Spider venoms: a rich source of acylpolyamines and peptides as new leads for CNS drugs
Georgina Estradaa, Elba Villegasb and Gerardo Corzo*a
aInstituto de Biotecnología, UNAM, Av. Universidad 2001, Cuernavaca, Morelos, 62210, México. E-mail: corzo@ibt.unam.mx
bCentro de Investigación en Biotecnología UAEM, Av. Universidad 2001, Cuernavaca, Morelos, 62210, México
First published on 6th December 2006
Abstract
Covering: up to 2006
Advances in NMR and mass spectrometry as well as in peptide biochemistry coupled to modern methods in electrophysiology have permitted the isolation and identification of numerous products from spider venoms, previously unexplored due to technical limitations. The chemical composition of spider venoms is diverse, ranging from low molecular weight organic compounds such acylpolyamines to complex peptides. First, acylpolyamines (<1000 Da) have an aromatic moiety linked to a hydrophilic lateral chain. They were characterized for the first time in spider venoms, and are ligand-gated ion channel antagonists, which block mainly postsynaptic glutamate receptors in invertebrate and vertebrate nervous systems. Acylpolyamines represent the vast majority of organic components from the spider venom. Acylpolyamine analogues have proved to suppress hippocampal epileptic discharges. Moreover, acylpolyamines could suppress excitatory postsynaptic currents inducing Ca2+ accumulation in neurons leading to protection against a brain ischemic insult. Second, short spider peptides (<6000 Da) modulate ionic currents in Ca2+, Na+, or K+ voltage-gated ion channels. Such peptides may contain from three to four disulfide bridges. Some spider peptides act specifically to discriminate among Ca2+, Na+, and K+ ion channel subtypes. Their selective affinities for ion channel subfamilies are functional for mapping excitable cells. Furthermore, several of these peptides have proved to hyperpolarize peripheral neurons, which are associated with supplying sensation to the skin and skeletal muscles. Some spider N-type calcium ion channel blockers may be important for the treatment of chronic pain. A special group of spider peptides are the amphipathic and positively charged peptides. Their secondary structure is α-helical and they insert into the lipid cell membrane of eukaryotic or prokaryotic cells leading to the formation of pores and subsequently depolarizing the cell membrane. Acylpolyamines and peptides from spider venoms represent an interesting source of molecules for the design of novel pharmaceutical drugs.
Georgina Estrada received a BSc in Pharmaceutical Chemistry from the University of Michoacan in 1999, and a MSc in Biochemistry from the Institute of Biotechnology-UNAM in 2003. She is currently pursuing doctoral research at the Institute of Biotechnology-UNAM. Her PhD studies, under the supervision of Dr Corzo, concerned the expression of scorpion and spider neuropeptides. Her research interests have involved the application of signal peptides that improve the in vitro folding of cysteine-rich proteins. |
Elba Villegas was born in Queretaro and received a BSc in Food Engineering from the University of San Luis Potosi in 1986, and an MSc in Chemical Engineering from the Metropolitan University of Mexico in 1991. Her PhD studies, under the supervision of Professor Stanley Gilliland at the Oklahoma State University, concerned secondary metabolite biosynthesis from lactic acid bacteria for the development of probiotics. Her research interests have involved the application of techniques in synthetic organic chemistry, protein chemistry and molecular biology of antimicrobial peptides. She is teaching at the Faculty of Biology-UAEM. |
Gerardo Corzo was born in Chiapas and studied Biochemical Engineering at the Metropolitan University of Mexico (BSc 1986), the National Autonomous University of Mexico (MSc 1993), and the Oklahoma State University (PhD 1997). After postdoctoral years at the Suntory Institute for Bioorganic Research (Osaka, Japan) with Professor Terumi Nakajima he began teaching at the Institute of Biotechnology-UNAM in protein chemistry. He has had a long interest in the discovery of natural products from insects and recombinant expression of neuropeptides. |
 From left to right: Gerardo Corzo, Georgina Estrada and Elba Villegas. |
1 Introduction
The biological and ecological diversity of spiders is immense: 39![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 112 spider species are grouped into 3618 genera and 110 families.1 The Araneae can be divided into two large groups: the Mesothelae and the Opisthothelae. The Mesothelae look rather ‘primitive’ as they retain a lot of original characteristics, and do not have any toxicological interest as venom glands are lacking.2 The Opisthothelae have venom glands as a new character. The Opisthothelae additionally contain two groups: mygalomorph and araneomorph spiders according to the position of their chelicera, which is the pair of fang-like appendages near the mouth of the spider used for grasping and piercing. The mygalomorph chelicera is articulate in a manner that enables movements of the appendages parallel to the body axis (Fig. 1), and the araneomorph chelicera moves at right angles to the body axis. The largest spiders (tarantulas, family Theraphosidae) belong to the mygalomorphs and, with the exception of the genus Atrax, are far from the most dangerous species. The name ‘tarantulas’ often used for them is greatly misleading, as tarantulas represent a large species of the family Lycosidae (wolf spiders) amongst the Araneomorphae. The Araneomorphae are the bulk of spiders, comprising about 32000 species. The most dangerous species are found in the araneomorphs and include black widows (Latrodectus spp., Theridiidae), violin or gaucho spiders (Loxosceles spp., Loxoscelidae) and banana spiders (Phoneutria spp., Ctenidae), which are responsible for many severe envenomation cases, and recorded mortality.3 Several common names do not clearly describe the family (or species) considered. For example, the name ‘funnel web spider’ is used for two entirely different kinds of spiders, the Dipluridae and Hexathelidae (Mygalomorphae) and for Agelenidae (Araneomorphae): this term just describes the form of the web.
112 spider species are grouped into 3618 genera and 110 families.1 The Araneae can be divided into two large groups: the Mesothelae and the Opisthothelae. The Mesothelae look rather ‘primitive’ as they retain a lot of original characteristics, and do not have any toxicological interest as venom glands are lacking.2 The Opisthothelae have venom glands as a new character. The Opisthothelae additionally contain two groups: mygalomorph and araneomorph spiders according to the position of their chelicera, which is the pair of fang-like appendages near the mouth of the spider used for grasping and piercing. The mygalomorph chelicera is articulate in a manner that enables movements of the appendages parallel to the body axis (Fig. 1), and the araneomorph chelicera moves at right angles to the body axis. The largest spiders (tarantulas, family Theraphosidae) belong to the mygalomorphs and, with the exception of the genus Atrax, are far from the most dangerous species. The name ‘tarantulas’ often used for them is greatly misleading, as tarantulas represent a large species of the family Lycosidae (wolf spiders) amongst the Araneomorphae. The Araneomorphae are the bulk of spiders, comprising about 32000 species. The most dangerous species are found in the araneomorphs and include black widows (Latrodectus spp., Theridiidae), violin or gaucho spiders (Loxosceles spp., Loxoscelidae) and banana spiders (Phoneutria spp., Ctenidae), which are responsible for many severe envenomation cases, and recorded mortality.3 Several common names do not clearly describe the family (or species) considered. For example, the name ‘funnel web spider’ is used for two entirely different kinds of spiders, the Dipluridae and Hexathelidae (Mygalomorphae) and for Agelenidae (Araneomorphae): this term just describes the form of the web. | ||
| Fig. 1 Araneomorph (a, b, and e) and mygalomorph (c, d, and f) spiders. (a) Argiope sp., (b) Nephila sp., (c) Machrotele gigas, (d) Heteroscodra maculata, (e) Cupennius salei, and (f) Brachypelma vagans. C. salei and B. vagans show their characteristic araneomorph (Ach) and mygalomorph (Mch) chelicera. | ||
Spider venoms contain molecules with a large range of molecular masses (0.1–14 KDa), and possess a high diversity of antagonists that disturb the function of ion channels and other cell receptors.4 Spider venoms are fast becoming recognized as essential tools for the study of cellular receptors (Fig. 2).
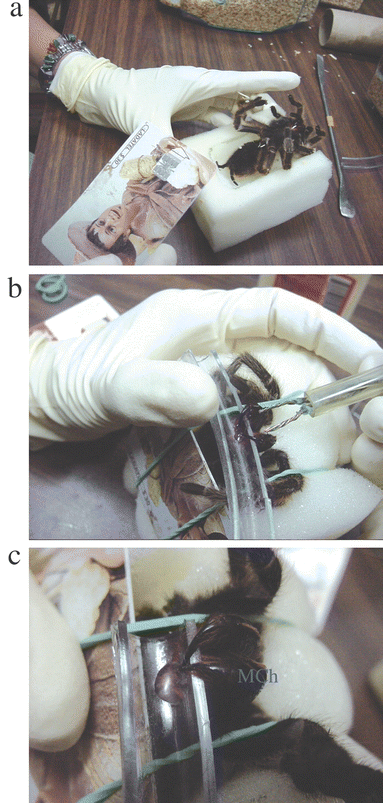 | ||
| Fig. 2 The venom milking process. (a) Brachypelma vagans is anesthetized with carbon dioxide, (b) electrical stimulation to the chelicera of B. vagans, (c) crude venom from B. vagans. To milk venom from a spider, the fangs of lightly anaesthetized spiders are inserted into thick, soft vinyl tubing, and a low voltage/current electrical shock is applied. | ||
Two major classes of molecules are present in spider venoms, the acylpolyamines and peptides (ion channel modifiers and pore-forming) (Fig. 3). These molecular categories represent around two thirds of the dry weight of the spider venom.5 Escoubas et al.6,7 have estimated a minimum of 600 different molecular masses in the hexathelid spider Atrax robustus. If one predicts that each spider venom contains approximately 300 compounds, then it can be calculated that spider venoms as a whole contain more than 11 million different compounds if one assumes a total of 39000 different spider species. This figure is quite large, so spiders have been named eight-legged pharmacists, and they have also been considered as an organism that uses natural combinatorial chemistry to produce their toxins.8 Few acylpolyamines and spider peptides have been described compared to that theoretical figure, and their names are quite arbitrary depending on the researcher who discovered and named them. The principal targets of acylpolyamines and peptides are insect receptors; however, because of the similarity of invertebrate receptors to the vertebrate ones, several spider molecules recognize and antagonize mammalian receptors. This work reviews the studies and structures of acylpolyamines and peptides from spider venoms that target mammalian receptors; so those molecules may have a potential therapeutic use.
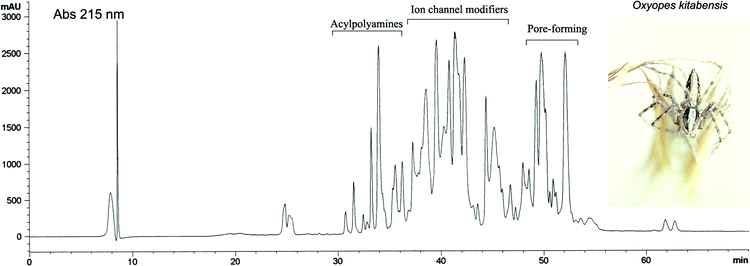 | ||
| Fig. 3 Reversed-phase chromatogram of the venom from the oxyopid spider Oxyopes kitabensis (inset). The chromatogram shows a typical protein profile of spider venom containing acylpolyamines and low molecular weight (2–8 KDa) peptides. | ||
2 Spider acylpolyamines
A great number of spider acylpolyamines have been discovered. These acylpolyamines are low molecular weight compounds, which are present only in the venom glands. These molecules are hydrophilic and provoke a fast paralysis of their prey.9 Acylpolyamines have been shown to have strong interactions with the neuromuscular junctions of insects. They were first found in the venom of theraposid spiders.10,11 Later, in 1982, the first biological records of an acylpolyamine with neurotoxic effects were recorded from acylpolyamines of the orb-web spider Nephila clavata (Joro spider). The first structure of a spider acylpolyamine was elucidated by Grishin et al.12 from the araneid spider Argiope lobata in 1986. Later, in 1988, a second spider acylpolyamine structure was determined by Budd et al.13 from the araneid spider Argiope trifasciata.Although acylpolyamines are extracted from the venom gland of spiders from different genera and different ecological niches, the structural similarity of acylpolyamines 1 from the venom of spiders is evident14 (Fig. 4). The common structure of spider acylpolyamines comprises an aromatic moiety at one end, and either a primary amino group, or a guanidine group at the other.11 The lipophilic core is either bound directly to the acylpolyamine through an amide bond or through an amino acid linker. The aromatic core is a 2,4-dihydroxyphenyl- or indol-3-acetyl-group with or without a hydroxyl group in the 4-position, although spider acylpolyamines with hydroxyl groups in the 4- and/or 6-position have been found.11,14
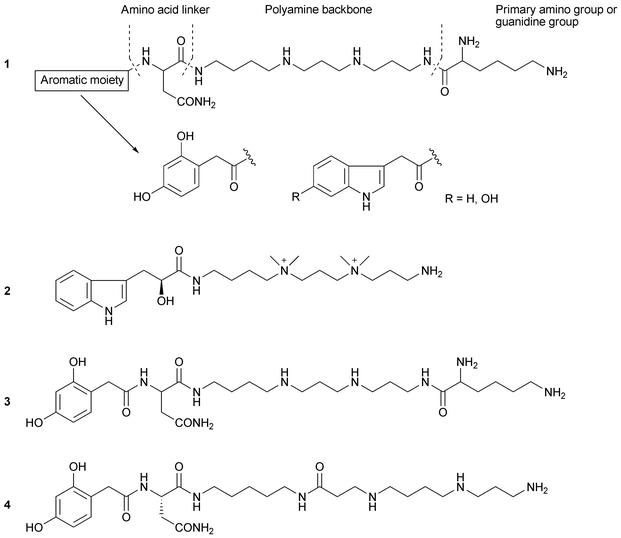 | ||
| Fig. 4 Structures of spider acylpolyamines. General structure 1, MG30 2, NPTX-594 3, and JSTX-3 4. | ||
Following the characterization of the structures, several native spider acylpolyamines and their analogues have been chemically synthesized15–20 for the purpose of obtaining sufficient material for biological assays.
