preferred, biologically important, metal for the patellamide family of cyclic peptides in sea squirts.25 Indeed, patellamide–copper conjugates may have evolved as special enzymes for specific oxidation and electron transfer processes in the marine environment.23–25
In order to probe in more detail the importance of oxazoline rings in patellamides for metal chelation, Comba et al.26 synthesised the analogues 23, 24, 25, and 26 of patellamide A (4) containing no oxazoline rings in their structures. Surprisingly, X-ray crystal analysis showed that the analogue 25 had a ‘square’ (‘saddle’) conformation similar to that formed in patellamide D (7), cf.16. Furthermore, the thiazole rings in 25 are approximately co-parallel, with the amide groups associated with the isoleucine co-planar with the thiazole rings. These features have suggested that 25 is partially preorganised for coordination to two Cu(II) ions. Separate comparative studies established that the other structures 23, 24 and 26 have ‘square’ conformations, similar to 25.
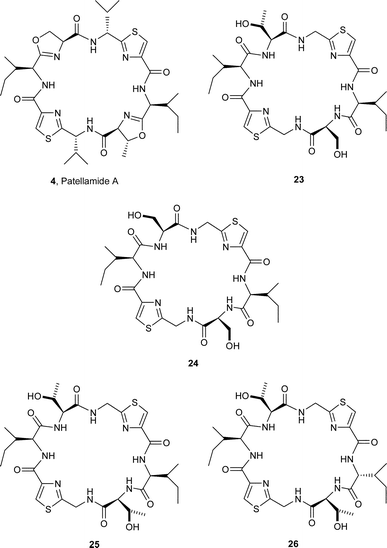
Each of the analogues 23, 24, and 26 was found to bind in a similar manner with Cu2+ ions, and all of them formed both mono- and di-nuclear complexes. Three mononuclear complexes were detected, with the doubly deprotonated complex [Cu(H4L)] predominating, followed by the mono-deprotonated complex [Cu(H5L)]+. In solution however, the dinuclear complex [Cu(H4L)(OH2)n] (L = 23, 24, 25 and 26; where n = 2,3 depending on whether the Cu(II) site is 5- or 6-coordinate) was found to be the most stable. The mononuclear complexes were found to convert slowly into this more stable dinuclear species and free ligand, on standing.27
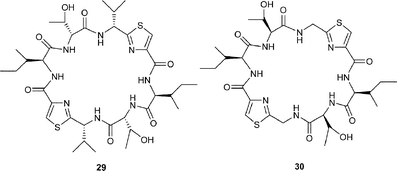
Calcium is another metal that occurs in high concentrations in vivo, and there is good evidence that sea-squirts have a mechanism for sequestering Ca2+ ions. In some contemporaneous studies to those mentioned above with the analogues 23–26 of patellamide A (4), Fairlie et al.28 were also able to establish that the affinity of certain azole-based cyclic peptides for Ca2+ was loosely associated with the number of azole rings within their macrocycles. Thus, complexation studies involving 1H NMR and CD spectroscopy showed that ascidiacyclamide 10 and its analogue 27 lacking oxazoline rings, and the cyclic peptide 28 devoid of both thiazole and oxazoline rings, formed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with Ca2+, but their affinity decreased with the increasing number of 5-membered rings in their structures i.e.30 > 27 > 10. It is not clear, however, whether this observed decrease in binding affinity is due to a decrease in stability of the calcium complexes arising from the reduced flexibility of the macrocycle, or to the decrease in the number of carbonyl oxygen donors available for chelation, or indeed a combination of the two.
:1 complexes with Ca2+, but their affinity decreased with the increasing number of 5-membered rings in their structures i.e.30 > 27 > 10. It is not clear, however, whether this observed decrease in binding affinity is due to a decrease in stability of the calcium complexes arising from the reduced flexibility of the macrocycle, or to the decrease in the number of carbonyl oxygen donors available for chelation, or indeed a combination of the two.
In parallel studies, the more flexible cyclic peptides 28 and 30, possessing differing numbers of carbonyl groups, (i.e. six and eight respectively), were found to have binding constants to Ca2+ of the same order, while the cyclic peptides 27, 29, 30, which each possess six carbonyls, had binding constants which ranged from log10K 2–5.7. The cyclic peptides 27 and 29 also differ in the chirality of their threonine residues, but since they have similar flexibility, their ability to bind calcium is dependent on the accessibility of the carbonyl oxygen atoms in the two different conformations. The ability of the peptides to bind calcium was found to be 28
∼
30 > 27 > 10 > 29, suggesting that their binding affinities were related to their conformational flexibility.29
Finally, a novel potassium complex of a cyclic peptide resulting from acid-catalysed hydrolysis (HClO4–MeOH) of the oxazoline rings in ascidiacyclamide 10 has been prepared by Gahan et al.30 X-ray crystal analysis of this bis-amino macrolide shows that the potassium ion is bound to the two nitrogen atoms of the thiazole rings and to the oxygen centres of the adjacent amide groups. Two perchlorate anions, positioned on either side of the plane of the macrocylic ring, are also associated with the potassium ion (Fig. 6).