We have reported the structure determination of MG30 2, an acylpolyamine from the hexathelid spider Macrothele gigas from South Japan, using two-dimensional NMR and MS analysis. The absolute configuration of MG30 was assigned after the enantioselective synthesis of (S)- and (R)-MG30. MG30 has two dimethyldialkyl ammonium salts in the main chain. Also, we have reported the chemical synthesis of NPTX-594 3 isolated from Nephila madagascariencis21 (Fig. 4).
2.1 Acylpolyamines and glutamate receptors
Acylpolyamines have been extensively studied in vertebrates. Acylpolyamine toxins from spiders block neuromuscular junctions targeting primarily ionotropic glutamate receptors.11 Because of their small size and aqueous solubility, acylpolyamines are plausible candidates for targeting glutamate receptors. The ionotropic receptors themselves are ligand-gated ion channels, i.e. on binding glutamate that has been released from a companion cell, charged ions such as Na+ and Ca2+ pass through a channel in the centre of the receptor complex. Since glutamate is the major excitatory neurotransmitter in the central nervous system (CNS), glutamate receptors play an important role in the mediation of excitatory synaptic transmission and they are the means by which neurons communicate with each other.22 There is considerable experimental and clinical evidence indicating that glutamate is involved in the pathogenesis of neuronal degeneration in the context of hypoxia/ischemia and trauma of the central nervous system, seizures and hypoglycemia.23,24 In addition, glutamate is thought to be involved in the pathogenesis of chronic neurodegenerative disorders, such as amyotrophic lateral sclerosis, Huntington's, Alzheimer's and Parkinson's disease.25 Functional glutamate receptors have also been identified in lung, muscle, pancreas and bone.26,27The ionotropic glutamate receptor family comprises N-methyl-D-aspartate (NMDA), the α-amino-3-hydroxy-5-methylisoxasole-4-propionic acid (AMPA) and kainate (KA) receptors.28 The latter two are also called non-NMDA receptors. Ionotropic glutamate receptors are involved in central synaptic pathways, serving different functions that are fulfilled by subtypes of these receptors acting in conjunction with each other.22 The development of specific agonists and antagonists for ionotropic glutamate receptors has potential for the development of novel drugs for neurological, mental and psychiatric disorders.22
The Joro spider toxins (JSTX) from the tetragnathid spider Nephila clavata are a mixture of acylpolyamines where the major constituent is JSTX-3 4.29 It was found that JSTX-3 blocks the postsynaptic glutamate receptors in mammalian central neurons.30–32 Acylpolyamines that antagonize glutamate cell receptors have subsequently been found in other spiders.33–38
In 1993, Konnerth and his group39 examined JSTX-3 action on the recombinant AMPA/KA receptors expressed in Xenopus oocytes and found that JSTX-3 acts as a subunit-specific blocker. Subunits forming a receptor channel with a linear current–voltage (I–V) relationship (GluR1/2, GluR2/3, and GluR6) were not affected, while receptor subunits with rectifying I–V relationships (GluR1, GluR3, GluR4, and GluR1/3) were blocked by JSTX-3. Using receptor-subunit mutants obtained by site-directed mutagenesis, they identified a single amino acid position (glutamine in the proposed second transmembrane domain) that was critical for the JSTX-3 block. Since several health disorders arise as a consequence of alteration of the function of Na+, K+ and Ca2+ channels via glutamate receptors, acylpolyamines could clearly act as antagonists and prevent ionic flux. Spider acylpolyamines could find a role as novel agents for regulating such ion channels. Nowdays, JSTX-3 is commercially available and it has subunit-specific activity on the glutamate receptors.39 Pharmacological studies involving JSTX-3 have given new insights into the molecular mechanisms of glutamate receptors in various nervous diseases.40
2.2 Acylpolyamines and the polyamine catabolism pathway
The polyamine catabolism pathway is important in preventing the toxic effects of excess polyamines on cells.41,42 Increases in polyamine concentration have been linked to certain types of cancer. For example, stimulation of human fibroblasts to cell proliferation by serum or epidermal growth factor was followed by an 18–100 fold increase in the uptake of putrescine.43 Moreover, brain tumours have been shown to have an increased rate of putrescine uptake.44 Inhibition of polyamine biosynthesis in cells in culture by α-difluoromethylornithine, an inhibitor of the enzyme that catalyzes the production of putrescine, causes a substantial depletion of intracellular putrescine and spermidine with resultant cell proliferation inhibition.45 Synthetic analogues have been sought that attempt to maximize interactions with the polyamine catabolism pathway.46,47 To explore aspects of this biology, an acylpolyamine analogue that resembles spider acylpolyamines was synthesized from spermidine. The resultant compound N1-[(N6-dansyl)-6 aminocaproyl] spermine (DACS, Fig. 5) 5 inhibits polyamine transport and could be used as a pharmaceutical agent for treating diseases related to polyamine transport such as cancer and post-angioplasty injury. This compound is able to displace polyamine uptake with a Ki of 8 nM.48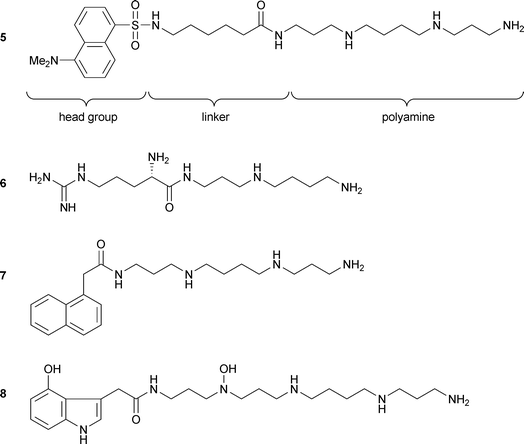 | ||
| Fig. 5 Structures of analogues of spider acylpolyamines and AG-505. DACS 5, L-Arg-3,4 6, Naspm 7, and AG-505 8. | ||
2.3 Acylpolyamines as neuroprotective agents
Ischemic insult triggers excessive excitatory synaptic activation followed by sustained Ca2+ influx through glutamate receptors, and elevation of intracellular Ca2+ has been implicated in the process of neuronal death.40,49,50 In events such as prolonged hypoxia and ischemia, which may or may not be associated with hypoglycaemia, neuronal damage, to varying degrees, is encountered. Ischemia typically occurs during heart attacks, but the damage incurred at these times is substantially limited to the heart tissues, and certain treatments have been developed.51 The use of polyamines as neuroprotective agents is concerned with the effects of more long-term ischemia on the brain, such as occurs with stroke patients or as a result of head injury. The severity of the ischemia depends on the nature of the stroke or injury, but, invariably, there is brain damage. Various neuroprotective agents are known which attempt to alleviate the problem of brain damage, but all of those currently known tend to be associated with adverse side effects.51,52 Other authors also described the neuroprotective effects of the N-type voltage sensitive calcium channel (VSCC) blocker conotoxin MVIIA, which has also been named SNX-111, but despite the neuroprotective effects of this compound, adverse side effects, in vivo, are observed.53 The venom of the funnel web spider Agelenopsis aperta contains a mixture of toxins including a fraction termed FTX which blocks P-type VSCC in rat Purkinje neurons and cortical synaptosomes. The active component of crude FTX, called FTX-3,3, has been isolated and identified. The electrophysiological properties of FTX-3,3 has been studied in some detail demonstrating that it preferentially blocks P-type VSCC but also blocks N- and neuronal L-type VSCC.54,55 Because of the potential involvement of VSCC in cellular Ca2+ loading during ischemic depolarization, these toxins, in modified form, could be useful neuroprotective agents. A synthetic compound based on the structure of FTX-3.3, was generated via solid support combinatorial chemistry from which, L-arginyl-3,4-spermidine (L-Arg-3,4) 6, emerged as a lead compound from an in vitro screen against hypoxia in organotypic brain slice cultures.56,57 Moreover, using slice-patch recording from CA1 pyramidal neurons, unusual slow excitatory postsynaptic potentials (EPSCs) that consist of non-NMDA currents were recorded only after ischemia.58 The abnormally slow EPSCs were effectively blocked by JSTX-3 and its analogue, 1-naphthylacetyl spermine (Naspm) 7. The results indicate that JSTX-3 are effective at blocking abnormal EPSCs that may induce Ca2+ accumulation leading to delayed neuronal death after transient ischemic insult.47 Similar neuroprotective action against ischemia by a variety of antagonists including JSTX-3 was reported in the corticostriatal brain slices.522.4 Acylpolyamines and pain
Sorkin et al.59 found that allodynia was blocked specifically by JSTX-3, suggesting that spinal mechanisms leading to tactile allodynia in this injury model act via a Ca2+-permeable AMPA linkage. They characterized the analgesic profile associated with the blockade of these spinal receptors by intrathecally delivered JSTX-3.60 JSTX-3 (5 mg) was given 1 h after carrageen-blocked induction of thermal hyperalgesia and mechanical allodynia. The behavioural effect of intrathecal Ca2+-permeable AMPA antagonists showed an important role for this spinal receptor in regulating hyperalgesic states induced by tissue injury and inflammation and revealed an action that is distinct from those observed with other glutamate receptor antagonists.40 Jones and Sorkin61 confirmed that intrathecal pre-treatment with NMDA receptor antagonists blocked development of spinal sensitization, whereas secondary mechanical allodynia evoked by thermal injury to the hind paw in the rat was not blocked by intrathecal pretreatment with NMDA receptor but blocked by antagonists to AMPA/KA and Ca2+-permeable AMPA/KA receptors. These findings suggest a role for these receptors in the development of spinal sensitization.Pogatzki et al.62 studied secondary hyperalgesia caused by gastrocnemius incision in the rat. Only secondary mechanical hyperalgesia was reversed by JSTX-3, while primary mechanical hyperalgesia and guarding behaviour were unchanged. The results indicate that spinal sensitization contributing to behaviours for secondary hyperalgesia after incision requires Ca2+-permeable AMPA/KA receptors.40 The data further demonstrate that behaviour for secondary mechanical hyperalgesia after incision can be inhibited without affecting the behaviour for primary mechanical hyperalgesia and guarding. Mechanisms for central sensitization causing secondary hyperalgesia in postoperative patients may therefore be separated from spontaneous pain and hyperalgesia that arises adjacent to the area of the incision.40
Moreover, acylpolyamine AG-505 8, from the agelenid spider Agelenopsis aperta, was able to block capsaicin receptor channels. These receptors are non-selective cation channels that integrate multiple noxious stimuli in sensory neurons.37
2.5 Acylpolyamines and nervous diseases
JSTX-3 was used to characterize Ca2+-permeable AMPA receptors in a variety of central neurons. By use of JSTX-3, Gu et al.63 showed that Ca2+-permeable AMPA receptors were synaptically localized on a subpopulation of dorsal horn neurons and the receptors provided a synaptically gated route of Ca2+ entry. Activation of these receptors strengthened synaptic transmission mediated by AMPA receptors. The study concluded that this pathway for postsynaptic Ca2+ influx may provide a new form of activity-dependent modulation of synaptic strength. In cultures of motor neurons from 15 day old rat embryos, Ca2+ influx via AMPA/KA receptors was responsible for the regulation of dendrite outgrowth when synaptic contacts from afferent neurons to motor neurons were made in the spinal cord.64JSTX-3 was also used to explore the selective death of motor neurons, which might be responsible for amyotrophic lateral sclerosis (ALS). Mutations in the Cu/Zn-superoxide dismutase (SOD-1) gene are responsible for a subset of familial ALS. Roy et al.65 showed that JSTX-3 reduced formation of SOD-1 proteinaceous aggregates and prevented death of motor neurons expressing SOD-1 mutants and concluded that normally nontoxic glutamatergic input, particularly via Ca2+-permeable AMPA/KA receptors, is a major factor in the vulnerability of motor neurons to the toxicity of SOD-1 mutants. Van Den Bosch and Robberecht66 compared the sensitivity of motor neurons and that of dorsal horn neurons to KA. Short exposure to KA resulted in the death of motor neurons, while dorsal horn neurons were unaffected. This selective motor neuron death was completely inhibited by JSTX-3.
2.6 Synthetic analogues of acylpolyamines
One of the disadvantages of native spider acylpolyamine toxins is their lack of selectivity for this family of ionotropic glutamate receptors. They can antagonize both NMDA and non-NMDA receptors. However, new synthetic analogues of acylpolyamines have the ability to antagonize a sub-family of the glutamate receptors that may serve as basis for future development of particular drugs for the treatment of neurological diseases.Among such acylated spermine derivatives, Naspm 7 was the most potent.67 In lobster neuromuscular synapses, the potency of Naspm for blocking of EPSPs was 1–1.5 orders less potent than JSTX-3, but the effect was reversible.68 In cultured hippocampal neurons, Naspm reversibly blocked both kainate- and quisqualate-activated inward currents in a non-competitive manner. The single channel conductance calculated from noise produced by quisqualate and kainate was considerably reduced by Naspm treatment. These results indicate that Naspm exerts its blocking action on non-NMDA receptor channels through effects on both single channel conductance and kinetics.69 Simpler synthetic spider acylpolyamine analogues have been sought to attempt to maximize interactions with either invertebrate neuromuscular synapses or vertebrate mammalian glutamate receptors.46,67
3 Spider peptides
Because of their nature, peptides from the venom of spiders are toxic to the specific spider's preys; however, they may not be toxic to other organisms, so we use the term toxin, peptide or peptide toxin to indicate a peptide obtained from the venom of spiders. More than 296 amino acid sequences70 and the three dimensional (3D) structures of nearly 30 spider peptides71 have been described. Table 1 shows the amino acid sequences and biological activity of several such peptides. Spider peptides belong to a structural family which has the inhibitory cystine knot (ICK), a motif that is formed by two disulfide bridges, a part of the backbone of the peptide and a third disulfide bridge going though this loop71,72 (Fig. 6). The amino acid variations of the exposed loops are particular since they bind specific voltage-gated and ligand-gated channels. Moreover, they can recognize an ion channel region far from the pore, and they induce a shift of channel opening to more depolarized potentials that alter the voltage dependent properties of K+, Na+ or Ca+ currents. In this sense, spider peptides can be considered as gating modifiers rather than gating blockers.73| Name | Amino acid sequences | aa | db | pI | Biological activity | Reference |
|---|---|---|---|---|---|---|
| a aa, number of amino acids; db, disulfide bridges; Protein pI were calculated using pK values of amino acids described at http://us.expasy.org/tools/pi_tool.html.142–144 | ||||||
| Voltage-gated | ||||||
| K+ blockers | ||||||
| Hanatoxin 1 | ECRYLFGGCKTTSDCCKHLGCKFRDKYCAWDFTFS | 35 | 3 | 8.3 | Kv2.1 (Shab-related) and Shab1-related channels expressed in X. laevis oocytes | 84 |
| Heteropodatoxin 1 | DCGTIWHYCGTDQSECCEGWKCSRQLCKYVIDW | 33 | 3 | 4.8 | Kv4.2 expressed in X. laevis oocytes | 85 |
| Phrixotoxin 1 | YCQKWMWTCDSARKCCEGLVCRLWCKKII | 29 | 3 | 8.9 | Kv4.2 and Kv4.3 in COS transfected cells and in X. laevis oocytes | 87 |
| ScTx1 | DCTRMFGACRRDSDCCPHLGCKPTSKYCAWDGTI | 33 | 3 | 7.8 | Kv2.1 and Kv2.2 in COS transfected cells | 111 |
| HmTx1 | ECRYLFGGCSSTSDCCKHLSCRSDWKYCAWDGTFS | 35 | 3 | 6.8 | Kv4 and Kv4.1 in COS transfected cells | 111 |
| SGTx1 | TCRYLFGGCKTTADCCKHLACRSDGKYCAWDGTF | 34 | 3 | 8.3 | Fast transient and delayed rectifier currents in rat cerebellum granular cells | 112 |
| PhTx3-1 | AECAAVYERCGKGYKRCCEERPCKCNIVMDNCTCKKFISE | 40 | 4 | 8.2 | A-type currents in GH3 cells | 113 |
| TLTx1 | AACLGMFESCDPNNDKCCPNRECNRKHKWCKYKLW | 35 | 3 | 8.6 | Gating modifier Kv4.2 | 90 |
| Ca2+ blockers | ||||||
| AgaIA | AKALPPGSVCDGNESDCKCYGKWHKCRCPWKWHFTGEGPCTCEKGMKHTCITKLHCPNKAEWGLDW-SPC | 69 | 5 | 8.4 | L-type in rat dorsal root ganglion neurons | 114–116 |
| AgaIIA | GCIEIGGDCDGYQEKSYCQCCRNNGFCS… incomplete | — | — | — | N-type in chick synaptosomes | 114,117 |
| AgaIIIA | SCIDIGGDCDGEKDDCQCCRRNGYCSCYSLFGYLKSGCKCVVGTSAEFQGICRRKARQCYNSDPDKCESHNKPKRR | 76 | 6 | 8.5 | L-, P/Q-, R-, N-type in rat brain synaptosomes and HEK293 transfected cells | 117–119 |
| AgaIVA | KKKCIAKDYGRCKWGGTPCCRGRGCICSIMGTNCECKPRLIMEGLGLA | 48 | 4 | 8.5 | P/Q-type and P-type currents in cerebellar Purkinje neurons | 117,120 |
| SNX-482 | GVDKAGCRYMFGGCSVNDDCCPRLGCHSLFSYCAWDLTFSD | 41 | 3 | 4.8 | R-type current in rat neurohypophyseal nerve terminals and L-type calcium channel in transfected HEK cells | 94 |
| SNX-325 | GSCIESGKSCTHSRSMKNGLCCPKSRCNCRQIQHRHDYLGKRKYSCRCS | 49 | 4 | 9.5 | N-type expressed in X. laevis oocytes | 101 |
| GSTxSIA | DCVRFWGKCSQTSDCCPHLACKSKWPRNICVWDGSV | 36 | 3 | 8.3 | N- and P/Q-type in rat hippocampal neurons | 96 |
| Huwentoxin-I | ACKGVFDACTPGKNECCPNRVCSDKHKWCKWKL | 33 | 3 | 8.9 | N-type expressed in prostaglandin E1 differentiated NG108-15 cells | 102,103 |
| Huwentoxin-X | KCLPPGKPCYGATQKIPCCGVCSHNKCT | 28 | 3 | 8.9 | N-type in rat dorsal root ganglion | 104 |
| DW13.3 | AECLMIGDTSCVPRLGRRCCYGAWCYCDQQLSCRRVGRKRECGWVEVNCKCGWSWSQRIDDWRADYSCKCPEDQ | 74 | 6 | 8.0 | P/Q-, N-, L-, and R-type in X. laevis oocytes | 105 |
| ω-PTx-IIA | SCINVGDFCDGKKDDCQCCRDNAFCSCSVIFGYKTNCRCEVGTTATSYGICMAKHKCGRQTTCTKPCLSKRCKKNH | 76 | 7 | 8.8 | P/Q-, N-, and R-type in BHK cell lines | 108 |
| PnTx3–6 | ACIPRGEICTDDCECCGCDNQCYCPPGSSLGIFKCSCAHANKYFCNRKKEKCKKA | 55 | 6 | 8.3 | Reversible inhibition of recombinant mammalian Ca+2 P,Q and R-types | 121 |
| Voltage-gated | ||||||
| Na+ blockers | ||||||
| Robustoxin | CAKKRNWCGKNEDCCCPMKCIYAWYNQQGSCQTTITGLFKKC | 42 | 4 | 8.9 | Blocks inactivation of Na+ currents in adult rat dorsal root ganglion neurons | 122,123 |
| Versutoxin | CAKKRNWCGKTEDCCCPMKCVYAWYNEQGSCQSTISALWKKC | 42 | 4 | 8.7 | Blocks inactivation of Na+ currents in adult rat dorsal root ganglion neurons | 124,125 |
| Huwentoxin-IV | ECLEIFKACNPSNDQCCKSSKLVCSRKTRWCKYQI | 35 | 3 | 8.9 | Block the pore of Na+ channels in adult rat dorsal root ganglion neurons | 126 |
| ProTx-I | ECRYWLGGCSAGQTCCKHLVCSRRHGWCVWDGTFS | 35 | 3 | 8.3 | Nav 1.8 and Kv 2.1 expressed in X. laevis oocytes, and T-type in HEK293 cells | 127 |
| ProTx-II | YCQKWMWTCDSERKCCEGMVCRLWCKKKW | 29 | 3 | 8.9 | Nav 1.8 and Kv 2.1 expressed in X. laevis oocytes | 127 |
| PnTx1 | AELTSCFPVGHECDGDASNCNCCGDDVYCGCGWGRWNCKCKVADQSYAYGICKDKVNCPNRHLWPAKVCKKPCRRNCGG | 79 | 6 | 8.2 | Reversible inhibition of recombinant Nav 1.2 | 128 |
| CcoTx1 | DCLGWFKSCDPKNDKCCKNYTCSRRDRWCKYDL | 33 | 3 | 8.6 | Nav 1.2 | 129 |
| PaurTx3 | DCLGFLWKCNPSNDKCRPNLVCSRKDKWCKYQI | 33 | 3 | 9.0 | Nav 1.2 | 129 |
| JZTX-I | ACGQFWWKCGEGKPPCCANFACKIGLYLCIWSP | 33 | 3 | 8.3 | TTX resistant Na+ channel a-like cardiac myocytes | 130 |
| JZTX-III | DGECGGFWWKCGRGKPPCCKGYACSKTWGWCAVEAP | 36 | 3 | 8.3 | TTX resistant Na+ channel site 4 rat cardiac myocytes | 131 |
| Magi4 | CGSKRAWCKEKKDCCCGYNCVYAWYNQQSSCERKWKYLFTGEC | 43 | 4 | 8.6 | Blocks inactivation of Na+ current mammals and insects. Receptor site 3 of Nav | 132 |
| Magi5 | GCKLTFWKCKNKKECCGWNACALGICMPR | 29 | 3 | 9.1 | Open Nav 1.2 at more negative potentials. Receptor site 4 of Nav | 132 |
| Magi6 | KCVDGSCDPYSSNAPRCCGSQICQCIFFVPCYCKYR | 36 | 4 | 8.2 | Modifies currents of brain Nav | 132 |
| HNTX-IV | ECLGFGKGCNPSNDQCCKSSNLVCSRKHRWCKYEI-NH2 | 35 | 3 | 8.6 | TTX sensitive Na+ channel | 133 |
| Ligand-gated | ||||||
| ASIC | ||||||
| PcTx1 | EDCIPKWKGCVNRHGDCCEGLECWKRRRSFEVCVPKTPKT | 40 | 3 | 8.7 | ASIC in sensory neurons from rat and ASIC1a expressed in X. laevis oocytes and COS cells | 134 |
| Mechano sensitive ion channels | ||||||
| GsMTx-4 | GCLEFWWKCNPNDDKCCRPKLKCSKLFKLCNFSSA | 35 | 3 | 8.9 | Stretch-activated channels in adult rat astrocytes | 135 |
| Glutamate uptake | ||||||
| PnTX3–4 | SCINVGDFCDGKKDCCQCDRDNAFCSCSVIFGYKTNCRCE | 40 | 4 | 5.0 | Inhibits glutamate uptake in rat synaptosomes | 136 |
| Pore-forming | ||||||
| Lycotoxin I | IWLTALKFLGKHAAKHLAKQQLSKL | 25 | 0 | 10.6 | Phospholipids, promotes efflux of Ca2+ from synaptosomes, red-blood cells | 137 |
| Lycotoxin II | KIKWFKTMKSIAKFIAKEQMKKHLGGE | 27 | 0 | 10.2 | Phospholipids, promotes efflux of Ca2+ from synaptosomes, red-blood cells | 137 |
| Cupiennin 1 | GFGALFKFLAKKVAKTVAKQAAKQGAKYVVNKQME | 35 | 0 | 10.3 | Antimicrobial analysis, red-blood cells | 138 |
| Oxyopinin 1 | FRGLAKLLKIGLKSFARVLKKVLPKAAKAGKALAKSMADENAIRQQNQ | 48 | 0 | 11.3 | Phospholipids, reduction of cell membrane resistance by opening non-selective ion channels in Sf9 cells | 5 |
| Oxyopinin 2a | GKFSVFGKILRSIAKVFKGVGKVRKQFKTASDLDKNQ | 37 | 0 | 10.8 | Phospholipids, reduction of cell membrane resistance by opening non-selective ion channels, red blood cells | 5 |
| Lycocitin 1 | GKLQAFLAKMKEIAAQTL-NH2 | 18 | 0 | 10.3 | Antimicrobial activity against Gram-positive and Gram-negative bacteria and fungi | 139 |
| Lycocitin 3 | KIKWFKTMKSLAKFLAKEQMKKHLGE | 26 | 0 | 10.2 | Antimicrobial activity against Gram-positive and Gram-negative bacteria and fungi | 139 |
| Latarcin 1 | SMWSGMWRRKLKKLRNALKKKLKGE | 25 | 0 | 11.7 | Antimicrobial activity against Gram-positive and Gram-negative bacteria and fungi, hemolitic | 140 |
| Latarcin 3 | SWKSMAKKLKEYMEKLKQRA | 20 | 0 | 10.1 | Antimicrobial activity against Gram-positive and Gram-negative bacteria and fungi, hemolitic | 140 |
| Latarcin 5 | GFFGKMKEYFKKFGASFKRRFANLKKRL | 28 | 0 | 11.1 | Antimicrobial activity against Gram-positive and Gram-negative bacteria and fungi, hemolitic | 140 |
| Gomesin | QCRRLCYKQRCVTYCRGR | 18 | 2 | 9.9 | Antimicrobial activity against Gram-positive and Gram-negative bacteria and fungi | 141 |
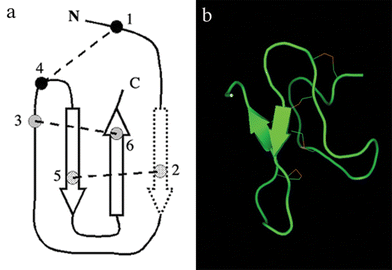 | ||
| Fig. 6 Schematic representation of the inhibitor cystine knot motif. (a) The dotted arrow represents a β-strand, which might be not found in some disulfide bridged spider peptides,74 (b) structure of paluIT1, an insecticidal spider peptide from Paracoelotes luctuosus (PDB 1V90). | ||