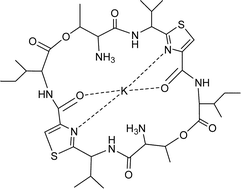 | ||
| Fig. 6 Schematic diagram of the K+ complex of ascidiacyclamide 10 after acid-catalysed opening of the two oxazoline rings. | ||
5 Assembly of cyclic peptides from azole-based amino acids
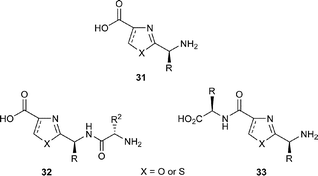
So studies have shown that azole-based cyclic peptides can form complexes with a number of metals! But what is the biological relevance of these metal–ligand associations? Also, can we use this knowledge to facilitate the assembly of natural and non-natural cyclic peptides? Indeed, may the latter have applications as membrane ion channel mimics, for example, or even for the development of macromolecular devices and scaffolds for protein mimics?Circumstantial evidence suggests that the biosynthesis of azole-based cyclic peptides occurs via i) assembly of a particular peptide chain from the constituent amino acids, followed by ii) macrolactamisation of an appropriate ω-amino acid to the cyclic peptide, and then iii) elaboration of the various azole rings by controlled sequences of cyclodehydrations and oxidations (Scheme 1). At some point in this sequence, epimerase enzymes intervene to invert certain stereocentres along the peptide backbone, no doubt, to lower the energies for some macrolactamisations e.g. the formation of the lissoclinamides 4 (2), 5 (3) and 7 (21). It is, however, tempting to contemplate alternative biosynthetic pathways to azole-based natural cyclic peptides involving the assembly of pre-formed azole amino acids, i.e. 31 or peptidyl-azoles, viz. 32 and 33. In this way, one could also envisage metal ions in the marine milieu acting as templates, bringing together the azole units in a coordinated fashion. Perhaps inspired by these possibilities, in recent years, a number of authors have examined the cyclooligomerisations of a range of peptidyl-azoles, which have given some credence to these speculations.
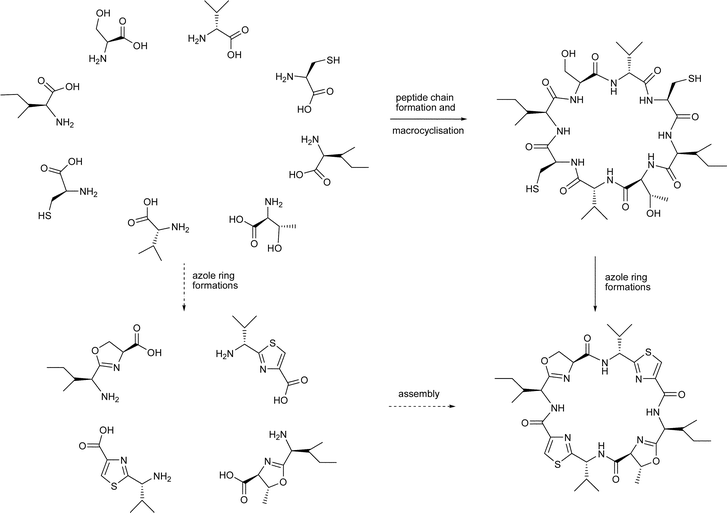 | ||
| Scheme 1 Assembly of cyclic peptides. | ||
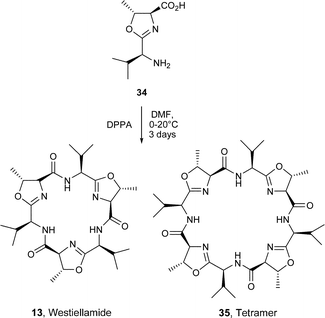
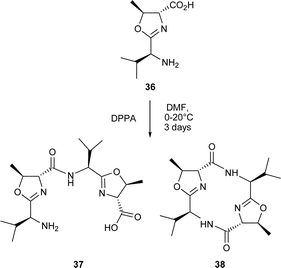
As early as 1992, Wipf and Miller31 described the cyclooligomersiation of the trans-oxazolidine 34 in the presence of DPPA which led to the C3-symmetric 18-membered hexapeptide westiellamide 13 in 20% yield. The formation of 13 was accompanied by the 24-membered cyclic tetramer 35, which was obtained in a similar yield. Interestingly, the cyclooligomersiation of the LD-epimer 36 corresponding to 34 gave substantial amounts of the 12-membered dimer 38, in addition to the anticipated 18- and 24-membered macrocycles, cf. 13 and 35. These differences in reactivity were rationalised on the basis of the lack of non-bonding interactions between the valine side chains in the acyclic intermediate 37 produced from the LD-epimer 36, thereby favouring formation of the smaller ring system 38.