A comparison of the surface features of ion channel gating modifier peptides of voltage-gated K+, Na+ and Ca2+ channels suggests that the combination of a hydrophobic patch and the surrounding charged residues may represent the structural basis for the specificity for the binding of spider peptides to cell membrane receptors.75–78 A remarkable characteristic of these peptides is the predominance of basic and anionic residues (Fig. 7). The basic residues appear to be important to recognition and binding to the ion channels and anionic residues seem to be central for biological activity.77,78 Spider peptides have similar structural and surface characteristics to the cone snail peptide MVIIA, which is the base of a potent analgesic against chronic pain already approved by the Food and Drug Administration (FDA). The 3D structures of spider peptides, all of which have been solved by Nuclear Magnetic Resonance (NMR),71 together with the recent crystallization of K+ ion channels, have facilitated structure–function studies of spider peptides and their receptors.79,80
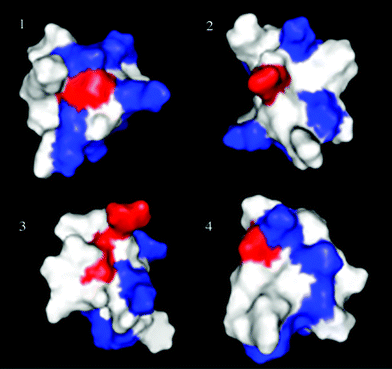 | ||
| Fig. 7 Three-dimensional structures of short peptide toxins from spider venoms and the N-type cone snail toxin MVIIA. Magi5 (Na+, PDB unpublished) 1, phrixotoxin (K+, PDB 1V7F) 2, ω-grammotoxin-SIA (Ca2+, PDB 1KOZ) 3, and MVIIA (Ca2+, PDB 1MVJ) 4. Three-dimensional structure data was obtained from the Protein Data Bank and the structures were displayed by the software PyMOL.81 Each panel represents an arbitrary view of the structure showing the basic (blue) and acidic (red) residues. | ||
3.1 Spider peptides and potassium channels
Potassium channels represent the largest and most diverse sub-group of ion channels and play a central role in regulating the membrane potential of cells. Members of this family play a critical role in cellular signalling processes regulating neurotransmitter release, heart rate, insulin secretion, neuronal excitability, epithelial electrolyte transport, smooth muscle contraction and cell volume regulation.82 There are several families of K+ channels in animals, including: (i) voltage activated channels (Kv); (ii) inward rectifier channels (Kirs), which have a higher conductance for K+ ions moving into the cell; and (iii) two-pore channels (K2P), a relatively new family of potassium channels that have two pore lining domains, each flanked by two transmembrane domains.82 More than 50 human genes encoding various K+ channel subunits have been cloned during the past decade,83 and the biophysical properties of the resulting channels and their modulation by ligands have been largely addressed.Unlike other animal peptide toxins such snake, bee, scorpion or sea anemone toxins, which block Kv1 or Kv3 channels, spider peptides target Kv2 and Kv4 channels, which are expressed in the CNS and in the cardiovascular system. Their selective affinities for Kv2 and/or Kv4 subfamilies are very useful for dissecting currents in neuronal and cardiac cells and for the determination of their contribution in physiological processes.73
Spider peptides have permitted, in particular, the study of new voltage activated channels. The first spider peptides found to block Kv were hanatoxins 1 and 2 (HaTx 1 and 2) from the Chilean theraposid Grammostola spatulata.84 HaTxs block the Kv2.1 (shab-related K+ channel) with a Kd of 42 nM. HaTxs are unrelated in their primary sequence to other K+ channel inhibitors such as Kv peptide toxins from scorpion venoms. Other Kv such as the shaker-related (Kv1-like), shaw-related (Kv3-like), and eag K+ channels are insensitive to HaTxs, whereas a shal-related (Kv4-like) K+ channel is sensitive to HaTxs.84
Similar to the HaTxs, the heteropodatoxins (HpTx 1–3) were isolated from the venom of the Malayan sparassid spider Heteropoda venatoria.85,86 The HpTxs prolonged the action-potential duration of isolated rat ventricular myocytes, suggesting that the peptides blocked K+ currents. The HpTxs block the Kv4.2 in a voltage-dependent manner, with fewer blocks at more positive potentials. In addition, the HpTxs slow the time course of current activation and inactivation and shift the voltage-dependence of current inactivation to more positive potentials.85
The phrixotoxins (PaTxs) were purified from the venom of the Chilean theraposid Phrixotrichus auratus87 (Table 1, Fig. 7). Phrixotoxins specifically block Kv4.3 and Kv4.2 currents with IC50 of 5 nM to 70 nM, by altering the gating properties of the channels, and they also block cardiac current Ito1 in isolated murine cardiomyocytes. The shaker (Kv1), shab (Kv2) and shaw (Kv3) subfamilies of currents were not inhibited by phrixotoxins.87 The Ito1 current is a rapidly activating and inactivating potassium current which is inhibited by 4-aminopyridine and class I antiarrhythmic drugs.88 The electrocardiography consequences of the in vivo inhibition of Ito1 were properly characterized by intravenous injections of PaTx1 in mice.87 It resulted in a dose-dependent decrease of the rapid component of the murine T wave and a subsequent prolongation of the QT interval arguing for a role of Ito1 that is paramount in shaping the early phase of the cardiac action potential re-polarization. Injection of higher doses of PaTx1 resulted in various transient heart rhythms and conduction problems including the occurrence of premature ventricular beats, ventricular tachycardia and different degrees of atrio-ventricular block.87 Thus, spider toxins that are specific for Kv4 channels are interesting tools for characterising the role of Ito1 in cardiac physiology. More recently, other spider peptides that inhibit voltage-dependent potassium channels in the shab (Kv2) and shal (Kv4) subfamilies were isolated from the venom of the African theraposids Stromatopelma calceata (ScTx1) and Heteroscodra maculata (HmTx1, HmTx2).89 ScTx1 is the first high-affinity inhibitor of the Kv2.2 channel subtype to be described, with an IC50 of 21.4 nM. ScTx1 also inhibits the Kv2.1 channels, with an IC50 of 12.7 nM, as well as Kv2.1/Kv9.3 heteromultimers that have been proposed to be involved in O2 sensing in pulmonary artery myocytes. Also, ScTx1, with a higher affinity (IC50 of 1 nM) and specificity on Kv4.2 (than Kv4.1 and Kv4.3) could further contribute to the characterization of Ito1 currents in cardiac electrophysiology. Similarly, TLTx1 from the theraposid spider Theraposa leblondi inhibits Kv4.2 channels, and no activity was observed for Kv1.3, Kv1.4, Kv2.1 or Kv3.4 channels.90 Other physiological studies have pointed out that spider peptides that block Kv4.2 and Kv4.3, also inhibit Ito1 currents, and induce hypertrophy of rat ventricular myocytes in vitro.86 These potassium channel modifiers emerge as promising molecules for the development of cardiac drugs.
3.2 Spider peptides and calcium channels
Voltage-sensitive calcium channels (VSCCs) play a crucial role in coupling the electrical activity of neurons to a variety of cellular processes such as neurotransmitter and hormone secretion, cell proliferation and gene expression.91 To date, at least five different types of VSCCs have been identified. The L-type Ca2+ channels are present mainly in the cardiovascular system, and are highly sensitive to non-peptide drugs such as dihydropyridines (DHP), phenylalkylamines and benzodiazepines. The non-L-type Ca2+ channels (T-, N-, P/Q-, and R-type) are insensitive to dihydropyridines; however, they can be antagonized by many peptide toxins from animal venoms. Such peptide drugs may therefore represent a novel generation of Ca2+ channel modulators with neuroprotective and analgesic properties.92 Although multiple subtypes of voltage-gated calcium channels exist, pharmacological and ion-channel gene knockdown approaches in animals have revealed N-type and T-type calcium channels to be particularly attractive molecular targets for the discovery and development of new analgesic drugs.93The venom of the North American agelenid spider Agelenopsis aperta was the source of the first peptide blockers of non-L VSCCs. The ω-agatoxins were categorized based on their neuromuscular effects on insects and the displacement of previously identified calcium channel blockers such as the conotoxins, in chick synaptosomes91 (Table 1).
SNX-482, a selective peptide antagonist of R-type and L-type VSCCs was isolated from the African theraposid Hysterocrates gigas.94 At low nanomolar concentrations (∼40 nM), SNX-482 blocked a native resistant (R-type) Ca2+ current in rat neurohypophyseal nerve terminals, but concentrations of 200–500 nM had no effect on R-type Ca2+ currents in several types of rat central neurons. Experiments involving chimeric channels combining structural features of R-type (α1E) and L-type (α1C) calcium channel subunits indicated that the presence of domains III and IV of the R-type current is a prerequisite for a strong gating inhibition. SNX-482 also has small but pronounced effects on L-type calcium channel gating95; so, SNX-482 has proven specificity of spider peptides to VSSCs in neurons. Another peptide, named GsTxSIA, was isolated from the venom of the Chilean theraposid G. spatulata,96 and blocks N-, P- and Q-type VSCCs. The three-dimensional structure of this toxin was determined by NMR spectroscopy (Fig. 7).97 Although GsTxSIA was originally identified as an inhibitor of voltage-gated Ca2+ channels, it also binds to K+ channels although with much lower affinity. Similar binding was observed for HaTx1, which binds to the voltage-sensing domains of both K+ and Ca2+ channels but with very different affinities. A comparison between HaTx1 and GsTxSIA structures identifies a conserved face containing a large hydrophobic patch surrounded by positively charged residues which appear to be important in binding to the receptor site on the channel, possibly located on or near the voltage sensor. This also suggests a conserved binding domain relating to the voltage sensing capability of ion channels of different ion selectivity as revealed by toxin binding.
Saegusa et al.98 reported that mice lacking N-type VSCCs showed suppressed responses to a painful stimulus that induces inflammation, and also showed reduced symptoms of naturopathic pain.98 Intrathecal administration of non-selective blockers of VSCCs has anti-nociceptive effects on responses to hot plate tests.99,100 Moreover, other pharmacological and ion-channel gene knockdown approaches in animals have revealed N-type and T-type calcium channels to be particularly attractive molecular targets for the discovery and development of new analgesic drugs.93 A relevant finding was anti-nociceptive effects of the N-type blocker MVIIA from the venom of the cone snail Conus magus. The FDA has accepted this peptide toxin as a drug against chronic pain under the name of ziconotide. Spider venoms also contain several peptide toxins that specifically affect the N-type VSCCs, and sooner or later a spider peptide with similar therapeutic characteristics may be on the pharmaceutical market; hence, interesting N-type VSCC peptide toxins have been elucidated. For example, SNX-325 has been isolated from the venom of the segrestiid spider Segestria florentina.101 At nanomolar concentrations (IC50 of 3–30 nM), SNX-325 is a selective blocker of the N-type but not cardiac L-, P/Q-, or R-type, calcium channels. Similarly, huwentoxin-I (HWTX-I) and huwentoxin-X (HWTX-X) from the venom of the Chinese theraposid Selenocosmia huwena102 inhibited N-type Ca2+ channels and had only very weak effects on L-type Ca2+ channels.103,104
Larger peptides (>8 KDa) with VSCC specificity have also been found in spider venoms. The peptide DW13.3 isolated from the venom of the filistatid spider Filistata hibernalis causes a potent but transient block of native calcium channels.105 DW13.3 had the highest affinity for P/Q-type, followed by N → L → R-type with no effect on T-type currents recorded from GH3 cells. A block produced a dose-response with a Hill coefficient of 1.0 for all calcium channel subtypes, suggesting a single molecular target. DW13.3 produced a partial block of both α1A (60%) currents and P-type (76%) currents in Purkinje cells. Selective exclusion of the P/Q-type channel ligand ω-conotoxin MVIIC (but not ω-agatoxin IVA) from its binding site in Purkinje neurons suggests that DW13.3 binds to a site close to the pore of the channel. Other large peptides, ω-phonetoxin-IIA (ω-PnTxIIA) and PnTx3–6 purified from the venom of the spider P. nigriventer,106,107 blocked P/Q-type and N-type calcium currents almost irreversibly at 3 nM (for ω-PnTxIIA), whereas R-type currents showed partial and readily reversible inhibition. Binding and displacement assays with mono 125I-ω-PTxIIA, demonstrated that rat brain synaptosomes displayed multiple classes of binding sites. High affinity binding of 125I-ω-PTxIIA was totally displaced by ω-PTxIIA (Ki = 100 pM), but only partially by ω-conotoxin GVIA (25% inhibition) and ω-conotoxin MVIIC (50% inhibition at 0.3 µM).108 ω-PnTxIIA has been tested as a new tool for anti-calcium channel autoantibody assays in Lambert–Eaton myasthenic syndrome ischemia.109 Also, it has been reported that large spider peptides such P. nigriventer neurotoxins may exert neuroprotection in in vitro models of brain ischemia.110
3.3 Spider peptides and sodium channels
Voltage-activated Na+ channels (Nav), together with K+ channels represent the physiological basis of signal transmission in the nervous system. Their coordinated function is essential for efficient impulse generation and propagation in the central and peripheral nervous system. There are at least 10 different genes encoding distinct sodium channels in mammals and one or two genes in invertebrate species.111 The structural diversity of sodium channels appears to be paralleled by their diversity in physiological and pharmacological properties.112 Sodium channel organization in the neuron might affect nerve activity, leading to sensory and motor dysfunction. In humans, this could contribute to the pathophysiological mechanism of several neurological diseases, such as multiple sclerosis, epilepsy, stroke, peripheral neuropathies and neuropathic pain.113 The Nav is the primary molecular target of numerous therapeutic drugs (e.g. local anesthetics, anticonvulsants and antiarrhythmics) and insecticides (e.g. pyrethroids). Scorpion, sea anemone and cone snail peptide toxins have significantly contributed to the understanding of the topology and pharmacology of sodium channels.114 Spider peptides as sodium channel ligands and current modifiers, could also substantially contribute to the understanding of the complexity of sodium channels and their physiological role in the nervous system.Since all subtypes of Nav share more than 70% homology and similar pharmacology, their responses to a variety of spider neurotoxins could be diverse. Two main categories of sodium channels have been characterized in mammals based on the use of tetrodotoxin 9 (TTX, Fig. 8). The TTX-sensitive channels are found essentially in mammalian brain and skeletal muscle (Nav1.1–1.3, 1.4, 1.6 and 1.7), and the TTX-resistant channels are found in heart (Nav1.5) and sensory neurons in peripheral ganglia (Nav1.8 and 1.9). Therefore, the ability of spider toxins to discriminate between Nav channel subtypes in mammalian tissues is of potential value for future rational design of novel therapeutics. Presently, the number of spider peptide toxins found to target the mammalian Nav channel is limited. However, some of them have shown interesting pharmacology. For example, JZTX-I and -III, from the Chinese theraposid Chilobrachys jingzhao, have been shown to selectively modulate cardiac Nav1.5 channels and may prove useful tools in discriminating this channel from other TTX-resistant channels such as Nav1.8 and Nav1.9 important in pain perception.115,116 Both ProTx-I and ProTx-II, from the theraposid Thrixopelma pruriens, were isolated on the basis of their ability to inhibit the TTX-resistant Nav1.8 channel, distributed in small diameter nociceptive dorsal root ganglion (DRG) neurons, in an attempt to find novel analgesic compounds.117 Unfortunately these peptide toxins also inhibit other Nav channel subtypes and VSCCs at similar concentrations. Therefore the hunt for a selective blocker of the Nav1.8 or 1.9 channels continues.
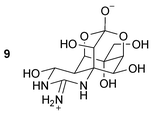 | ||
| Fig. 8 Structure of tetrodotoxin (TTX). | ||
The first sodium channel spider peptides isolated that target mammalian receptors were δ-atracotoxin Ar1 (robustoxin) and δ-atracotoxin Hv1 (versutoxin), from the venoms of the Australian hexathelid spiders Atrax robustus and Hadronyche versuta, respectively.118,119 The Australian funnel web spiders A. robustus and H. versuta are dangerous species, with respiratory failure observed after human envenomation by A. robustus.120 δ-Atracotoxins Ar1 and Hv1 have the unique characteristic of a cysteine triplet at residues 14–16 (Table 1). They consist of a small, triple-stranded, antiparallel β-sheet and several reverse turns. δ-Atracotoxins Ar1 and Hv1 slow the inactivation of TTX-sensitive sodium channels,121,122 and bind to site 3 of both invertebrate and vertebrate Nav, displacing with nanomolar affinity the scorpion α-toxins.122 They show no structural homology with either sea anemone or α-scorpion peptide toxins that modify the inactivation kinetics of voltage-gated sodium channels by also interacting with the site 3 of sodium channels. Nevertheless, δ-atracotoxins Ar1 and Hv1 contain charged residues that are topologically related to those implicated in the binding of sea anemone and α-scorpion peptide toxins to mammalian voltage-gated sodium channels, suggesting similarities in their mode of binding with these channels,123–125 Because of their huge affinity to Nav, amino acid mutagenesis of robustoxin and versutoxin offers a valuable strategy for information on Nav pharmacology. However, chemical or recombinant syntheses of these peptides are extremely difficult due to their cystine network.
Another structurally distinct group of spider peptides, from the Brazilian ctenid spider Phoneutria nigriventer, also targets sodium channels.126 This series of peptides from the toxic fraction PnTx1 of the venom of P. nigriventer produced reversible inhibition of recombinant Nav1.2 interacting with microsites of conotoxins.127 Moreover, two peptide toxins (Magi4 and 5) were isolated from the hexathelid spider Macrothele gigas. Competitive binding assays using several 125I-labelled peptide toxins clearly demonstrated the specific binding affinity of Magi4 and Magi5 to site 3 and site 4 of the rat sodium channel. Magi4 blocks the inactivation of the Nav, and Magi5 opens Nav at more negative potentials128 (Fig. 7). Recently, another sodium channel neurotoxin from the venom of the theraposid spider Selenocosmia huwena, which has been named huwentoxin-IV, was described.129 It specifically inhibits the neuronal TTX-sensitive Nav DRG neurons (IC50 of 30 nM), while having no significant effect on the TTX-resistant sodium channels. According to these results, and since it suppresses the peak sodium current without altering the activation or inactivation, huwentoxin-IV is proposed to bind at site 1 of the sodium channel through a mechanism quite similar to that of TTX. Similarly, hainantoxin-IV (HNTX-IV) can specifically inhibit the neuronal TTX-sensitive sodium channels.130 The recently described ceratotoxins (CcoTx1–2) and phrixotoxin 3 (PaurTx3) are peptides from the theraposids Ceratogyrus cornuatus and Phrixotrichus auratus respectively that affect Nav. CcoTx1 and CcoTx2, differing by only one amino acid, are potent modulators of different Nav subtypes from the central nervous system, except for Nav1.3, which is only affected by CcoTx2. PaurTx3 is one of the most potent peptide modulators of voltage-gated sodium channels described thus far from spider venom, modulating Nav1.2 with an IC50 of 0.6 nM.131 All those peptides are shorter than robustoxin and versutoxin; so, they are relatively easy to synthesize chemically because of their less complicated disulfide pattern.
3.4 Glutamate receptors
As mentioned above, glutamate receptors mediate fast excitatory transmission at synapses in the brain and spinal cord.132 The toxin phoneutriatoxin 3–4 (PhTx3–4) from the ctenid spider P. nigriventer has been reported to inhibit the uptake of glutamate in a time-dependent manner and to lead to a decrease in the Ca2+-independent release of glutamate133 (Table 1). Based on results obtained with manipulation of the redox state of cysteine residues in rat synaptosomes and in PhTx3–4, it was suggested that the effect is due to interactions that involve cysteines both in the peptide toxin and in the glutamate transporters. However, the specificity of PhTx3–4 for glutamate transporters is unknown. The peptide PhTx4–7 from the same spider seems to produce similar effects.134,135 Until now only these two peptides have been reported to affect glutamate receptors as well as the acylpolyamines.3.5 Peptides acting on acid-sensing ion channels (ASIC) and mechanosensitive ion channels (MS)
ASIC channels are associated with nociception, taste transduction, and perception of extracellular pH fluctuations in the brain.136 These channels play a central role in epilepsy or brain ischemia.137 A novel peptide (PcTx1) from the venom of the South American theraposid Psalmopoeus cambridgei, blocks, with an IC50 of 0.9 nM, a particular subclass of ASIC that are expressed in both central nervous system neurons and sensory neurons in dorsal root ganglia138 (Table 1). PcTx1 being the first high affinity and highly selective pharmacological agent for this novel class of ionic channels will be instrumental in fully characterizing the physiological role of these channels.Compared to voltage-dependent or ligand-gated ion channels that have been extensively studied over the last twenty years, there is little knowledge available on the structure and function of MS channels that constitute the third major group of ion channels classified according to their gating mechanism.139 MS channels also called SACs (stretch-activated ion channels) open and close in response to mechanical stimuli. GsMTx-4, a spider peptide from the Chilean theraposid G. spatulata, produced a complete block of SACs in outside-out patches in both adult astrocytes and cardiocytes and appeared specific since it had no effect on whole-cell voltage-sensitive currents140 (Table 1). Moreover, in isolated ventricular cells from a rabbit dilated cardiomyopathy model, GsMTx-4 produced a near complete block of the volume-sensitive cation-selective current, but did not affect the anion current. GsMTx-4 is the first peptide that specifically blocks stretch-activated currents although with a rather low affinity (650 nM). GsMtx-4 could be the first of a new class of antiarrhythmic agents to be directed against atrial fibrillation.141,142
3.6 Non-selective spider peptides
Spider peptides such ProTx-I and ProTx-II, from the venom of the theraposid Thrixopelma pruriens, inhibit the TTX-resistant Nav1.8; however they have shown promiscuity across ionic channel families. ProTx-I also shifts the voltage-dependence of activation of T-type VSCCs with an IC50 of 50 nM without affecting the voltage-dependence of inactivation, and also inhibits Kv 2.1 channels with 10-fold less potency than its potency on Nav channels. The non-selective actions of protoxins (ProTxI and II) illustrate the issue of cross-reactivity due to potentially conserved structures within domains of voltage-gated ion channels. However, target cross-reactivity also exemplifies a current issue in research aimed at determining the ion channel or receptor target of spider and other peptide neurotoxins. Many pharmacological studies investigate only a limited range of targets emphasizing the fact that these peptide toxins may act with greater efficacy at other sites. Since several spider peptide toxins present promiscuity among ion channels these potential drugs could induce an autoimmune response to peptidic drugs; so they must be dosed carefully during application.1433.7 Pore-forming peptides
Amphipathic and cationic α-helical peptides are broadly found in animals as part of their biological defense system.144 The first report of cytolytic activity found in the crude venoms of spiders was that from the venom of the Chinese lycosid spider Lycosa singoriensis145; however, no primary structures were reported. Several structures of spider peptides with pore-forming activities have been identified: lycotoxins I and II from the venom of the lycosid spider Lycosa carolinensis,146 cupiennins from the venom of the ctenid spider Cupiennius salei,147,148 oxyopinins from the venom of the oxyopid spider Oxyopes kitabensis,5 and lycocitins from lycosid Lycosa singoriensis.149 Cupiennins and oxyopinins are the largest pore-forming peptides isolated from the venom of a spider thus far (Table 1). A hydrophobic N-terminal chain region and a C-terminus composed preferentially of polar and charged residues characterize both cupiennins and oxyopinins. Oxyopinins can be divided into two peptide groups: oxyopinin 1 includes a proline at position 24, indicating a possible flexible hinge region that could separate two segments of α-helices in the peptide. Oxyopinins 2a, 2b, 2c and 2d exhibit highly similar sequences. Both peptide groups are characterized by a 6- to 9-fold repeat of three to four amino acid residues, always starting with lysine or arginine. This pattern is similar to the trans-membrane segments of several voltage-gated ion channels.150,151 The membrane lytic activity of lycotoxins, cupiennins and oxyopinins reduces ion and voltage gradients across cell membranes, promotes efflux of ions from synaptosomes, and causes hemolysis of erythrocytes.5,146,148 Recently, seven novel antimicrobial peptides were isolated from the zodariid spider Lachesana tarabaevi. These peptides were named latarcins and two of them are the shortest antimicrobial peptides known152 (Table 1).All pore-forming peptides from spiders inhibit the growth of Gram-positive (Staphylococcus aureus, Bacillus subtilis), Gram-negative (Escherichia coli, Pseudomonas aeruginosa) bacteria and fungi (Candida albicans) at micromolar concentrations. So far, all species of the family Lycosidae tested seem to contain cytolytic peptides.153 Besides the chemical structures already elucidated there is an indication of other pore forming peptides from other species. For example, some spider species that contain pore-forming effects in their venoms are from the spider families amaurobiidae (Paracoelotes luctuosus), araneidae (Neoscona arabesca and Eriophora edax), lycosidae (Oculycosa supermirabilis, Lycosa helluo, Lycosa sp. and Trochosa sp.), oxyopidae (Peucetia viridans and Oxyopes sp.), salticidae (Phylaeus chrysops, Phidippus ardens, Phidippus johnsoni and Phidippus octopunctatus), thomisidae (Xysticus acerbus), theridiidae (Latrodectus hesperus), zodariidae (Lachesana sp. and Nenilinea sp.,) and theraposidae (Aphonopelma sp.).153–155 However, several spiders seem not to contain cytolytic peptides in their venom suggesting that the pore-forming molecules have only a membrane depolarizing activity aimed at the cells and tissues of the spider's prey similar to other components of the venom such neurotoxins.