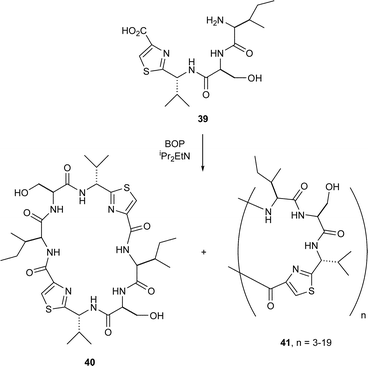
Later, Fairlie et al.32 described the somewhat indiscriminate cyclooligomerisation of the thiazole tetrapeptide 39 in the presence of BOP–DIPEA in DMP, which led to a range of cycloligomers of constitution 41 (n = 3–19). At low monomer concentrations however (i.e. 10−2–10−4 M), the predominant product was the dimer 40.
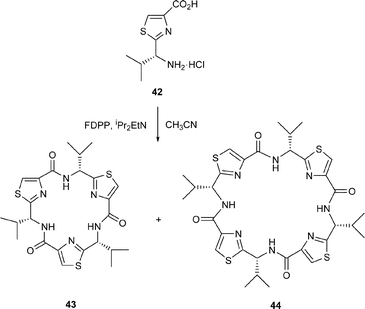
In contemporaneous studies, our own research group carried out a systematic study of the cyclooligomerisations of a range of azole-based amino acids, some of which were conducted in the presence of metal ions, leading to concise syntheses of a variety of novel thiazole and oxazole-based cyclic peptides. Thus, in our first studies we showed that the D-valine-derived thiazole 42 underwent cyclooligomerisation in the presence of FDPP and iPr2EtN (Hünigs base) in acetonitrile at 10–50 nM concentration, to give a 5 : 2 mixture of the cyclic trimer 43 and the cyclic tetramer 44 in a combined yield of 80–90%; with DMF as solvent the yields approach 98%, with only trace amounts of higher oligomers (cyclic pent-, hex- hept-, oct- and nonamers) being detected by mass spectrometry.33 X-Ray crystal structure analysis, in combination with 1H NMR measurements, established that the macrocycles 43 and 44 were nearly planar with all of their isopropyl groups on the same face of the molecules. The X-ray structure of the C3-symmetric trimer 43 (Fig. 7) also showed that the amide NH and thiazole nitrogen atoms are all orientated towards the centre of the macrocycle, forming a network of bifurcated H-bonds, which rigidifies the molecule.
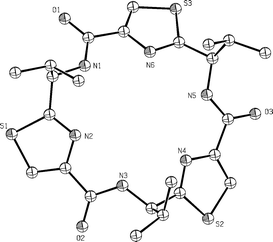 | ||
| Fig. 7 X-Ray structure of the cyclic trimer 43. | ||
In a similar study, the cyclooligomerisation of the alanine-derived thiazole 45a, in the presence of FDPP, led to a 9 : 2 mixture of the cyclic trimer 46a and the cyclic tetramer 47a, in 55% overall yield. The enhanced formation of trimer over tetramer from 36a, compared to 42, supports the notion that as the size of the side-chain, R, increases, unfavourable transannular interactions increase in the cyclic trimers. This feature is reflected in the conformations of the linear peptide precursors derived from 42 and 45a, immediately prior to their cyclisations, with the precursor derived from 45a, containing less bulky side chains, undergoing cyclisation at a faster rate. In cognisance with this suggestion, none of the cyclic trimer 46b was obtained when the bulky phenylalanine derived thiazole 45b was treated with FDPP in acetonitrile. Interestingly, however, when an enantiomerically enriched sample of 45b (ee = 70%) was used in the cyclooligomerisation, the diastereoisomeric cyclic trimer 48, together with the cyclic tetramer 49 were the major products isolated. The formation of the trimer 48, in this instance, is attributed to the contamination of the phenylalanine-based thiazole 45b with its enantiomer. In accord with this supposition, treatment of a racemic sample of the valine-derived thiazole 42 with FDPP in the presence of Hünigs base gave a mixture of all the possible expected racemic cyclic trimers 43 and 50 and the racemic cyclic tetramers 44, 51, 52 and 53 in a combined yield of 75%.
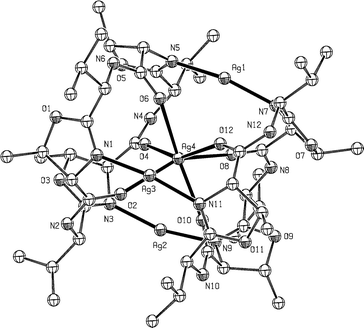
In complementary studies, the presence of metal ions during the cyclooligomerisation of the valine-derived thiazole 42 was shown to influence the relative amounts of the cyclic trimer 43 and the cyclic tetramer 44 products formed in the reaction.34 Thus, the presence of larger metal ions, e.g. Cd2+, was shown to enhance the formation of the cyclic tetramer 44, whereas smaller alkali metal ions favoured the production of the cyclic trimer 43. This feature can be attributed to a templating effect of the metal ion, since the size of the cyclic products formed was related to the ionic radius of the metal template (Table 1). During these metal-influenced cyclooligomerisation studies, an interesting and novel crystalline complex, containing five silver atoms in a sandwich arrangement, was characterised by X-ray crystallography from the association of the cyclic tetramer 44 and AgBF4 (Fig. 8).
| Metal | Yield | Trimer to tetramer ratio (43 : 44) | Metal | Yield | Trimer to tetramer ratio (43 : 44) |
|---|---|---|---|---|---|
| No metal | 95% | 3 : 1 | NiCl2 | 78% | 7 : 3 |
| LiBF4 | 57% | 4 : 1 | CuCl2 | — | — |
| NaBF4 | 56% | 3 : 1 | Cu(BF4)2 | — | — |
| KBF4 | 91% | 7 : 3 | CoCl2 | — | — |
| CsBF4 | 93% | 3 : 1 | AgBF4 | 57% | 2 : 1 |
| ZnCl2 | 81% | 4 : 1 | NH4PF6 | 30% | 11 : 2 |
| CdCl2 | 41% | 2 : 1 | Zn(BF4)2 | 42% | 4 : 1 |
| Hg(ClO4)2 | 35% | 3 : 1 | Zn(OAc)2 | — | — |
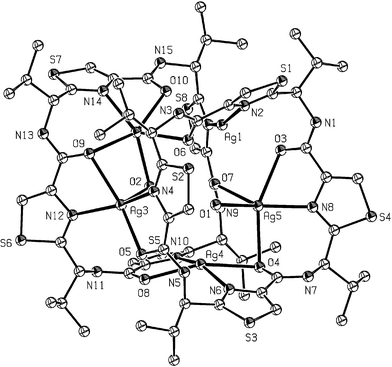 | ||
| Fig. 8 X-Ray crystal structure of a silver sandwich complex of the cyclic tetramer 44. | ||
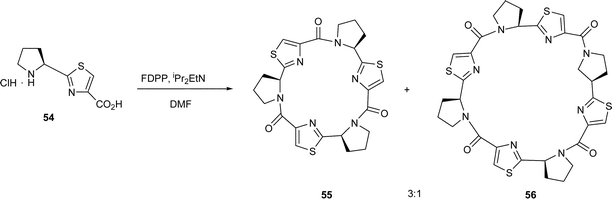
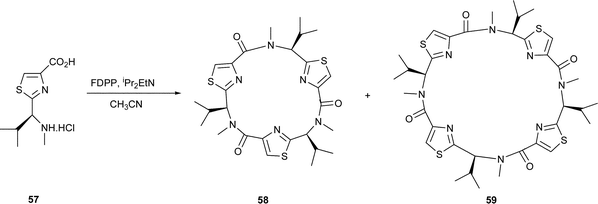
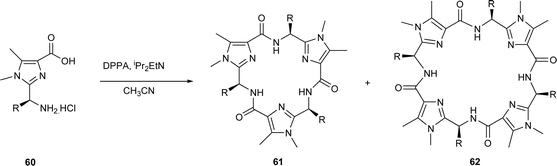
The cyclooligomerisations of amino acid–based thiazoles is clearly a useful synthetic route to libraries of novel, non-natural cyclic peptides. Indeed, the scope for the method has also been demonstrated with proline35 and N-methylvaline thiazole36 amino acids, i.e. 54 and 57, leading to similar cyclic trimers, viz. 55 and 58, and to cyclic tetramers, viz. 56 and 59, and Habenhauer et al.37 have used the method to prepare unusual cyclic peptides containing imidazole rings, viz. 60
→
61 and 62.