Gomesin is the only cysteine-rich, cationic cytolytic peptide, with homology to tachyplesin and polyphemusin from horseshoe crabs, found in spiders. Gomesin was isolated from the hemolymph of the theraposid Acanthoscurria gomesiana135 (Table 1). The solution structure of gomesin has been determined, and it consists of a two-strand antiparallel beta sheet connected by a non-canonical beta turn. Since gomesin contains only two cystines. A comparison between the structures of gomesin and other cysteine-rich cytolytic peptides from arthropods highlights several common features in the distribution of hydrophobic and hydrophilic residues, where one face of the beta sheet is completely hydrophilic, while the other face is hydrophobic.156 The mode of action of gomesin is suspected to be also by pore formation but by a different mechanism to that proposed for the α-helical peptides.
A peculiar characteristic in some latarcins152 is that the C-terminus is as hydrophobic as the N-terminus; so, these peptides may insert into cellular membranes from either the C- or the N-terminus.
3.8 Potential uses of spider venoms in agriculture
The higher specific toxicity of spider peptides for insect over mammalian receptors has led to the proposal of using them as biopesticides, using transgenic baculoviruses as delivery vectors.162 Investigations of spider venoms for the identification of biological entities with commercial potential have focused primarily on the agrochemical sector. The ultimate goal of these activities has been the search for chemical constituents, which interact selectively with invertebrate species to induce paralysis and or death with minimal mammalian toxicological properties. Spider peptides might be useful for defining new insecticidal targets or for pioneering novel biopesticides.163 One approach has been the creation of insect-specific baculoviruses164 that have been proposed as an attractive alternative to chemical pest control. Recombinant viruses expressing scorpion and spider peptide toxins could for example lead to reductions in crop damage.162,165,166 Moreover, only cupiennins and oxyopinins, the largest pore-forming peptides from spiders, have shown pronounced insecticidal activity; therefore, pore-forming peptides may have a synergistic action with neurotoxins thereby facilitating prey capture.5 Cupiennins and oxyopinins could have the potential to increase the resistance of some plants to insect predation. The morphology of the transgenic plant Nicotiana tabacum was unaffected when it expressed the pore-forming esculentin,167 which is similar in structure to oxyopinins. Challenging Nicotiana tabacum with bacterial or fungal phytopathogens demonstrated enhanced resistance up to the second plant generation. Moreover, these transgenic plants displayed insecticidal properties.167 Pore-forming peptides from spiders also synergistically enhanced the activity of insecticidal neurotoxins.5 Application of mixtures containing oxyopinin 1 and insecticidal peptides to insect larvae increased the lethal effect observed. This result suggests that linear amphipathic peptides and neuropeptides cooperate to kill pest larvae efficiently.5 The need for high insect specificity makes venomous arthropods such as spiders, an attractive source of new lead compounds, as they have evolved highly selective toxins to target insect prey. However, it is prudent to be careful in promoting the use of pore-forming peptides to enhance the defense of food crops. Since most of the pore-forming peptides affect cell membranes, ingestion of the peptides by human or animals may cause unpleasant gastrointestinal problems.4 Conclusions
Thus far, the venom composition of approximately 0.1% of the 39000 spider species has been studied. Table 2 summarizes both the spider species that have been investigated and some of the molecules that have been obtained from their venoms with activity towards mammalian receptors. Acylpolyamines and peptides are constantly produced in the venom of spiders resulting in a rich diversity of chemical compounds with high potency and selectivity for multiple cellular targets. Therefore, the venom of spiders is a novel and rich source of molecules for designing prototypical drugs that could regulate calcium, sodium, and/or potassium ion channels (with emphasis on drugs that block or modulate channels of central neurons). Acylpolyamines and peptides from spiders could be used to design agents, which act on ligand-gated or voltage-gated proteins in cell membranes to alter the activity of ionic channels indirectly and also to alter the efficacy of low molecular weight pharmaceutical agents that produce similar effects. Also, they are valuable tools to increase the understanding of ionic channel structure, properties and function. The ability of spider acylpolyamines and peptides to discriminate between several ion channel subtypes in mammalian tissues is of potential value for the future rational design of novel drugs. Researchers can take advantage of this pre-optimized chemical library as a diversified source of molecular probes for identifying differences in cellular targets for novel therapeutics. The development of efficacious and selective molecules from the venom of spiders capable of being employed in a recombinant expression or used to design non-peptide drugs could pave the way to new pharmaceutical products.
| Spider | Peptide/polyamine | Targeta | Spider | Peptide/polyamine | Targeta |
|---|---|---|---|---|---|
| a Na+, Ca2+, and K+, sodium, calcium and potassium ion channels; PLM, Phospholipid membranes; MS, mechano-sensitive ion channels; ASIC, acid-sensing ion channels; GLUP, glutamate uptake; GLU, glutamate receptors. References to the toxins are given throughout the text and in Table 1. | |||||
| Suborder Mygalomorphae | Suborder Araneomorphae | ||||
| Fam. Theraphosidae | Fam. Tetragnathidae | ||||
| Chilobrachys jingzhao | JZTX-I | Na+ | Nephila clavata | JSTX-3 | GLU |
| JZTX-III | Na+ | Fam. Nephilidae | |||
| Ceratogyrus cornatus | CcoTx1–3 | Na+ | Nephila madagascariencis | NPTX-594 | GLU |
| Grammostola spatulata (now G. rosea) | Hanatoxin 1–2 | K+ | Fam. Sicariidae | ||
| GsMTx-2, 4 | MS | Loxosceles intermedia | LiTx1–3 | Na+ | |
| GSTxSIA | K+, Ca2+ | Fam. Agelenidae | |||
| Phrixotrichus auratus | Phrixotoxin 1–2 | K+ | Agelenopsis aperta | ω-AgaI–IVA | Ca2+ |
| PaurTx3 | Na+ | AG-505 | GLU | ||
| Stromatopelma calceata | ScTx1 | K+ | Fam. Segestriidae | ||
| Heteroscodra maculata | HmTx1–2 | K+ | Segestria florentina | SNX-325 | Ca2+ |
| Scodra griseipes (now Stromatopelma calceata) | SGTx1 | K+ | Fam. Filistatidae | ||
| Filistata hibernalis | DW13.3 | Ca2+ | |||
| Thrixopelma pruriens | ProTx-I,II | Na+, Ca2+ | Fam. Ctenidae | ||
| Psalmopoeus cambridgei | PcTx1 | ASIC | Phoneutria nigriventer | PnTx3–6 | Ca2+ |
| Selenocosmia huwena (now Ornithoctonus huwena) | Huwentoxin-I | Ca2+ | PnTX3–4 | GLUP | |
| Huwentoxin-IV | Na+ | PhTx1–3 | K+ | ||
| Huwentoxin-X | Ca2+ | ω-PTx-IIA | Ca2+ | ||
| Hysterocrates gigas | SNX-482 | Ca2+ | PnTx1 | Na+ | |
| Acanthoscurria gomesiana | Gomesin | PLM | Cupiennus salei | Cupiennin 1–4 | PLM |
| Selenocosmia hainana | HNTxIV,V | Na+ | Fam. Lycosidae | ||
| Theraposa leblondi | TLTx1 | K+ | Lycosa carolinensis | Lycotoxin I,II | PLM |
| Fam. Hexathelidae | Lycosa singoriensis | Lycocitin 1–3 | PLM | ||
| Hadronyche versuta | δ-ACTX-Hv1 | Na+ | Fam. Oxyopidae | ||
| Atrax robustus | δ-ACTX-Ar1 | Na+ | Oxyopes kitabensis | Oxyopinin 1,2a–b | PLM |
| Macrothele gigas | Magi4–6 | Na+ | Fam. Zodariidae | ||
| MG30 | GLU | Lachesana tarabaevi | Latarcin 1–7 | PLM | |
| Missulena bradleyi | MSTX-Mb1 | Na+ | Fam. Sparassidae | ||
| Heteropoda venatoria | HpTx1–3 | K+ | |||
5 Acknowledgements
We thank H. Clement and Dr A. Alagon for providing the spider pictures. Also, we thank Dr R. Norton for the Magi5 coordinates. This work was supported in part by a grant from the Dirección General de Asuntos del Personal Académico (DGAPA-UNAM) IN226006.6 References
- N. I. Platnick, The World Spider Catalog, Version 7.0, American Museum of Natural History, New York, 2006, online at http://research.amnh.org/entomology/spiders/catalog/index.html Search PubMed.
- J. Haupt, Zoologica (Stuttgart), 2003, 154, 1–101 Search PubMed.
- E. Grishin, Eur. J. Biochem., 1999, 264, 276–280 CrossRef CAS.
- P. Escoubas, S. Diochot and G. Corzo, Biochimie, 2000, 82, 893–907 CrossRef CAS.
- G. Corzo, E. Villegas, F. Gomez-Lagunas, L. D. Possani, O. S. Belokoneva and T. Nakajima, J. Biol. Chem., 2002, 277, 23627–23637 CrossRef CAS.
- P. Escoubas, B. Sollod and G. F. King, Toxicon, 2006, 47, 650–663 CrossRef CAS.
- P. Escoubas, Toxicon, 2006, 47, 609–613 CrossRef CAS.
- P. Escoubas and L. Rash, Toxicon, 2004, 43, 555–574 CrossRef CAS.
- M. E. Adams, E. E. Herold and V. J. Venema, J. Comp. Physiol., A, 1989, 164, 333–342 CrossRef CAS.
- F. G. Fisher and H. Bohm, Liebigs Ann. Chem., 1957, 603, 232 CrossRef.
- K. Stromgaard, K. Andersen, P. Krogsgaard-Larsen and J. W. Jaroszewski, Mini Rev. Med. Chem., 2001, 1, 317–338 Search PubMed.
- E. V. Grishin, T. M. Volkova, A. S. Arsen'ev, O. S. Reshetova and V. V. Onoprienko, Bioorg. Khim., 1986, 12, 1121–1124 Search PubMed.
- T. Budd, P. Clinton, A. Dell, I. R. Duce, S. J. Johnson, D. L. Quicke, G. W. Taylor, P. N. R. Usherwood and G. Usoh, Brain Res., 1988, 448, 30–39 CrossRef CAS.
- M. Hisada, T. Fujita, H. Naoki, Y. Itagaki, H. Irie, M. Miyashita and T. Nakajima, Toxicon, 1998, 36, 1115–1125 CrossRef CAS.
- Y. Hashimoto, T. Yasuhara, Y. Endo, K. Shudo, Y. Aramaki, N. Kawai and T. Nakajima, Tetrahedron Lett., 1987, 28, 3511–3514 CrossRef CAS.
- T. Teshima, T. Wakamiya, Y. Aramaki, T. Nakajima, N. Kawai and T. Shiba, Tetrahedron Lett., 1987, 28, 3509–3510 CrossRef CAS.
- T. Shinada, Y. Nakagawa, K. Hayashi, G. Corzo, T. Nakajima and Y. Ohfune, Amino Acids, 2003, 24, 293–301 Search PubMed.
- K. Nihei, M. J. Kato, T. Yamane, M. S. Palma and K. Konno, Bioorg. Med. Chem. Lett., 2002, 12, 299–302 CrossRef CAS.
- F. Wang, S. Manku and D. G. Hall, Org. Lett., 2000, 2, 1581–1583 CrossRef CAS.
- S. T. Moe, D. L. Smith, Y. Chien, J. L. Raszkiewicz, L. D. Artman and A. L. Mueller, Pharm. Res., 1998, 15, 31–38 CrossRef CAS.
- T. Wakamiya, T. Kinoshita, Y. Hattori, Y. Yamaguchi, H. Naoki, G. Corzo and T. Nakajima, Bull. Chem. Soc. Jpn., 2004, 77, 331–340 CrossRef CAS.
- J. C. Watkins and D. E. Jane, Br. J. Pharmacol., 2006, 147(Suppl 1), S100–108 CrossRef CAS.
- P. Quintana, S. Alberi, D. Hakkoum and D. Muller, Eur. J. Neurosci., 2006, 23, 975–983 CrossRef.
- D. Giuliani, C. Giaroni, E. Zanetti, L. Canciani, P. Borroni, S. Lecchini and G. Frigo, Neurochem. Int., 2006, 48, 191–200 CrossRef CAS.
- K. R. Gogas, Curr. Opin. Pharmacol., 2006, 6, 68–74 CrossRef CAS.
- D. J. Mason, L. J. Suva, P. G. Genever, A. J. Patton, S. Steuckle, R. A. Hillam and T. M. Skerry, Bone, 1997, 20, 199–205 CrossRef CAS.
- A. J. Patton, P. G. Genever, M. A. Birch, L. J. Suva and T. M. Skerry, Bone, 1998, 22, 645–649 CrossRef CAS.
- G. L. Collingridge and R. A. Lester, Pharmacol. Rev., 1989, 41, 143–210 Search PubMed.
- N. Kawai, A. Niwa and T. Abe, Brain Res., 1982, 247, 169–171 CrossRef CAS.
- N. Kawai, A. Miwa, M. Saito, H. S. Pan-Hou and M. Yoshioka, J. Physiol. (Paris), 1984, 79, 228–231 CAS.
- N. Akaike, N. Kawai, N. I. Kiskin, E. M. Kljuchko, O. A. Krishtal and A. Y. Tsyndrenko, Neurosci. Lett., 1987, 79, 326–330 CrossRef CAS.
- M. Saito, N. Kawai, A. Miwa, H. Pan-Hou and M. Yoshioka, Brain Res., 1985, 346, 397–399 CrossRef CAS.
- Y. Aramaki, T. Yasuhara, T. Higashijima, M. Yoshioka, A. Miwa, N. Kawai and T. Nakajima, Proc. Jpn. Acad., 1986, 62, 359–562 Search PubMed.
- E. K. Michaelis, N. Galton and S. L. Earley, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 5571–5574 CrossRef CAS.
- P. N. R. Usherwood, I. R. Duce and P. Boden, J. Physiol. (Paris), 1984, 79, 241–245 CAS.
- M. E. Adams, R. L. Carney, F. E. Enderlin, E. T. Fu, M. A. Jarema, J. P. Li, C. A. Miller, D. A. Schooley, M. J. Shapiro and V. J. Venema, Biochem. Biophys. Res. Commun., 1987, 148, 678–683 CrossRef CAS.
- T. Kitaguchi and K. J. Swartz, Biochemistry, 2005, 44, 15544–15549 CrossRef CAS.
- J. W. Lin, B. Rudy and R. Llinas, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 4538–4542 CAS.
- M. Blaschke, B. U. Keller, R. Rivosecchi, M. Hollmann, S. Heinemann and A. Konnerth, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6528–6532 CrossRef CAS.
- N. Kawai, Toxin Rev., 2005, 24, 271–287 Search PubMed.
- N. Seiler, Pharmacol. Ther., 2005, 107, 99–119 CrossRef CAS.
- N. Seiler, Can. J. Physiol. Pharmacol., 1987, 65, 2024–2035 CAS.
- P. Pohjanpelto, J. Cell Biol., 1976, 68, 512–520 CrossRef CAS.
- N. Volkow, S. S. Goldman, E. S. Flamm, H. Cravioto, A. P. Wolf and J. D. Brodie, Science, 1983, 221, 673–675 CrossRef CAS.
- T. A. Ratko, C. J. Detrisac, K. V. Rao, C. F. Thomas, G. J. Kelloff and R. C. Moon, Anticancer Res., 1990, 10, 67–72 CAS.
- M. Raditsch, M. Geyer, H. R. Kalbitzer, W. Jahn, J. P. Ruppersberg and V. Witzemann, Eur. J. Biochem., 1996, 240, 416–426 CAS.
- H. Tsubokawa, K. Oguro, T. Masuzawa, T. Nakaima and N. Kawai, J. Neurophysiol., 1995, 74, 218–225 Search PubMed.
- N. M. J. Vermeulin, C. L. O’Day, H. K. Webb, M. R. Burns and D. Bergstrom, US Pat., 6646149, 2003 Search PubMed.
- H. Benveniste, M. B. Jorgensen, N. Diemer and A. J. Hansen, Acta Neurol. Scand., 1988, 78, 529–536 CrossRef CAS.
- D. W. Choi, Trends Neurosci., 1988, 11, 465–467 CrossRef CAS.
- A. Schurr, Curr. Drug Targets, 2004, 5, 603–618 Search PubMed.
- P. Calabresi, D. Centonze, L. M. Cupini, C. Costa, F. Pisani and G. Bernardi, Neurobiol. Dis., 2003, 12, 82–88 CrossRef CAS.
- K. Valentino, R. Newcomb, T. Gadbois, T. Singh, S. Bowersox, S. Bitner, A. Justice, D. Yamashiro, B. B. Hoffman and R. Ciaranello, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7894–7897 CAS.
- R. Llinas, M. Sugimori, J. W. Lin and B. Cherksey, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 1689–1693 CAS.
- J. R. Dupere, E. Moya, I. S. Blagbrough and M. M. Usowicz, Neuropharmacology, 1996, 35, 1–11 CrossRef CAS.
- B. Morrison, 3rd, A. K. Pringle, T. McManus, J. Ellard, M. Bradley, F. Signorelli, F. Iannotti and L. E. Sundstrom, Br. J. Pharmacol., 2002, 137, 1255–1268 CrossRef CAS.
- A. K. Pringle, B. Morrison, 3rd, M. Bradley, F. Iannotti and L. E. Sundstrom, Naunyn-Schmiedeberg’s Arch. Pharmacol., 2003, 368, 216–224 CrossRef CAS.
- H. Tsubokawa, K. Oguro, H. P. C. Robinson, T. Masuzawa and N. Kawai, NeuroReport., 1995, 6, 527–531 Search PubMed.
- L. S. Sorkin, T. L. Yaksh and C. M. Doom, NeuroReport., 1999, 10, 3523–3526 Search PubMed.
- L. S. Sorkin, T. L. Yaksh and C. M. Doom, Anesthesiology, 2001, 95, 965–973 CrossRef CAS.
- T. L. Jones and L. S. Sorkin, J. Pharmacol. Exp. Ther., 2004, 310, 223–229 CrossRef CAS.
- E. M. Pogatzki, J. S. Niemeier, L. S. Sorkin and T. J. Brennan, Pain, 2003, 105, 97–107 CrossRef CAS.
- J. G. Gu, C. Albuquerque, C. J. Lee and A. B. MacDermott, Nature, 1996, 381, 793–796 CrossRef CAS.
- F. Metzger, S. Wiese and M. Sendtner, J. Neurosci., 1998, 18, 1735–1742 CAS.
- J. Roy, S. Minotti, L. Dong, D. A. Figlewicz and H. D. Durham, J. Neurosci., 1998, 18, 9673–9684 CAS.
- L. Van Den Bosch and W. Robberecht, Brain Res. Bull., 2000, 53, 383–388 CrossRef CAS.
- T. Asami, H. Kagechika, Y. Hashimoto, K. Shudo, A. Miwa and N. N. T. Kawai, Biomed. Res., 1989, 10, 185–189 CAS.
- N. Kawai, Neurosci. Res., 1991, 12, 3–12 CrossRef CAS.
- Y. Sahara, H. P. C. Robinson, A. Miwa and N. Kawai, Neurosci. Res., 1991, 10, 200–210 CrossRef CAS.
- S. Kozlov and E. Grishin, Toxicon, 2005, 46, 672–686 CrossRef CAS.
- G. Ferrat and H. Darbon, Toxin Rev., 2005, 24, 359–381 Search PubMed.
- P. K. Pallaghy, K. J. Nielsen, D. J. Craik and R. S. Norton, Protein Sci., 1994, 3, 1833–1839 CrossRef CAS.
- S. Diochot, Toxin Rev., 2005, 24, 289–312 Search PubMed.
- G. F. King, H. W. Tedford and F. Maggio, J. Toxicol. Toxin Rev., 2002, 21, 359–389 Search PubMed.
- H. Takahashi, J. I. Kim, H. J. Min, K. Sato, K. J. Swartz and I. Shimada, J. Mol. Biol., 2000, 297, 771–780 CrossRef CAS.
- C. Bernard, C. Legros, G. Ferrat, U. Bischoff, A. Marquardt, O. Pongs and H. Darbon, Protein Sci., 2000, 9, 2059–2067 CAS.
- G. Corzo and P. Escoubas, Cell. Mol. Life Sci., 2003, 60, 2409–2426 CrossRef CAS.
- G. Corzo, P. Escoubas, E. Villegas, I. Karbat, D. Gordon, M. Gurevitz, T. Nakajima and N. Gilles, Biochemistry, 2005, 44, 1542–1549 CrossRef CAS.
- Y. Li-Smerin and K. J. Swartz, J. Gen. Physiol., 2000, 115, 673–684 CrossRef CAS.
- S. Y. Lee and R. MacKinnon, Nature, 2004, 430, 232–235 CrossRef CAS.
- W. L. DeLano, The PyMOL molecular graphics system, 2002, DeLano Scientific, San Carlos, CA, USA, http://pymol.sourceforge.net/ Search PubMed; H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 Search PubMed , http://www.rcsb.org/pdb/.
- C. C. Shieh, M. Coghlan, J. P. Sullivan and M. Gopalakrishnan, Pharmacol. Rev., 2000, 52, 557–594 Search PubMed.
- G. Yellen, Nature, 2002, 419, 35–42 CrossRef CAS.
- K. J. Swartz and R. MacKinnon, Neuron, 1995, 15, 941–949 CrossRef CAS.
- M. C. Sanguinetti, J. H. Johnson, L. G. Hammerland, P. R. Kelbaugh, R. A. Volkmann, N. A. Saccomano and A. L. Mueller, Mol. Pharmacol., 1997, 51, 491–498 CAS.
- Z. Kassiri, C. Zobel, T. T. Nguyen, J. D. Molkentin and P. H. Backx, Circ. Res., 2002, 90, 578–585 Search PubMed.
- S. Diochot, M. D. Drici, D. Moinier, M. Fink and M. Lazdunski, Br. J. Pharmacol., 1999, 126, 251–263 CrossRef CAS.
- S. W. Yeola and D. J. Snyders, Cardiovasc. Res., 1997, 33, 540–547 CrossRef CAS.
- P. Escoubas, G. Corzo, B. J. Whiteley, M. L. Celerier and T. Nakajima, Rapid Commun. Mass Spectrom., 2002, 16, 403–413 CrossRef CAS.
- J. Ebbinghaus, C. Legros, A. Nolting, C. Guette, M. L. Celerier, O. Pongs and R. Bahring, Toxicon, 2004, 43, 923–932 CrossRef CAS.
- M. E. Adams and B. M. Olivera, Trends Neurosci., 1994, 17, 151–155 CrossRef CAS.
- J. Striessnig, M. Grabner, J. Mitterdorfer, S. Hering, M. J. Sinnegger and H. Glossmann, Trends Pharmacol. Sci., 1998, 19, 108–115 CrossRef CAS.
- J. G. McGivern, Drug Discovery Today, 2006, 11, 245–253 CrossRef CAS.
- R. Newcomb, B. Szoke, A. Palma, G. Wang, X. Chen, W. Hopkins, R. Cong, J. Miller, L. Urge, K. Tarczy-Hornoch, J. A. Loo, D. J. Dooley, L. Nadasdi, R. W. Tsien, J. Lemos and G. Miljanich, Biochemistry, 1998, 37, 15353–15362 CrossRef CAS.
- E. Bourinet, S. C. Stotz, R. L. Spaetgens, G. Dayanithi, J. Lemos, J. Nargeot and G. W. Zamponi, Biophys. J., 2001, 81, 79–88 CrossRef CAS.
- R. A. Lampe, P. A. Defeo, M. D. Davison, J. Young, J. L. Herman, R. C. Spreen, M. B. Horn, T. J. Mangano and R. A. Keith, Mol. Pharmacol., 1993, 44, 451–460 CAS.
- K. Takeuchi, E. Park, C. Lee, J. Kim, H. Takahashi, K. Swartz and I. Shimada, J. Mol. Biol., 2002, 321, 517–526 CrossRef CAS.
- H. Saegusa, T. Kurihara, S. Zong, A. Kazuno, Y. Matsuda, T. Nonaka, W. Han, H. Toriyama and T. Tanabe, EMBO J., 2001, 20, 2349–2356 CrossRef CAS.