In other studies from our laboratories, the synthesis of libraries of mixed azole cyclic peptides, from cyclisations of mixtures of thiazole and oxazole-based amino acids, has also been achieved.38 Indeed, some of these studies have led to concise syntheses of various cyclic peptidic natural products. Remarkably, when a 1 : 1 : 1 mixture of the oxazole amino acid 63 and the thiazole-amino acids 42 and 45a in CH3CN was reacted in the presence of FDPP–DIPEA, a reasonably selective assembly of the amino acids ensued leading to the formation of the natural product dendroamide A (64) in 23% yield (Scheme 2).39 The positional isomer 65 of dendroamide A was produced in a similar yield (22%), together with the four thiazole-based cyclic trimers 66a, 66b, 67a and 67b in a combined yield of 30%. In a similar manner, the coupling of a mixture of 42, 68 and 69 in the presence of FDPP–DIPEA, produced equal amounts of the natural product nostocyclamide 70 and the cyclic trimers 71, 72, and 73 in a combined yield of 65% (Scheme 3).40
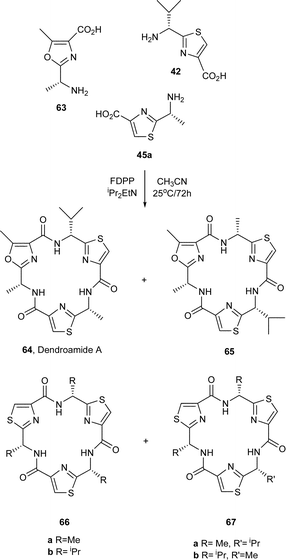 | ||
| Scheme 2 Synthesis of dendroamide A (64) via self-assembly of amino acid based azoles. | ||
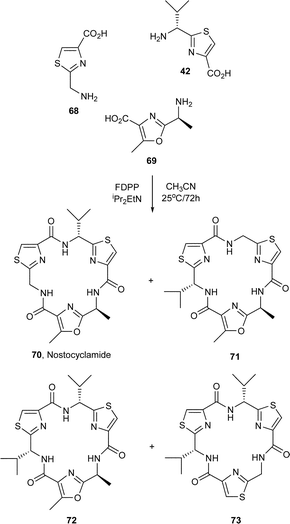 | ||
| Scheme 3 Synthesis of nostocyclamide (70) via self-assembly of amino acid based azoles. | ||
The high-yielding and selective cyclooligomerisations of ‘simple’ substituted azole-based amino acids has been developed by Fairlie et al.41 and by ourselves,42 leading to the synthesis of a variety of novel tubular and cage cyclic peptide structures. These structures represent new classes of supramolecular peptides which could have applications as membrane ion channel mimics, or as scaffolds for protein mimics, or even in catalysis.43
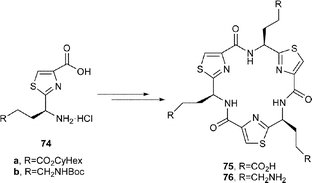
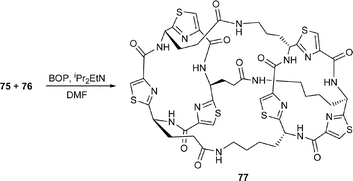
Thus, Farilie et al.42 synthesised the cyclic trimers 75 and 76 from the corresponding thiazole amino acids 74a and 74b following cyclooligomersation, chromatographic separation of the resulting trimers, and deprotection of the CO2-cHex and NHR protective groups. A coupling reaction between 75 with 76, using BOP–DIPEA then produced the C3-symmetric cylinder (or tube) 77 in 47% yield. In a similar manner, the cone-shaped compound 80 was prepared by reaction of 1,4,7-triazacyclononane 79 with the tris-bromide 78, produced in three straightforward steps from the tris-carboxylic acid 75.
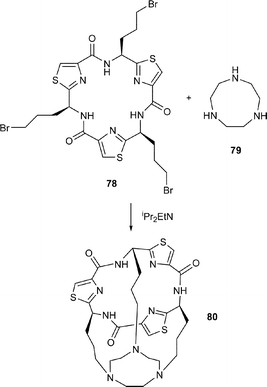
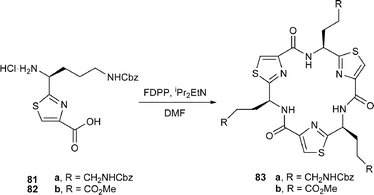
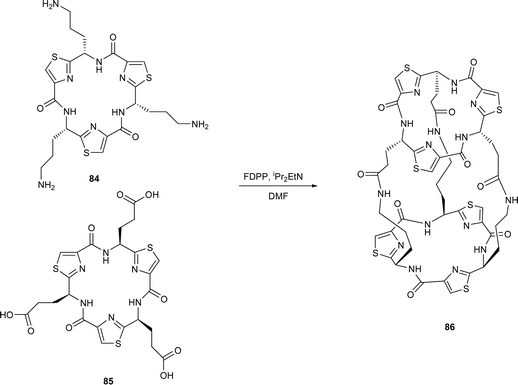
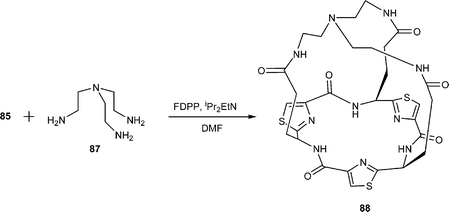
In contemporaneous investigations, our own research group42 prepared the L-ornithine and L-glutamic acid thiazole amino acids 81 and 82 respectively. Treatment of 81 and 82 separately with FDPP–DIPEA in DMF first gave the corresponding cyclic trimers 83a and 83b respectively, which were then deprotected to their free amine and carboxylic acid, 84 and 85 respectively. When a mixture of the tris-amine 84 and the tris-carboxylic acid 85 was treated with FDPP and iPr2EtN under high dilution in DMF, the tubular C3-symmetric structure 86 was obtained as a solid in 30% yield. A corresponding reaction between the tricarboxylic acid 85 and tris(aminoethyl) amine 87 in the presence of FDPP and iPr2NEt gave the cage structure 88, also as a solid, in 40% yield.
6 Summary and outlook
Since our earlier review, in 1993, significant advances have been made in establishing the relationships between metals and azole-based cyclic peptides found in ascidians (‘sea squirts’). Indeed, the isolation and characterisation of several cyclic peptide–metal complexes, both in vivo and in vitro, has given considerable credence to earlier suggestions that (a) metals could be involved in the biological assembly of azole-based cyclic peptides, and (b) cyclic peptide–metal conjugates probably evolved to carry out specific biological functions, i.e. biological oxidation and electron transfer processes, in the marine milieu. Clearly there are other aspects of the biology and ecological functions of these ligand–metal conjugates that await further exploration, and it is hoped that the next generation of enquiring chemists will be lured into this exciting multidisciplinary area of research, and uncover these additional mysteries.7 Acknowledgements
We thank our group of collaborators, some of whom are mentioned in the reference section, and the group of sponsors, i.e. EPSRC, AstraZeneca, Merck Sharpe & Dohme, SmithKline Beecham (now GSK), and Pfizer Ltd, who have provided generous support for our research in this area over the past decade.8 References
- J. P. Michael and G. Pattenden, Angew. Chem., Int. Ed. Engl., 1993, 32, 1 CrossRef.
- For a recent review of synthetic studies of marine cyclic peptides, see: Y. Hamada and T. Shioiri, Chem. Rev., 2005, 105, 4441 Search PubMed.
- (a) J. M. Wasylyk, J. E. Biskupiak, C. E. Costello and C. M. Ireland, J. Org. Chem., 1983, 48, 4445 CrossRef CAS; (b) B. M. Degnan, C. J. Hawkins, M. F. Lavin, E. J. McCaffrey, D. L. Parry, A. L. van den Brenk and D. J. Watters, J. Med. Chem., 1989, 32, 1349 CrossRef CAS; (c) F. J. Schmitz, M. B. Ksebati, J. S. Chang, J. L. Wang, M. B. Hossain, D. van der Helm, M. H. Engel, A. Serban and J. A. Silfer, J. Org. Chem., 1989, 54, 3463 CrossRef CAS; (d) C. J. Hawkins, M. F. Lavin, K. A. Marshall, A. L. van den Brenk and D. J. Watters, J. Med. Chem., 1990, 33, 1634 CrossRef CAS; (e) L. A. Morris, J. J. K. van den Bosch, K. Versluis, G. S. Thompson and M. Jaspars, Tetrahedron, 2000, 56, 8345 CrossRef CAS.
- (a) C. M. Ireland, A. R. Durso, R. A. Newman and M. P. Hacker, J. Org. Chem., 1982, 47, 1807 CrossRef CAS; (b) M. A. Rashid, K. R. Gustafson, J. H. Cardellina, II and M. R. Boyd, J. Nat. Prod., 1995, 58, 594 CrossRef CAS; (c) L. A. McDonald and C. M. Ireland, J. Nat. Prod., 1992, 55, 376 CrossRef CAS.
- C. M. Ireland and P. J. Scheuer, J. Am. Chem. Soc., 1980, 102, 5688 CrossRef CAS; D. F. Sesin, S. J. Gaskell and C. M. Ireland, Bull. Soc. Chim. Belg., 1986, 95, 853 CAS ; cf. D. E. Williams and R. E. Moore, J. Nat. Prod., 1989, 52, 732 Search PubMed.
- Y. Hamamoto, M. Endo, M. Nakagawa, T. Nakanishi and K. Mizukawa, J. Chem. Soc., Chem. Commun., 1983, 323 RSC; Y. Hamada, S. Kato and T. Shioiri, Tetrahedron Lett., 1985, 26, 3223 CrossRef CAS.
- (a) B. M. Degnan, C. J. Hawkins, M. F. Lavin, E. J. McCaffrey, D. L. Parry and D. J. Watters, J. Med. Chem., 1989, 32, 1354 CrossRef CAS; (b) M. P. Foster, G. P. Concepción, G. B. Caraan and C. M. Ireland, J. Org. Chem., 1992, 57, 6671 CrossRef CAS; (c) L. J. Perez and D. J. Faulkner, D. J., J. Nat. Prod., 2003, 66, 247 CrossRef CAS.
- (a) T. W. Hambley, C. J. Hawkins, M. F. Lavin, A. L. van den Brenk and D. J. Watters, Tetrahedron, 1992, 48, 341 CrossRef CAS; (b) M. P. Prinsep, R. E. Moore, I. A. Levine and G. M. L. Patterson, J. Nat. Prod., 1992, 55, 140 CrossRef CAS.
- A. Rudi, L. Chill, M. Aknin and Y. Kashman, J. Nat. Prod., 2003, 66, 575 CrossRef CAS.
- see (a) C. E. Salomon and D. J. Faulkner, J. Nat. Prod., 2002, 65, 689 CrossRef CAS; (b) E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisan, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315 CrossRef CAS.
- (a) T. Ishida, M. Inoue, Y. Hamada, S. Kato and T. Shioiri, T., J. Chem. Soc., Chem. Commun., 1987, 370 RSC; (b) T. Ishida, M. Tanaka, M. Nabae, M. Inoue, S. Kato, Y. Hamada and T. Shioiri, T., J. Org. Chem., 1998, 53, 107; (c) T. Ishida, Y. In, M. Doi, M. Inoue, Y. Hamada and T. Shioiri, Biopolymers, 1992, 32, 131 CrossRef CAS.
- Y. In, M. Doi, M. Inoue, T. Ishida, Y. Hamada and T. Shioiri, Chem. Pharm. Bull., 1993, 41, 1686 CAS; Y. In, M. Doi, M. Inoue, T. Ishida, Y. Hamada and T. Shioiri, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 432 CrossRef.
- F. J. Schmitz, M. B. Ksebati, J. S. Chang, J. L. Wang, M. B. Hossain, D. van der Helm, M. H. Engel, A. Serban and J. A. Silfer, J. Org. Chem., 1989, 54, 3463 CrossRef CAS.
- T. Ishida, Y. In, F. Shinozaki, M. Doi, D. Yamamoto, Y. Hamada, T. Shioiri, M. Kamigauchi; and M. Sugiura, J. Org. Chem., 1995, 60, 3944 CrossRef CAS.
- G. Abbendante, D. P. Fairlie, L. R. Gahan, G. R. Hanson, G. K. Pierens and A. L. van den Brenk, J. Am. Chem. Soc., 1996, 118, 10384 CrossRef CAS.
- P. Wipf, P. C. Fritch, S. J. Geib and A. M. Sefler, J. Am. Chem. Soc., 1998, 120, 4105 CrossRef CAS.
- (a) D. B. Carlisle, Proc. R. Soc. London, Ser. B, 1968, 171, 31 CrossRef CAS; (b) H. Michibata, Comp. Biochem. Physiol., 1984, 78A, 285 Search PubMed; (c) J. H. Swinehart, W. R. Biggs, D. J. Halko and N. C. Schroeder, Biol. Bull., 1974, 146, 302 Search PubMed; (d) E. P. Levine, Science, 1961, 133, 1352 CrossRef CAS; (e) G. W. Rayner-Canham, M. van Roode and J. Burke, Inorg. Chim. Acta, 1985, 106, L37 CrossRef CAS.
- A. L. van den Brenk, K. A. Byriel, D. P. Fairlie, L. R. Gahan, G. R. Hanson, C. J. Hawkins, A. Jones, C. H. L. Kennard, B. Moubaraki and K. S. Murray, Inorg. Chem., 1994, 33, 3549 CrossRef CAS.
- A. L. van den Brenk, K. A. Byriel, D. P. Fairlie, L. R. Gahan, G. R. Hanson, C. J. Hawkins, A. Jones, C. H. L. Kennard, B. Moubaraki and K. S. Murray, Inorg. Chem., 1994, 33, 3549 CrossRef CAS.
- (a) P. Wipf, S. Venkatraman, C. P. Miller and S. J. Geib, Angew. Chem., Int. Ed. Engl., 1994, 22, 1516 CrossRef; (b) P. Wipf, C. P. Miller and C. M. Grant, Tetrahedron, 2000, 56, 9143 CrossRef CAS.
- C. J. Hawkins, Pure Appl. Chem., 1988, 60, 1267 CrossRef CAS.
- D. J. Freeman, G. Pattenden, A. F. Drake and G. Siligardi, J. Chem. Soc., Perkin Trans. 2, 1998, 129 RSC.
- (a) L. A. Morris, M. Jaspars, J. J. Kettenes-van den Bosch, K. Versluis, A. J. R. Heck, S. M. Kelly and N. C. Price, Tetrahedron, 2001, 57, 3185 CrossRef CAS; (b) L. A. Morris, B. F. Milne, G. S. Thompson and M. Jaspars, J. Chem. Soc., Perkin Trans. 2, 2002, 1072 RSC.
- L. G. Øndahl, N. Sokolenko, G. Abbenante, D. P. Fairlie, G. R. Hanson and L. R. Gahan, J. Chem. Soc., Dalton Trans., 1999, 8, 1227 RSC.
- For other, related studies see: (a) L. A. Morris, B. F. Milne, M. Jaspars, J. J. Kettenes-van den Bosch, K. Versluis, A. J. R. Heck, S. M. Kelly and N. C. Price, Tetrahedron, 2001, 57, 3199 CrossRef CAS; (b) L. A. Morris, B. F. Milne, G. S. Thompson and M. Jaspars, J. Chem. Soc., Perkin Trans. 2, 2002, 1072 RSC.
- P. V. Bernhardt, P. Comba, D. P. Fairlie, L. R. Gahan, G. R. Hanson and L. Lötzbeyer, Chem. Eur. J., 2002, 8, 1527 CrossRef CAS.
- A. L. van den Brenk, J. D. A. Tyndall, R. M. Cusack, A. Jones, D. P. Fairlie, L. R. Gahan and G. R. Hanson, J. Inorg. Biochem., 2004, 98, 1857 CrossRef CAS.
- R. M. Cusack, L. Grøndahl, G. Abbenante, D. P. Fairlie, L. R. Gahan, G. R. Hanson and T. W. Hambley, J. Chem. Soc., Perkin Trans. 2, 2000, 323 RSC.
- R. M. Cusack, L. Grøndahl, D. P. Fairlie, L. R. Gahan and G. R. Hanson, J. Chem. Soc., Perkin Trans. 2, 2002, 556 RSC.
- A. L. van den Brenk, D. P. Fairlie, L. R. Gahan, G. R. Hanson and T. W. Hambley, Inorg. Chem., 1996, 35, 1095 CrossRef CAS.
- (a) P. Wipf, C. P. Miller and C. M. Grant, Tetrahedron, 2000, 56, 9143 CrossRef CAS; (b) P. Wipf and C. P. Miller, J. Am. Chem. Soc., 1992, 114, 10975 CrossRef CAS.
- N. Sokolenko, G. Abbenante, M. J. Scanlon, A. Jones, L. R. Gahan, G. R. Hanson and D. P. Fairlie, J. Am. Chem. Soc., 1999, 121, 2603 CrossRef CAS.
- (a) A. Bertram, A. J. Blake, J. S. Hannam, K. A. Jolliffe, F. Gonzlez-López de Turiso and G. Pattenden, Synlett, 1999, 1723 CAS; (b) A. Bertram, A. J. Blake, F. Gonzlez-López de Turiso, J. S. Hannam, K. A. Jolliffe, G. Pattenden and M. Skae, Tetrahedron, 2003, 59, 6979 CrossRef CAS.
- A. J. Blake, J. S. Hannam, K. A. Jolliffe and G. Pattenden, Synlett, 2000, 1515 CAS.
- S. Jayaprakash, G. Pattenden, M. S. Viljoen and C. Wilson, Tetrahedron, 2003, 59, 6637 CrossRef CAS.
- L. Dudin, G. Pattenden, M. S. Viljoen and C. Wilson, Tetrahedron, 2005, 61, 1257 CrossRef CAS.
- G. Haberhauer and F. Rominger, Tetrahedron Lett., 2002, 43, 6335 CrossRef CAS.
- A. Bertram, N. Maulucci, O. M. New, S. M. M. Nor and G. Pattenden, in preparation.
- (a) A. Bertram and G. Pattenden, Synlett, 2000, 1519 CAS; (b) A. Bertram and G. Pattenden, Heterocycles, 2002, 58, 521 CrossRef CAS.
- A. Bertram and G. Pattenden, Synlett, 2001, 1873 CrossRef CAS.
- Y. Singh, N. Sokolenko, M. J. Kelso, L. R. Gahan, G. Abbenante and D. P. Fairlie, J. Am. Chem. Soc., 2001, 123, 333 CrossRef CAS.
- (a) G. Pattenden and T. Thompson, Chem. Commun., 2001, 717 RSC; (b) G. Pattenden and T. Thompson, Tetrahedron Lett., 2002, 43, 2450.
- (a) Y. Singh, M. J. Stoermer, A. J. Lucke, M. P. Glenn and D. P. Fairlie, Org. Lett., 2002, 4, 3367 CrossRef CAS; (b) D. Mink, S. Mecozzi and J. Rebek, Tetrahedron Lett., 1998, 39, 5709 CrossRef CAS; (c) G. Haberhauer, L. Somogyi and J. Rebek, Tetrahedron Lett., 2000, 41, 5013 CrossRef CAS.
Footnote |
| † The following abbreviations have been used: DPPA: (PhO)2P(O)N3; BOP: (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate; DIPEA: iPr2EtN (Hünigs base); DMF: HCON(CH3)2; FDPP: Ph2P(O)OC6F5. |
| This journal is © The Royal Society of Chemistry 2007 |