- W. Rajendra, A. Armugam and K. Jeyaseelan, Toxicon, 2004, 44, 1–17 CrossRef CAS.
- S. V. Reddy and T. L. Yaksh, Neuropharmacology, 1980, 19, 181–185 CrossRef CAS.
- R. Newcomb, A. Palma, J. Fox, S. Gaur, K. Lau, D. Chung, R. Cong, J. R. Bell, B. Horne, L. Nadasdi and J. Ramachandran, Biochemistry, 1995, 34, 8341–8347 CrossRef CAS.
- P. A. Zhou, X. J. Xie, M. Li, D. M. Yang, Z. P. Xie, X. Zong and S. P. Liang, Toxicon, 1997, 35, 39–45 CrossRef CAS.
- K. Peng, X. D. Chen and S. P. Liang, Toxicon, 2001, 39, 491–498 CrossRef CAS.
- Z. Liu, J. Dai, L. Dai, M. Deng, Z. Hu, W. Hu and S. Liang, J. Biol. Chem., 2006, 281, 8628–8635 CrossRef CAS.
- K. G. Sutton, C. Siok, A. Stea, G. W. Zamponi, S. D. Heck, R. A. Volkmann, M. K. Ahlijanian and T. P. Snutch, Mol. Pharmacol., 1998, 54, 407–418 CAS.
- A. C. Cassola, H. Jaffe, H. M. Fales, S. Castro-Afeche, F. Magnoli and J. Cipolla-Neto, Pfluegers Arch., 1998, 436, 545–552 CrossRef CAS.
- L. B. Vieira, C. Kushmerick, M. E. Hildebrand, E. Garcia, A. Stea, M. N. Cordeiro, M. Richardson, M. V. Gomez and T. P. Snutch, J. Pharmacol. Exp. Ther., 2005, 314, 1370–1377 CrossRef CAS.
- R. G. dos Santos, C. Van Renterghem, N. Martin-Moutot, P. Mansuelle, M. N. Cordeiro, C. R. Diniz, Y. Mori, M. E. De Lima and M. Seagar, J. Biol. Chem., 2002, 277, 13856–13862 CrossRef CAS.
- N. Martin-Moutot, L. Haro, R. G. Santos, Y. Mori and M. Seagar, Neurobiol. Dis., 2006, 22, 57–63 CrossRef CAS.
- A. C. Pinheiro, R. S. Gomez, A. R. Massensini, M. N. Cordeiro, M. Richardson, M. A. Romano-Silva, M. A. Prado, L. D. Marco and M. V. Gomez, Neurochem. Int., 2006, 49, 543–547 CrossRef CAS.
- G. F. Lopreato, Y. Lu, A. Southwell, N. S. Atkinson, D. M. Hillis, T. P. Wilcox and H. H. Zakon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7588–7592 CrossRef CAS.
- S. G. Waxman, T. R. Cummins, J. A. Black and S. Dib-Hajj, Novartis Found. Symp., 2002, 241, 34–51 Search PubMed ; discussion 51–60.
- S. D. Novakovic, R. M. Eglen and J. C. Hunter, Trends Neurosci., 2001, 24, 473–478 CrossRef CAS.
- S. Cestele and W. A. Catterall, Biochimie, 2000, 82, 883–892 CrossRef CAS.
- Y. Xiao, J. Tang, Y. Yang, M. Wang, W. Hu, J. Xie, X. Zeng and S. Liang, J. Biol. Chem., 2004, 279, 26220–26226 CrossRef CAS.
- Y. Xiao, J. Tang, W. Hu, J. Xie, C. Maertens, J. Tytgat and S. Liang, J. Biol. Chem., 2005, 280, 12069–12076 CAS.
- R. E. Middleton, V. A. Warren, R. L. Kraus, J. C. Hwang, C. J. Liu, G. Dai, R. M. Brochu, M. G. Kohler, Y. D. Gao, V. M. Garsky, M. J. Bogusky, J. T. Mehl, C. J. Cohen and M. M. Smith, Biochemistry, 2002, 41, 14734–14747 CrossRef CAS.
- D. D. Sheumack, R. Claassens, N. M. Whiteley and M. E. Howden, FEBS Lett., 1985, 181, 154–156 CrossRef CAS.
- M. R. Brown, D. D. Sheumack, M. I. Tyler and M. E. Howden, Biochem. J., 1988, 250, 401–405 CAS.
- W. C. Hodgson, Clin. Exp. Pharmacol. Physiol., 1997, 24, 10–17 CrossRef CAS.
- G. M. Nicholson, M. Willow, M. E. Howden and T. Narahashi, Pfluegers Arch., 1994, 428, 400–409 CrossRef CAS.
- M. J. Little, H. Wilson, C. Zappia, S. Cestele, M. I. Tyler, M. F. Martin-Eauclaire, D. Gordon and G. M. Nicholson, FEBS Lett., 1998, 439, 246–252 CrossRef CAS.
- J. I. Fletcher, B. E. Chapman, J. P. Mackay, M. E. Howden and G. F. King, Structure, 1997, 5, 1525–1535 CrossRef CAS.
- P. K. Pallaghy, D. Alewood, P. F. Alewood and R. S. Norton, FEBS Lett., 1997, 419, 191–196 CrossRef CAS.
- M. D. Temple, M. G. Hinds, D. D. Sheumack, M. E. Howden and R. S. Norton, Toxicon, 1999, 37, 485–506 CrossRef CAS.
- M. N. Cordeiro, C. R. Diniz, A. C. Valentim, V. R. von Eickstedt, J. Gilroy and M. Richardson, FEBS Lett., 1992, 310, 153–156 CrossRef CAS.
- N. Martin-Moutot, P. Mansuelle, G. Alcaraz, R. G. Dos Santos, M. N. Cordeiro, M. E. De Lima, M. Seagar and C. Van Renterghem, Mol. Pharmacol., 2006, 69, 1931–1937 CrossRef CAS.
- G. Corzo, N. Gilles, S. Honoo, E. Villegas, L. Dai, T. Nakajima and J. Haupt, FEBS Lett., 2003, 547, 43–50 CrossRef CAS.
- K. Peng, Q. Shu, Z. Liu and S. Liang, J. Biol. Chem., 2002, 277, 47564–47571 CrossRef CAS.
- D. Li, Y. Xiao, X. Xu, X. Xiong, S. Lu, Z. Liu, Q. Zhu, M. Wang, X. Gu and S. Liang, J. Biol. Chem., 2004, 279, 37734–37740 CrossRef CAS.
- F. Bosmans, L. Rash, S. Zhu, S. Diochot, M. Lazdunski, P. Escoubas and J. Tytgat, Mol. Pharmacol., 2006, 69, 419–429 CAS.
- K. Williams, Biochem. J., 1997, 325, 289–297 CAS.
- H. J. Reis, M. A. Prado, E. Kalapothakis, M. N. Cordeiro, C. R. Diniz, L. A. De Marco, M. V. Gomez and M. A. Romano-Silva, Biochem. J., 1999, 343, 413–418 CrossRef CAS.
- R. A. Mafra, S. G. Figueiredo, C. R. Diniz, M. N. Cordeiro, J. D. Cruz and M. E. De Lima, Brain Res., 1999, 831, 297–300 CrossRef CAS.
- H. J. Reis, M. V. Gomez, E. Kalapothakis, C. R. Diniz, M. N. Cordeiro, M. A. Prado and M. A. Romano-Silva, NeuroReport, 2000, 11, 2191–2194 Search PubMed.
- R. Waldmann and M. Lazdunski, Curr. Opin. Neurobiol., 1998, 8, 418–424 CrossRef CAS.
- E. W. McCleskey and M. S. Gold, Annu. Rev. Physiol., 1999, 61, 835–856 CrossRef CAS.
- P. Escoubas, J. R. De Weille, A. Lecoq, S. Diochot, R. Waldmann, G. Champigny, D. Moinier, A. Menez and M. Lazdunski, J. Biol. Chem., 2000, 275, 25116–25121 CrossRef CAS.
- B. Martinac, Cell Physiol. Biochem., 2001, 11, 61–76 Search PubMed.
- T. M. Suchyna, J. H. Johnson, K. Hamer, J. F. Leykam, D. A. Gage, H. F. Clemo, C. M. Baumgarten and F. Sachs, J. Gen. Physiol., 2000, 115, 583–598 CrossRef CAS.
- F. Bode, F. Sachs and M. R. Franz, Nature, 2001, 409, 35–36 CrossRef CAS.
- F. Sachs and R. A. Lampe, US Pat., 5968838, 1998 Search PubMed.
- E. Bourinet and G. W. Zamponi, Curr. Top. Med. Chem., 2005, 5, 539–546 CrossRef CAS.
- D. Andreu and L. Rivas, Biopolymers, 1998, 47, 415–433 CrossRef CAS.
- K. Xu, Y. Ji and X. Qu, Acta Zool. Sinica, 1989, 35, 300–305 Search PubMed.
- L. Yan and M. E. Adams, J. Biol. Chem., 1998, 273, 2059–2066 CrossRef CAS.
- S. Haeberli, L. Kuhn-Nentwig, J. Schaller and W. Nentwig, Toxicon, 2000, 38, 373–380 CrossRef CAS.
- L. Kuhn-Nentwig, M. Dathe, A. Walz, J. Schaller and W. Nentwig, FEBS Lett., 2002, 527, 193–198 CrossRef CAS.
- B. A. Budnik, J. V. Olsen, T. A. Egorov, V. E. Anisimova, T. G. Galkina, A. K. Musolyamov, E. V. Grishin and R. A. Zubarev, J. Mass Spectrom., 2004, 39, 193–201 CrossRef CAS.
- O. S. Belokoneva, E. Villegas, G. Corzo, L. Dai and T. Nakajima, Biochim. Biophys. Acta, 2003, 1617, 22–30 CAS.
- L. Kuhn-Nentwig, Cell. Mol. Life Sci., 2003, 60, 2651–2668 CrossRef CAS.
- S. A. Kozlov, A. A. Vassilevski, A. V. Feofanov, A. Y. Surovoy, D. V. Karpunin and E. V. Grishin, J. Biol. Chem., 2006, 281, 20983–20992 CrossRef CAS.
- E. Villegas and G. Corzo, Toxin Rev., 2005, 24, 345–357 Search PubMed.
- E. Cohen and G. B. Quistad, Toxicon, 1998, 36, 353–358 CrossRef CAS.
- I. Kazakov, A. B. Nenilin, P. B. Usmanov and B. A. Tashmukhamedov, Nauchn. Dokl. Vyssh. Shk. Biol. Nauk., 1985, 2, 30–33 Search PubMed.
- N. Mandard, P. Bulet, A. Caille, S. Daffre and F. Vovelle, Eur. J. Biochem., 2002, 269, 1190–1198 CrossRef CAS.
- O. Cirioni, A. Giacometti, R. Ghiselli, G. Dell'Acqua, Y. Gov, W. Kamysz, J. Lukasiak, F. Mocchegiani, F. Orlando, G. D'Amato, N. Balaban, V. Saba and G. Scalise, Circulation, 2003, 108, 767–771 CrossRef CAS.
- Genaera Corporation, http://www.genaera.com/.
- S. P. Concannon, T. D. Crowe, J. J. Abercrombie, C. M. Molina, P. Hou, D. K. Sukumaran, P. A. Raj and K. P. Leung, J. Med. Microbiol., 2003, 52, 1083–1093 CrossRef CAS.
- H. Ulvatne, Am. J. Clin. Dermatol., 2003, 4, 591–595 Search PubMed.
- E. Andres and J. L. Dimarcq, Rev. Med. Interne, 2004, 25, 629–635 CrossRef CAS.
- G. G. Prikhod'ko, M. Robson, J. W. Warmke, C. J. Cohen, M. M. Smith, P. Wang, V. Warren, G. Kaczorowski, L. H. T. Van Der Ploeg and L. K. Miller, Biol. Control, 1996, 7, 236–244 CrossRef.
- G. Corzo, P. Escoubas, M. Stankiewicz, M. Pelhate, C. P. Kristensen and T. Nakajima, Eur. J. Biochem., 2000, 267, 5783–5795 CrossRef CAS.
- K. Hoover, R. Herrmann, H. Moskowitz, B. C. Bonning, S. S. Duffey, B. F. McCutchen and B. D. Hammock, Pestic. Outlook, 1996, 7, 21–27 Search PubMed.
- B. F. McCutchen, P. V. Choudary, R. Crenshaw, D. Maddox, S. G. Kamita, N. Palekar, S. Volrath, E. Fowler, B. D. Hammock and S. Maeda, Biotechnology, 1991, 9, 848–852 Search PubMed.
- P. R. Hughes, H. A. Wood, J. P. Breen, S. F. Simpson, A. J. Duggan and J. A. Dybas, J. Invertebr. Pathol., 1997, 69, 112–118 CrossRef.
- D. Ponti, M. L. Mangoni, G. Mignogna, M. Simmaco and D. Barra, Biochem. J., 2003, 370, 121–127 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |