Quinoline, quinazoline and acridone alkaloids
Joseph P.
Michael
*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Wits 2050, South Africa. E-mail: jmichael@chem.wits.ac.za
First published on 3rd January 2007
Abstract
Covering: July 2004 to June 2005. Previous review: Nat. Prod. Rep., 2005, 22, 627
This review covers the isolation, structure determination, synthesis and biological activity of quinoline, quinazoline and acridone alkaloids from plant, microbial and animal sources; 134 references are cited.
1 Quinoline alkaloids
1.1 Quinoline alkaloids from rutaceous plants: occurrence, characterisation and biological activity
The secondary metabolites of Ruta chalepensis, among them a number of furo[2,3-b]quinoline alkaloids , have been itemised in a review in which the plant's medicinal uses are also described.1 A review with a similar focus deals with Taiwanese members of the genus Zanthoxylum (= Fagara).2 A survey of 288 natural products and 101 plant species with documented antileishmanial activity includes several quinoline alkaloids and rutaceous plants.3Table 1 contains a list of new quinoline and quinolinone alkaloids isolated from plants belonging to the Rutaceae, as well as novel sources of known alkaloids .4–21 Most of the new compounds bear prenyl or modified prenyl substituents on the quinoline nucleus.
| Speciesa | Alkaloid b | Ref. |
|---|---|---|
| a All plant species are members of the Rutaceae. b Only new alkaloids and new records for a given species are listed in the Table; in many cases, the presence of several previously isolated alkaloids was confirmed. Structures of known alkaloids , if not specifically numbered, may be found in previous reviews in this series. c New alkaloids . | ||
| Almeidea rubra | rel-(7R,8R)-7-Acetoxy-8-[(E)-3-hydroxy-3-methylbut-1-enyl]-4,8-dimethoxy-5,6,7,8-tetrahydrofuro[2,3-b]quinoline c 14 | 4 |
| Isodutadrupine | ||
| Isokokusagine c 17 | ||
| Isoskimmianine c 18 | ||
| Kokusagine 8 | ||
| Casimiroa pubescens | Casimiroine | 5 |
| Dictamnus albus | Haplopine | 6 |
| Robustine | ||
| Esenbeckia almawillia | N-Methyl-3,3-diprenylquinoline-2,4-dione 1 | 7,8 |
| Evodia lepta | Dictamnine 74 | 9 |
| Evolitrine 22 | ||
| Glycosmis arborea | Acutifolin | 10 |
| Glycocitlone C | ||
| Isodictamnine 19 | ||
| Iso-γ-fagarine 20 | ||
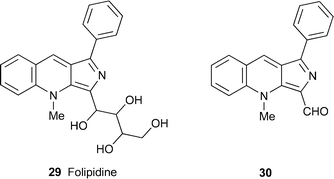
| Haplophyllum foliosum and H. pedicellatum | (–)-Folipidine c 29 | 11 |
| Orixa japonica | Edulinine | 12 |
| γ-Fagarine 21 | 13 | |
| Isoplatydesmine | 12 | |
| Lunidonine | ||
| Orixalone A c 4 | 13 | |
| Orixalone B c 5 | ||
| Orixalone C c 6 | ||
| (+)-Orixalone D c 7 | ||
| Preskimmianine | 12 | |
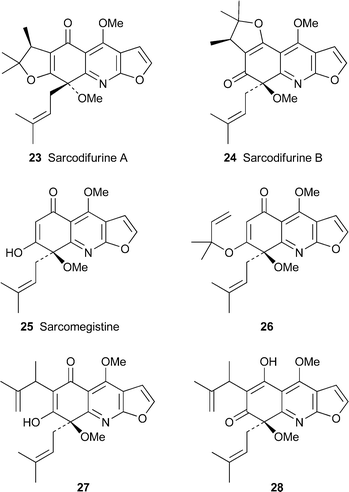
| Sarcomelicope follicularis | (–)-Sarcodifurine A c 23 | 14 |
| (+)-Sarcodifurine B c 24 | ||
| Skimmia laureola | (–)-Acetylribalinine c 2 | 15 |
| (+)-Ribaliprenylene c 3 | ||
| Teclea natalensis | Dictamnine | 16 |
| 4,7-Dimethoxy-8-prenyloxyfuro[2,3-b]quinoline 13 | ||
| Flindersiamine | ||
| (+)-Tecleanatalensine A (= tecleoxine) 12 | ||
| Tecleanatalensine B c 11 | ||
| Zanthoxylum beecheyanum (= Z. arnottianum) | Flindersine 63 | 17 |
| N-Methylatanine | ||
| N-Methylflindersine 62 | ||
| Toddaquinoline | ||
| Zanthobungeanine | ||
| Zanthodioline | ||
| Zanthoxylum budrunga (= Z. rhetsa) | γ-Fagarine | 18 |
| N-Methylflindersine | ||
| Zanthoxylum ekmanii | Dictamnine | 19 |
| Skimmianine 16 | ||
| Zanthoxylum integrifoliolum | γ-Fagarine | 20 |
| Zanthoxylum nitidum | Haplopine | 21 |
Essential oils obtained by steam distillation of the trunk bark, wood and roots of Amazonian Esenbeckia almawillia contained, as expected, a range of monoterpenes and sesquiterpenes; but an unexpected component, isolated in substantial amounts, was the known alkaloid N-methyl-3,3-diprenylquinoline-2,4-dione1—apparently the first time that such a compound has been found in essential oils.7 Its 1H and 13C NMR spectra were fully assigned, as were the spectra of derivatives in which either or both of the 4-oxo and 3-prenyl substituents were reduced.8 In the new alkaloids acetylribalinine2 and ribaliprenylene3, isolated from the leaves of the Pakistani plant Skimmia laureola, a 3-prenyl substituent has participated in cyclisation with the 2-hydroxy tautomer of a quinoline-2,4-dione to give a pyrano[2,3-b]quinolin-4-one ring system.15 Full spectroscopic details were reported for both alkaloids ; but their absolute configurations, although presented as shown, seem not to have been unambiguously determined.
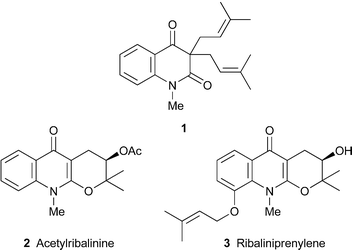
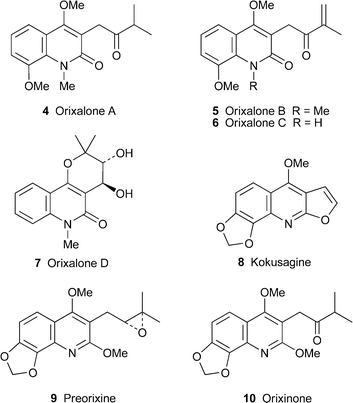
An extract from the stems of Orixa japonica, a prolific source of prenylated and related quinoline alkaloids , has yielded four additional members of this group.12 The hemiterpenoid substituent at C-3 in orixalones A, B and C, 4–6, has undergone a characteristic modification by oxidation to a ketone, while in (+)-orixalone D7 it has become part of a pyrano[3,2-c]quinolin-2-one system. The trans configuration of the diol in 7 was inferred from the large coupling constant (J 7.7 Hz) of the carbinol protons and the absence of a nuclear Overhauser effect (NOE) between them. However, the absolute configuration of the alkaloid was not determined. Orixalone A strongly inhibited the production of nitric oxide (NO) in stimulated murine macrophage RAW 264.7 cells at micromolar concentrations, whereas the known alkaloids kokusagine8, preorixine9 and orixinone10, isolated from O. japonica in the same study, had no effect at comparable concentrations. Since 4 was not significantly cytotoxic to the cells at the effective concentrations for NO inhibition, it may have clinical potential as a cancer-preventive agent by suppressing the excessive synthesis of NO.
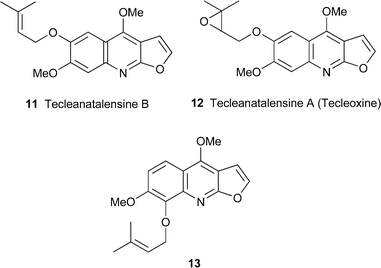
Although furo[2,3-b]quinoline alkaloids are not unusual in the genus Teclea, they have not previously been encountered in the southern and eastern African shrub Teclea natalensis, which has hitherto yielded only acridone alkaloids . Among the five furo[2,3-b]quinolines recently extracted from leaves of the plant were the new compound tecleanatalensine B11 and the known but uncommon alkaloids (+)-12 and 13, all of which were thoroughly characterised by 1H and 13C NMR spectroscopy and other standard spectroscopic techniques.16 Compound 12, given the name tecleanatalensine A in this report, was also claimed as a new alkaloid ; however, it had been reported a year earlier22 (cf.ref. 23a) as a metabolite of Teclea nobilis and given the name tecleoxine, which should thus take precedence. Its absolute configuration was not determined.
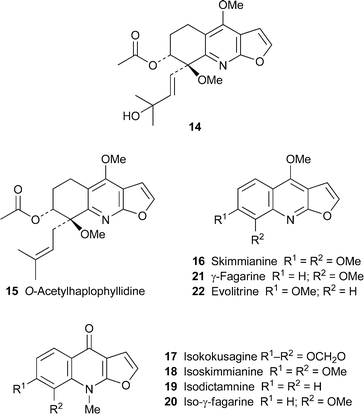
Screening of leaf extracts from the Brazilian plant Almeidea rubra for trypanocidal activity resulted in the isolation of 14, a new representative of the rare 5,6,7,8-tetrahydrofuro[2,3-b]quinoline alkaloids .4 Spectroscopic characterisation included HSQC and HMBC correlations to establish the nature and position of the substituents. While the alkaloid's absolute configuration was not determined, the trans arrangement of the 7-acetoxy and 8-methoxy groups was inferred from the similarity of its chemical shifts with those of 7-O-acetylhaplophyllidine15, a previously characterised Almeideametabolite . Also isolated in the present study were kokusagine8, skimmianine16 and their N-methyl isomers isokokusagine17 and isoskimmianine18. Although no mention was made of the fact, this appears to be the first time that 17 and 18 have been found as natural products; they have previously been prepared by the well-studied ‘iso rearrangement’ of 4-methoxyfuro[2,3-b]quinolines, which entails treatment of alkaloids such as 8 and 16 with iodomethane24 (cf.ref. 23b). Alkaloids 8, 14 and 16 showed moderate in vitro trypanocidal activity against the trypomastigote forms of the parasite Trypanosoma cruzi, which is responsible for American trypanosomiasis and Chagas' disease, but the iso-alkaloids were too unstable to be tested. Another two naturally occurring iso-alkaloids, isodictamnine19 and iso-γ-fagarine20, were isolated from stems of Glycosmis arborea together with γ-fagarine21 and several other quinoline, acridone and carbazole alkaloids .10 These three alkaloids showed inhibitory activity towards the induction of Epstein–Barr virus early antigen (EBV-EA) in Raji cells by the tumour promoter 12-O-tetradecanoylphorbol 13-acetate, and are thus potentially useful as chemoprotective agents in chemical carcinogenesis. γ-Fagarine, evolitrine22 and six related furoquinolines also inhibited human phosphodiesterase 5 (hPDE5A), a hydrolytic enzyme that regulates the intracellular levels of cGMP and influences vascular smooth muscle tone;6 while skimmianine and γ-fagarine showed useful cytotoxicity towards the murine leukemia P-388 cell line (ED50 <4 µg ml–1).20
The genus Sarcomelicope continues to yield new metabolites with surprising structures. Sarcodifurines A23 and B24, extracted from the New Caledonian species S. follicularis,14 also turned out to be 8-methoxy-8-prenyl derivatives of the furo[2,3-b]quinoline class, but with the additional fusion of an extraordinary 2,2,3-trimethyldihydrofuran ring. The structures, supported by spectroscopic data, were also confirmed by X-ray diffraction studies, which revealed the relative stereochemistries shown in the structural diagrams. A plausible biogenesis of these alkaloids is via another Sarcomelicopealkaloid , sarcomegistine25, which is thought to undergo O-alkylation at O-7 (or perhaps O-5) with a prenyl unit to give a 1,1-dimethylallyl ether such as 26. An ‘abnormal’ Claisen rearrangement to 27 or its alternative enol tautomer 28 followed by cyclisation would then give the observed alkaloids . The new compounds showed no significant biological activity when tested against a range of human pathogenic strains of yeasts.
Even more bizarre is the new alkaloid (–)-folipidine29, isolated from Haplophyllum foliosum and H. pedicellatum.11 This compound is quite unlike any other rutaceous quinoline alkaloid in possessing an unprecedented pyrrolo[3,4-b]quinoline nucleus and a highly hydroxylated aliphatic substituent. The gross structure was deduced from a comprehensive suite of spectroscopic techniques, among them a range of two-dimensional NMR experiments from which the connectivity of the unusual skeleton was deduced. In addition, all four alcohol protons gave distinct signals when the 1H NMR spectrum was recorded in deuteriated dimethyl sulfoxide; the proton of the primary alcohol appeared as a triplet, those of the three secondary alcohols were doublets, and all underwent exchange with deuterium oxide. Chemical transformations included the formation of a tetraacetate, and the production of aldehyde 30, named folipidinal, upon oxidation with sodium periodate. However, the relative and absolute configurations in the tetrahydroxybutyl substituent were not established. Folipidine showed no antitumour activity towards HeLa and HCT 116 cancer cells, although the plants' total alkaloid fraction (which also contains skimmianine and dubinidine) was active.
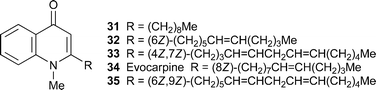
The preparative isolation of several known N-methylquinolin-4-one alkaloids bearing saturated or unsaturated hydrocarbon chains at C-2 from extracts of the Chinese medicinal herb Evodia rutaecarpa by high-speed countercurrent chromatography has been reported.25HPLC -electrospray mass spectrometry has been used for the simultaneous analysis of protoberberine, indoloquinazoline and quinolinone alkaloids in oriental Coptis-Evodia herbal preparations.26 The five familiar alkaloids 31–35 isolated after bioassay -guided fractionation from E. rutaecarpa and characterised by NMR spectroscopy were found to inhibit leukotriene biosynthesis in a bioassay using human polymorphonuclear granulocytes (IC50 12.1, 10.0, 10.1, 14.6 and 12.3 µM, respectively).27
1.2 Quinoline alkaloids from rutaceous plants: synthesis
2-Phenylquinoline 36, one of the simplest Galipeaalkaloids , continues to turn up among the products prepared during the course of various methodological studies (Scheme 1). The cross-coupling of phenylmagnesium bromide with 2-chloroquinoline37 gave the alkaloid in 74% yield when the reaction was catalysed by cobalt(II) acetylacetonate in dioxane at 50 °C.28 The product was obtained in 94% yield by heating acetophenone with 2-aminobenzyl alcohol38 in dioxane in the presence of the oxidation catalyst [Ru(DMSO)4]Cl2 (2 mol%) and benzophenone as a hydrogen scavenger .29 This oxidative cyclisation has also been accomplished in 89% yield with molecular oxygen and ruthenium-grafted hydrotalcite, a multifunctional heterogeneous catalyst , in toluene at 100 °C.30 In a reductive procedure, reaction of the malononitrile-2-nitrochalcone adduct 39 with titanium tetrachloride and zinc in boiling tetrahydrofuran afforded 2-phenylquinoline in 78% yield.31 Finally, the three-component condensation of aniline with benzaldehyde and phenyl vinyl sulfide to give 2-phenyl-4-phenylthio-1,2,3,4-tetrahydroquinoline40 was effected with ytterbium(III) triflate in dichloromethane at ambient temperature.32 Oxidation of 40 with solid-supported periodate and thermolysis of the intermediate sulfoxide then gave the alkaloid in 23% overall yield. Numerous unnatural analogues of 2-phenylquinoline were also made by these five versatile procedures, which should thus be suitable for preparing other simple 2-arylquinoline alkaloids . Of related interest is the oxidation of 2-aryl-2,3-dihydroquinolin-4(1H)-ones such as 41 with ferric chloride hexahydrate in boiling methanol to give 2-aryl-4-methoxyquinolines, among them the natural product 2-phenyl-4-methoxyquinoline42, which was obtained in 78% yield.33![Reagents and conditions: i, PhMgBr (3 equiv.), Co(acac)2 (0.1 equiv.), dioxane, 50 °C, 30 min; ii, [Ru(DMSO)4]Cl2 (2 mol%), Ph2CO (1 equiv.), KOH (1 equiv.), dioxane, 80 °C; iii, Ru-grafted hydrotalcite–NEt3 (3 mol% in Ru), O2 (1 atm), PhMe, 100 °C, 20 h; iv, TiCl4 (4 equiv.), Zn (8 equiv.), THF, reflux, 3 h, then 39, rt, 4 h; v, Yb(OTf)3 (0.05 equiv.), MgSO4, CH2Cl2, rt, 18 h; vi, IO4– on Amberlyst, dioxane–H2O, rt, 4 h, then 80 °C, 18 h; vii, FeCl3·6H2O (2.5 equiv.), MeOH, reflux 2.5 h.](/image/article/2007/NP/b509528j/b509528j-s1.gif) | ||
| Scheme 1 Reagents and conditions: i, PhMgBr (3 equiv.), Co(acac)2 (0.1 equiv.), dioxane, 50 °C, 30 min; ii, [Ru(DMSO)4]Cl2 (2 mol%), Ph2CO (1 equiv.), KOH (1 equiv.), dioxane, 80 °C; iii, Ru-grafted hydrotalcite–NEt3 (3 mol% in Ru), O2 (1 atm), PhMe, 100 °C, 20 h; iv, TiCl4 (4 equiv.), Zn (8 equiv.), THF, reflux, 3 h, then 39, rt, 4 h; v, Yb(OTf)3 (0.05 equiv.), MgSO4, CH2Cl2, rt, 18 h; vi, IO4– on Amberlyst, dioxane–H2O, rt, 4 h, then 80 °C, 18 h; vii, FeCl3·6H2O (2.5 equiv.), MeOH, reflux 2.5 h. | ||
Two recent syntheses of 43, the unnatural (S)-(+)-enantiomer of the Galipeaalkaloid angustureine, are shown in Scheme 2. The key step in the approach of Ma and co-workers was the N-phenylation of the optically active β-amino ester 44 with iodobenzene in the presence of copper(I) iodide to give the β-amino acid 45.34 The remaining steps in their short and efficient route entailed N-methylation of 45, formation of the second ring by an intramolecular Friedel–Crafts reaction on intermediate 46 and final defunctionalisation of the 2,3-dihydroquinolin-4(1H)-one 47 with hydrogen over palladium on carbon. In the approach by Arisawa and co-workers,35 Mitsunobu reaction between the p-toluenesulfonamide 48 and (R)-alcohol 49 produced the metathesis precursor (S)-50, which was smoothly converted into the dihydroquinoline 51 in 92% yield when treated with the second-generation Grubbs catalyst. Catalytic hydrogenation followed by cleavage of the toluenesulfonamide yielded (S)-(–)-52, an unspecified enantiomer of which is itself a known natural product from G. officinalis. Treatment with iodomethane completed the synthesis of (S)-(+)-angustureine43. In two unrelated articles, it was shown that catalytic hydrogenation of 2-pentylquinoline53 over the homogeneous iridium catalyst [Ir(COD)Cl]2 yielded the (R)-enantiomer of 43 in high enantiomeric excess (ee) when the reaction was performed in the presence of either the 3,3′-bipyridyl-bis(phosphine) ligand (R)-5436 or the (S,SP)-ferrocenyloxazoline-phosphine ligand 55.37
![Reagents and conditions: i, PhI, CuI, K2CO3, DMF–H2O (20 : 1), 110 °C, 48 h; ii, PhI, CuI, K2CO3, l-proline, DMF, 80 °C, then aq. K2CO3; iii, MeI, Ag2O, DMF, rt, 3 h, then 60 °C, overnight; iv, NaOH, MeOH–H2O (1 : 1), 60 °C, 2 h; v, SOCl2, CH2Cl2, rt, 2 h; vi, AlCl3, CH2Cl2, rt, 24 h; vii, H2 (101 kPa), 10% Pd/C, AcOH, rt, overnight; viii, DEAD, PPh3, THF, 0 °C, then rt, 2 h; ix, 50 (0.01 M in CH2Cl2), Grubbs II catalyst (5 mol%), 50 °C, 1 h; x, H2 (1 atm), PtO2, MeOH, rt, 12 h; xi, Na-anthracene, (CH2OMe)2, –65 °C, 10 min, then H2O; xii, MeI, K2CO3, THF, reflux, 10 h; xiii, H2 (700 psi), [Ir(COD)Cl]2 (0.005 mol), ligand 54 (0.011 mol), I2 (0.05 mol), THF, rt, 20 h; xiv, H2 (600 psi), [Ir(COD)Cl]2 (0.005 mol), ligand 55 (0.011 mol), I2 (0.05 mol), PhMe, rt, 12–16 h.](/image/article/2007/NP/b509528j/b509528j-s2.gif) | ||
| Scheme 2 Reagents and conditions: i, PhI, CuI, K2CO3, DMF–H2O (20 : 1), 110 °C, 48 h; ii, PhI, CuI, K2CO3, L-proline, DMF, 80 °C, then aq. K2CO3; iii, MeI, Ag2O, DMF, rt, 3 h, then 60 °C, overnight; iv, NaOH, MeOH–H2O (1 : 1), 60 °C, 2 h; v, SOCl2, CH2Cl2, rt, 2 h; vi, AlCl3, CH2Cl2, rt, 24 h; vii, H2 (101 kPa), 10% Pd/C, AcOH, rt, overnight; viii, DEAD, PPh3, THF, 0 °C, then rt, 2 h; ix, 50 (0.01 M in CH2Cl2), Grubbs II catalyst (5 mol%), 50 °C, 1 h; x, H2 (1 atm), PtO2, MeOH, rt, 12 h; xi, Na-anthracene, (CH2OMe)2, –65 °C, 10 min, then H2O; xii, MeI, K2CO3, THF, reflux, 10 h; xiii, H2 (700 psi), [Ir(COD)Cl]2 (0.005 mol), ligand 54 (0.011 mol), I2 (0.05 mol), THF, rt, 20 h; xiv, H2 (600 psi), [Ir(COD)Cl]2 (0.005 mol), ligand 55 (0.011 mol), I2 (0.05 mol), PhMe, rt, 12–16 h. | ||
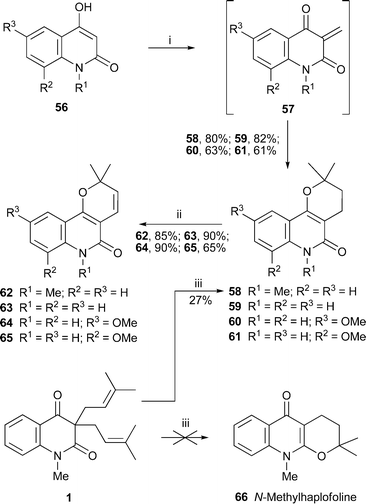
A short synthesis of four pyrano[3,2-c]quinolinone alkaloids by Mohan and co-workers entailed condensation of the 4-hydroxyquinolin-2(1H)-ones 56 with paraformaldehyde in the presence of 3,3-dimethylacrylic acid38 (Scheme 3). The reaction probably proceeds through the 3-methylene intermediates 57, which undergo an inverse electron demand Diels–Alder reaction with the acid followed by thermal decarboxylation to give the tricyclic products 58–61 in moderate to good yields (61–82%). The syntheses of N-methylflindersine62, flindersine63, haplamine64 and 8-methoxyflindersine65 were completed by dehydrogenation with DDQ. Also of interest is another fortuitous synthesis of 58 by treatment of N-methyl-3,3-diprenylquinoline-2,4-dione1 (cf. Section 1.1 above) with a solution of perchloric acid in acetic acid.8 According to a literature precedent, the expected product should have been the linearly fused pyrano[2,3-b]quinolinone alkaloid N-methylhaplofoline66, but the angularly fused product 58 (herein named N-methylisohaplofoline) was obtained instead in 27% yield. Thorough one- and two-dimensional NMR spectra corroborated its structure.
 | ||
Scheme 3
Reagents and conditions: i, (CH2O)n, Me2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2H, dioxan, 120 °C, 4.5 h; ii, DDQ, C6H6, reflux, 15 h; iii, HClO4 (20% in AcOH), 2.5 h. CHCO2H, dioxan, 120 °C, 4.5 h; ii, DDQ, C6H6, reflux, 15 h; iii, HClO4 (20% in AcOH), 2.5 h. | ||
The isolation of evolitrine22 from twigs of Evodia lunu-ankenda by bioassay -guided fractionation for anti-inflammatory activity inspired Lal et al. to undertake the synthesis of the alkaloid and several analogues for further testing.39 Starting from the known furo[2,3-b]quinoline-3,4-(2H,9H)-dione67, they devised an efficient new procedure for making the chloro derivative 68 by treatment with phosphorus oxychloride and Aliquat 336 in the absence of solvent (Scheme 4). The reaction consistently gave yields of 70–80% even with large batches of 50–60 g. Reduction of the chloroketone with sodium borohydride followed by dehydration produced the key intermediate 69, from which the chlorine atom could be displaced with a methoxide ion to produce evolitrine. Similarly, 69 and related variants bearing other substituents on the aromatic ring reacted with a variety of alkoxides and primary or secondary amines to afford a range of products 70 and 71, among others. Although many of these synthetic analogues showed anti-inflammatory activity in inhibiting carrageenan-induced edema in rats, evolitrine itself proved to be the most effective compound (57% inhibition at a dosage of 20 mg kg–1), rivalling the control drug indomethacin in its activity without producing toxic symptoms, cardiovascular effects or weight loss. It also showed significant inhibition of adjuvant arthritis in rats. The alkaloid is thus a very promising lead for the development of novel anti-inflammatory drugs.
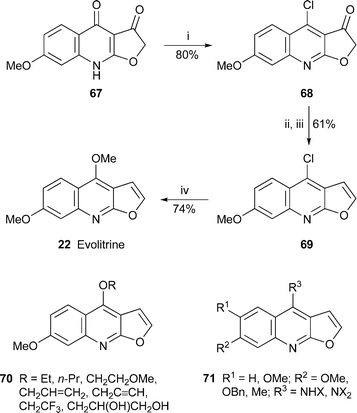 | ||
| Scheme 4 Reagents and conditions: i, POCl3 (2 equiv.), Aliquat 336 (10 mol%), rt, 48 h; ii, NaBH4, MeOH, 0 °C, then rt, 1 h; iii, KHSO4, dioxan, reflux, 3 h; iv, NaOMe, MeOH, reflux, 3 h. | ||
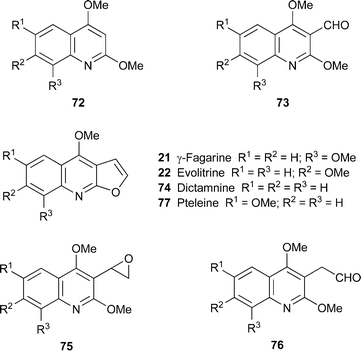
Directed ortho-metallation of 2,4-dimethoxyquinolines 72 with n-butyllithium followed by reaction with N,N-dimethylformamide to give the 3-formylquinolines 73 was the first step in a new synthesis of several furo[2,3-b]quinoline alkaloids .40 When intermediate 73 (R1 = R2 = R3 = H) was treated with dimethylsulfonium methylide, dictamnine74 and the epoxide 75 (R1 = R2 = R3 = H) were isolated in yields of 10% and 34%, respectively. The epoxide could be transformed into dictamnine in 70% yield by heating with polyphosphoric acid (PPA) at 125–130 °C. However, reaction of the remaining monomethoxylated intermediates 73 (R1, R2 or R3 = OMe) with the sulfonium ylide produced mixtures of the corresponding epoxides 75 and their rearranged products, the homologated aldehydes 76, in yields of approximately 50% each. Both 75 and 76 could be converted into the target alkaloids (γ-fagarine21, evolitrine22 and pteleine77; yields 30–80%) when heated with PPA.
1.3 Quinoline alkaloids from non-rutaceous plants
Two recent reviews deal with aspects of the synthesis and chemical transformations of the historically important, and perpetually topical, Cinchonaalkaloids , celebrated for their antimalarial properties. Hoffmann and Frackenpohl have surveyed some of the remarkable chemical transformations undergone by quinine78 and related alkaloids , including cinchonidine79, quinidine80 and cinchonine81.41 The reactions described include a variety of cleavages, epimerisation at C-9 and rearrangements of the quinuclidine moiety; and applications include the synthesis of novel ligands for catalytic asymmetric synthesis and the preparation of functionalised quinuclidines as useful chiral building blocks for synthesis. The review by Kaufman and Rúveda, which concentrates on quinine itself, briefly outlines the historical aspects of the alkaloid's isolation and structure determination before giving a lucid account of classical and contemporary efforts to realise its total synthesis.42 To date, three successful syntheses, by the groups of Woodward, Stork and Kobayashi, have been achieved, and these are described in detail.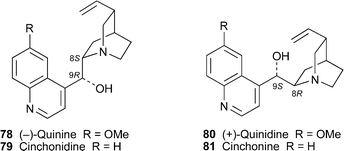
Cinchona alkaloids have also been found in Remijia species. Recent investigations of the Peruvian tree Remijia peruviana (Rubiaceae) have led to the isolation of the novel alkaloids (+)-N-acetyldeoxycinchonicinol82, (+)-N-acetylcinchonicinol83, (–)-acetylcupreine84 and (+)-N-ethylquinine85, the possible artifact cinchonine hydrochloride and the known quinoline alkaloids (+)-cinchonine, (–)-cupreine86, quinine and quinidine as well as some novel quinuclidine-substituted indoles.43,44 Complete 1H and 13C shift assignments based on two-dimensional NMR spectroscopic methods were reported for the new alkaloids , full assignments for the quinoline moieties apparently being deduced for the first time. As one might have expected, the 1H NMR spectra of the N-acetylated alkaloids 82 and 83 showed the presence of amide rotamers ; for 82, for example, the ratio of Z–Erotamers was 1 : 2 when the spectrum was recorded in chloroform at 299 K. Miscellaneous tests for biological activity showed that cupreine, acetylcupreine and, most notably, cinchonine affected the feeding behaviour of Leptinotarsa decemlineata (the Colorado potato beetle), while acetylcupreine was cytotoxic to insect Sf9 and mammalian CHO cells.44 While only quinine itself showed trypanocidal activity towards T. cruzi, acetylcupreine and cinchonine hydrochloride were especially cytotoxic towards a range of tumour cell lines.
Although isomers of 3-hydroxyquinine have been detected as oxidatively biotransformed metabolites of quinine in human plasma and urine, the assignment of the (3S)-configuration to the major diastereomer 87 has thus far been tentative. This assignment has now been confirmed by X-ray crystallography of the 9-acetyl derivative 88 of a synthetic sample prepared by oxidation of quinine.45
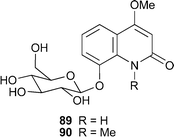
Two new representatives of the rare quinoline alkaloid glycosides , (–)-4-methoxy-8-(β-D-glucopyranosyloxy)quinolin-2(1H)-one89 and (–)-1-methyl-4-methoxy-8-(β-D-glucopyranosyloxy)quinolin-2(1H)-one90, were isolated from aerial parts of the Chinese medicinal plant Echinops gmelinii (Compositae).46 Characterisation included full NMR spectroscopic analysis, with HMBC correlations between the anomeric proton of the sugar and the quinolinone signal at C-8 indicating the site of attachment of the two moieties. In addition, hydrolysis of both alkaloids with dilute aqueous hydrochloric acid yielded glucose, which was identified by comparison with an authentic sample. The aglycone of 90 is itself a known alkaloid , folifidine, but the aglycone of 89 has not yet been reported as a natural product.
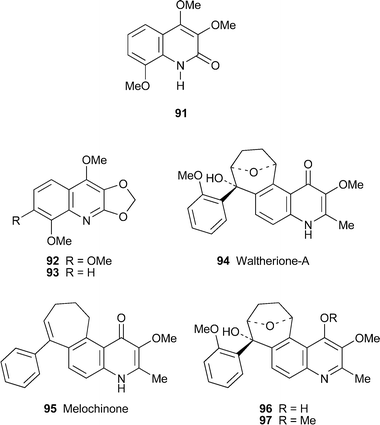
The known alkaloids 3,4,8-trimethoxyquinolin-2(1H)-one91 and 2,3-methylenedioxy-4,7,8-trimethoxyquinoline92 were isolated from Brazilian specimens of the perennial herb Sebastiania corniculata (family Euphorbiaceae, tribe Hipomaneae) together with the novel compound 2,3-methylenedioxy-4,8-dimethoxyquinoline93.47 The structures of all three were substantiated by spectroscopic methods. This appears to be the first record of quinoline alkaloids in the genus Sebastiania. The more unusual alkaloid (–)-waltherione A94, obtained from the root bark of the South American medicinal plant Waltheria douradinha (Sterculiaceae),48 contains a cyclohepta[f]quinolinone core previously found only in melochinone95, another metabolite of the Sterculiaceae first reported over 30 years ago.49 The structure and relative stereochemistry of 94 were elucidated on the basis of extensive one- and two-dimensional NMR spectra; in addition, AM1 calculations supported the assignment of the relative configurations at the three stereogenic centres. Furthermore, the 1NMR spectrum recorded in deuteriated DMSO revealed temperature-dependent tautomerism between the keto and enol forms 94 and 96, the latter readily giving a methyl ether 97 upon treatment with diazomethane. Waltherione A proved to be inactive when tested against a range of bacteria, whereas its methyl ether showed moderate activity against both Gram-positive and Gram-negative bacteria.
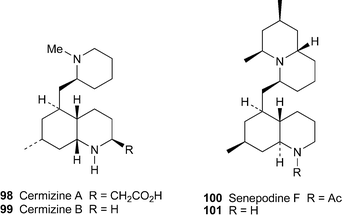
A recent review in this Journal gives a fine account of the structurally fascinating alkaloids , among them several complex decahydroquinolines, isolated from club mosses of the genus Lycopodium (Lycopodiaceae).50 Among the new metabolites isolated from L. cernuum are (+)-cermizine A98 and (–)-cermizine B99, two novel decahydroquinoline alkaloids of the phlegmarane class.51 Two-dimensional NMR methods established the structures and relative stereochemistry of the alkaloids , while NOESY correlations indicated the unusual cis ring junction in the decahydroquinoline ring system, and the comparative resistance to rotation between the two heterocyclic components. All phlegmarane-type alkaloids hitherto isolated possess a trans ring junction such as that found in (–)-senepodine F100, another new alkaloid isolated from a different club moss, L. chinense.52 This compound was also characterised spectroscopically, although broadening of NMR spectroscopic signals due to slowly interconverting amide rotamers made complete assignment difficult. However, hydrolysis of senepodine F with aqueous hydrochloric acid produced the deacetyl derivative 101, which gave well-resolved signals that facilitated a full assignment of the alkaloid's structure and relative stereochemistry.
1.4 Quinoline alkaloids from fungal and microbial sources
An important review on thiopeptide antibiotics brings together half a century of widely scattered literature on this group of structurally complex, highly modified macrocyclic peptides, most of which are produced by actinomycetes of the genus Streptomyces.53 The prototypical example is thiostrepton102, which contains, among other features, the same 7,8-dihydroquinoline core (102, outlined fragment) that is found in related antibiotics such as the thiopeptins, the siomycins and the Micromonospora metabolites Sch 18640 and Sch 40832. The review covers the isolation, structural elucidation, biosynthesis and biological activity of these and related natural products containing other heterocyclic cores. Also included is a brief survey of successful total syntheses of some of the antibiotics, notably the recently communicated synthesis of thiostrepton itself by Nicolaou and co-workers.54 The major challenge in this synthesis was, of course, the assembly of the macrocyclic peptide components; for our present purpose, it is sufficient to point out that the dihydroquinoline core was derived from quinaldic acid103, which was converted in several steps into the chiral epoxide 104. Treatment with the allyl ester of L-isoleucine in the presence of lithium perchlorate in acetonitrile at 60 °C afforded the building block 105 in 69% yield.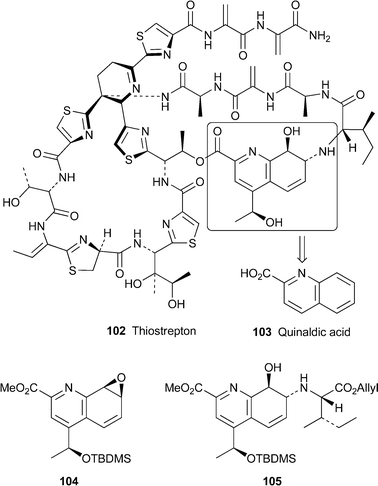
Intercellular communication in Pseudomonas aeruginosa bacterial colonies is mediated by a variety of ‘signalling’ molecules, many of which belong to a family of 4-hydroxy-2-alkylquinolines (HAQs) or their quinolone tautomers. The biosynthesis of these molecules, briefly alluded to in last year's review (cf.ref. 23c), has been further elucidated by means of feeding experiments with isotopically labelled precursors.55 GC-MS analysis showed that the incorporation of anthranilic acid labelled with 15N into the HAQ products 106, principally the 2-heptyl and 2-nonyl congeners, was efficient (ca. 66%), while the incorporation of acetate singly or doubly labelled with 13C was also substantial. Detailed experiments provided evidence for a metabolic pathway involving ‘head-to-head’ condensation of anthranilic acid 107 with suitably activated β-keto fatty acids 108; the 4-hydroxy group of the products originates from anthranilic acid, while decarboxylation of 108 occurs after condensation. It also appears that the fatty acids —principally decanoic and dodecanoic acids—are at least in part derived from a common pool involved in the biosynthesis of rhamnolipids.
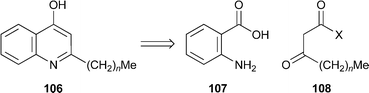
Quinolobactin
109, a simple siderophore produced in iron-deficient cells of a mutant strain of Pseudomonas fluorescens, has been prepared in 60% overall yield by treating xanthurenic acid110 with thionyl chloride in methanol, displacing the 4-chloro substituent from the intermediate 111 with a methoxide ion, and finally hydrolysing the methyl ester with aqueous sodium hydroxide.56 The protonation constants of the product were determined by means of potentiometric titrations, and the stability constants for complexation with ferric ions were determined by spectrophotometric and potentiometric methods. The metal complex, the structure of which has not previously been determined, is of the form FeL2, with typical coordination of 8-hydroxyquinolinate around the metal and no evidence of coordination by the carboxylate group.
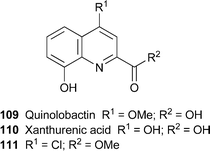
Fungi belonging to the genus Penicillium produce a variety of secondary metabolites formally derived from anthranilic acid. Viridicatin112 and viridicatol113, two well-known quinolin-2-ones of fungal origin, have now been detected in no fewer than 121 strains of P. crustosum from widely differing ecological niches and geographic regions, including subtropical, temperate and Arctic habitats; in the latter case, cultured fungal specimens obtained from glacier ice, sea ice and sea water all produced the metabolites.57 Full NMR spectroscopic data have been reported for the first time for a much rarer compound, (–)-peniprequinolone114, which was isolated from P. janczewskii, an endophytic fungus found in the phloem of the Chilean gymnosperm Prumnopitys andina.58 The novel compound quinolactacide115 was extracted from the fermentation broth of Penicillium citrinum Thom F 1539 isolated from a Japanese soil sample, and characterised by the usual complement of spectroscopic techniques.59 This compound showed excellent insecticidal activity against green peach aphids (Myzus persicae), and induced 88% mortality at a concentration of 250 ppm.
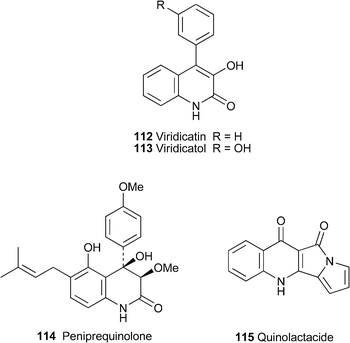
Quinolactacide has obvious structural similarities to the quinolactacins, two of which (quinolactacins A1 and A2) were reported from another strain of P. citrinum in 2001 but incompletely characterised at the time.60 A subsequent enantioselective synthesis of (+)-quinolactacin A261 (cf.ref. 23d) established the absolute stereochemistry shown in 116; it was also suggested that quinolactacin A1 was the epimer at C-1′. (+)-Quinolactacin A2 has now also been prepared from N-methyl isatoic anhydride 117 and the (2S,3S)-isoleucine derivative 118 by the short route shown in Scheme 5.62 A Friedländer-type annulation between the two precursors was effected by treatment with DBU in the presence of molecular sieves to give the quinolin-4-one 119 in 42% yield. Acidic hydrolysis of the Boc protecting group led to spontaneous cyclisation, giving 116 and a diastereomer in a ratio of 8 : 1. The spectroscopic and optical data of the major isomer matched those reported for (+)-quinolactacin A2. The spectra of the epimer , the formation of which was promoted by prolonged contact with silica gel, protic solvents or acidic media, could similarly be matched with those of natural quinolactacin A1. The inference is that the two quinolactacins are epimeric at C-3, not at C-1′ as was previously suggested. The equilibration possibly proceeds through an intermediate such as 120, and quinolactacin A1 should thus have the structure 121.
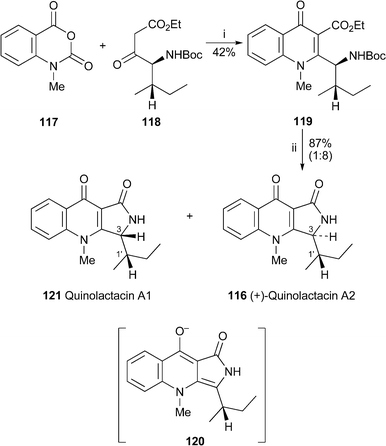 | ||
| Scheme 5 Reagents and conditions: i, DBU, 3 Å molecular sieves, CH2Cl2, 20 h; ii, TFA, EtOH, H2O, 2 h. | ||
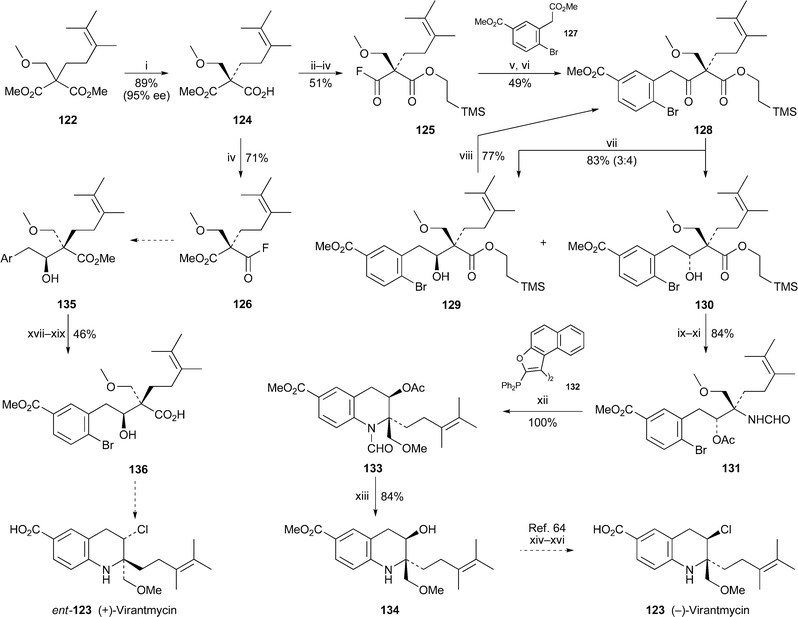
Enzyme-mediated desymmetrisation of the substituted malonic ester 122 with porcine liver esterase was the opening gambit in a stereodivergent synthesis of both enantiomers of the Streptomycesmetabolite virantmycin 123 by Back and Wulff63 (Scheme 6). The half-ester (S)-124, obtained in 89% yield and 95% ee, could be converted into either of the two pseudo-enantiomeric acyl fluorides 125 and 126, which are the precursors of (–)-123 and (+)-123, respectively. To complete the synthesis of natural (2R,3R)-(–)-virantmycin, the enolate of diester 127 was acylated with 125 and the resulting β-keto ester was hydrolysed and decarboxylated by the Krapcho procedure to give 128. A poorly stereoselective reduction of the ketone with sodium borohydride afforded the separable alcohol diastereomers 129 and 130 in a ratio of 3 : 4; fortunately, the undesired epimer 129 could be recycled by oxidation back to the ketone 128. Transformation of the major isomer 130 into the formamide 131 set the stage for the crucial construction of the tetrahydroquinoline nucleus by intramolecular aryl amination. This conversion, rendered especially difficult by the high degree of steric hindrance in the system, was eventually accomplished in quantitative yield with Pd2(dba)3 in the presence of the unusual ligand BINAPFu132. Hydrolysis of the acetate and N-formyl groups of the tetrahydroquinoline product 133 then afforded the alcohol 134, which is the enantiomer of an intermediate that featured in a previous synthesis of unnatural (+)-virantmycin by Morimoto and Shirahama64 (cf.ref. 23e). Application of Morimoto's protocol to 134 (a procedure that entails replacement of the alcohol with a chloride ion with retention of configuration via an intermediate aziridine) yielded (–)-virantmycin123, the NMR spectra and specific rotation of which were comparable with those reported in the literature. In a similar manner, the acyl fluoride 126 was converted into the alcohol 135 and thence into (+)-virantmycinent-123via carboxylic acid 136.
 | ||
| Scheme 6
Reagents and conditions: i, porcine liver esterase, DMSO + pH 8.0 phosphate buffer (1 : 4), rt, 7 d; ii, TMSCH2CH2OH, DCC, DMAP, CH2Cl2, rt, 3 d; iii, aq. KOH (10%), MeOH, 45 °C, 15 h; iv, cyanuric fluoride, py, CH2Cl2, 0 °C, 1 h; v, LiHMDS, + 127, Et2O, 0 °C, 10 min, then 125, Et2O, 0 °C, then rt, 20 h; vi, aq. NaCl (10%), DMSO, 125 °C, 20 h; vii, NaBH4, MeOH, 0 °C, 2.5 h; viii, PCC, CH2Cl2, rt, 3 d; ix, Ac2O, DMAP, py, rt, 3 d; x, Bu4NF in THF (1.0 M), rt, 1 h; xi, Ph2P(O)N3, NEt3, DMAP, PhMe, reflux, 2.25 h, then NaBH4, THF, rt, 12.5 h; xii, ligand 132, Pd2(dba)3, Cs2CO3, PhMe, 90 °C, 6.5 h; xiii, NaOH in MeOH (0.13 M), rt, 24 h; xiv, DEAD, PPh3, THF, rt; xv, NaOH, MeOH, reflux; xvi, Et4NCl, TFA, CH2Cl2, –15 °C; xvii, aq. KOH (10%), MeOH, rt, 20 h; xviii, n-PrSH, NaH, HMPA, rt, 4.75 h; xix, SOCl2, MeOH, –10 °C, 1 h, then reflux, 3 h. | ||
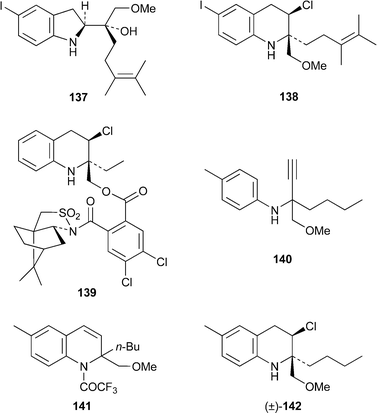
The first synthesis of natural (–)-virantmycin, by Kogen and co-workers, was communicated in 200365 (cf.ref. 23d). A comprehensive paper containing extensions to this work as well as full experimental details has now been published.66 The focus of the synthesis is the stereospecific rearrangement of the indoline-2-methanol 137 with tributylphosphine and tetrachloromethane to give product 138 as a single isomer in 45% yield. The new paper contains a further 18 examples of this rearrangement, most of them mediated by triphenylphosphine; the new virantmycin analogues, obtained in yields of 31–65%, bear a range of substituents at C-2 and/or on the aromatic ring. The (2R,3R)-absolute configuration of one of the model products was verified by X-ray structural analysis of the camphorsultam derivative 139. In another study of interest, the N-propargylaniline 140 was cyclised with copper(I) chloride and trifluoroacetic anhydride to give the 1,2-dihydroquinoline 141 in 63% yield.67 Addition of chlorine followed by selective dechlorination with sodium cyanoborohydride–zinc chloride and hydrolysis of the N-trifluoroacetyl substituent gave the racemic virantmycin analogue (±)-142 in an overall yield of 39%.
Anachelin
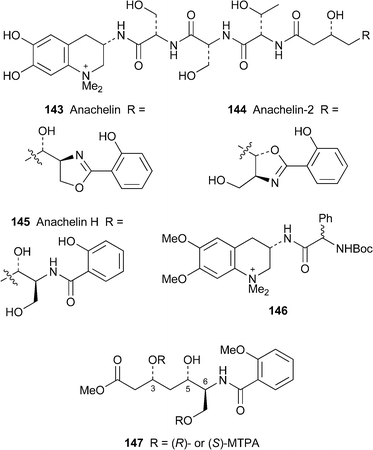
143 and anachelin-2144, siderophores isolated some years ago from the freshwater cyanobacterium Anabaena cylindrica,68,69 are intriguing hybrid metabolites containing alkaloid -like, peptide and polyketide fragments. The alkaloid moiety is the unique 3-amino-6,7-dihydroxy-1,1-dimethyl-1,2,3,4-tetrahydroquinolinium (Dmaq) system, which is linked through its 3-amino group to a hydrophilic tripeptide sequence (L-Ser-D-Ser-L-Thr) and thence to a unit derived from 6-amino-3,5,7-trihydroxyheptanoic (Atha) and salicylic acids. Both termini—the catechol-containing Dmaq segment and the 2-(2-hydroxyphenyl)oxazoline ring in 143 and 144—are responsible for the binding and transport of iron, while the atypical amino acid D-Ser in the middle of the molecule is thought to resist hydrolysis by endogenous proteases. Although the absolute configurations in the tripeptide segment of the anachelins were relatively easy to ascertain by standard degradation and analytical methods, the stereochemistries of the remaining four stereogenic centres could not be assigned. Okada and co-workers have now established the absolute configurations of both anachelins by means of selective spectroscopic, functionalisation and degradation studies.70 Knowing that both anachelins gave the same salicylamide product 145 (also known as anachelin H68) upon hydrolysis of the oxazoline rings, the authors assumed that the two alkaloids would have identical absolute configurations in their Dmaq and Atha segments. The equatorial disposition of the nitrogen substituent in the Dmaq unit of 143 was revealed by J values and NOESY correlations, while acidic hydrolysis of 144 and analysis of chemical shift differences in the (R)- and (S)-Boc-phenylglycine Dmaq derivatives 146 indicated that the stereogenic centre has the (S)-absolute configuration. The syn relative stereochemistry of the hydroxyl groups in the Atha segment of 143 was established by NMR spectroscopic studies of its acetonide derivative; while selective NMR investigations of 144 showed the substituents on the oxazoline ring to be trans . Finally, analysis of the bis(S)- and bis(R)-Mosher's esters of a degradation product 147 from the Atha unit of 143 allowed the assignment of the absolute stereochemistry at C-3, C-5 and C-6 as R, S and S, respectively. The authors concluded that the absolute configurations of both alkaloids were as shown in 143 and 144, with L-serine being a plausible biogenetic source of the N-terminus of the Atha residue.
The absolute configuration of the anachelins has also been substantiated by the synthesis of anachelin H145, the hydrolysis product obtained from both anachelin and anachelin-2, and some of its diastereomers . In a preliminary study, Gademann and Bethuel coupled the air-sensitive L-DOPA derivative 148 with a protected serine building block, and then oxidatively cyclised the intermediate 149 with dianisyltellurium oxide to give the unstable dihydroxy tetrahydroquinolinium product 150 in 80% yield71 (Scheme 7). That this type of cyclisation, which proceeds via an ortho-quinone, might be biomimetic was demonstrated by the conversion of 148 itself into 151 by treatment with a catechol oxidase (specifically, a mushroom tyrosinase) in the presence of oxygen. Intermediate 150 was converted in a further four steps into the alkaloid -tripeptide assemblage 152, which was designed to lend itself to a flexible strategy for coupling with various diastereomers of the Atha-salicylamide fragment 153. The first synthetic anachelin H diastereomer produced in this way had the (3R,5R,6S)-configuration. In a follow-up publication,72 Gademann and Bethuel devised a more efficient synthesis of the L-DOPA derivative 148, and achieved its cyclisation to the anachelin chromophore 151 with dianisyltellurium oxide. The yield after O-benzylation of this unstable catechol was 40%. Attachment of the appropriate amino acid residues then gave the pivotal intermediate 152. L-Serine provided the N-terminus for the construction of four different Atha-salicylamide diastereomers , viz. (3R,5R,6S)-, (3S,5R,6S)-, (3S,5S,6S)- and (3R,5S,6S)-153, the last of which showed a similar pattern of NMR signals to those in anachelin H itself. When this isomer was coupled to the amine terminus of 152 and the protecting groups were removed, the synthetic product, characterised with chloride as a counter-ion, proved to be spectroscopically and chromatographically identical to an authentic sample of anachelin H145.
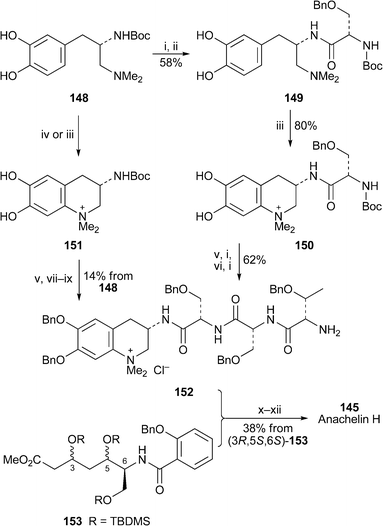 | ||
| Scheme 7 Reagents and conditions: i, TFA, CH2Cl2; ii, Boc-L-Ser(OBn)OH, EDC, HOBt; iii, dianisyltellurium oxide, CH2Cl2; iv, mushroom tyrosinase, O2; v, BnBr, Cs2CO3, Me2CO; vi, Boc-L-Thr(OBn)-D-Ser(OBn)OH, EDC, HOBt; vii, HCl, dioxane; viii, Boc-L-Ser(OBn)OCO2Bui, NMM, DMF, then HCl, dioxane; ix, Boc-L-Thr(OBn)-D-Ser(OBn)OCO2Bui, NMM, DMF, then HCl, dioxane; x, (3R,5S,6S)-153 + NaOH, MeOH, THF, then ClCO2Bui, NMM, DMF, then 152; xi, H2, Pd/C, AcOH, MeOH; xii, HCl (1%) in MeOH. | ||
1.5 Quinoline alkaloids from animals
cis-Decahydroquinoline 195A (pumiliotoxin C) 154, the best known and most widespread member of the decahydroquinoline family of amphibian skin alkaloids , remains a popular target for synthesis. Among the interesting features in a new route to the racemic compound by Girard et al.73 is the unusual use of an N-phenyl protecting group, which was attached to the commercially available piperidin-4-one ketal 155 by treatment with bromobenzene and the complex superbase prepared from sodium amide and sodium tert-butoxide (Scheme 8). The electrochemical oxidation of intermediate 156 in the presence of a cyanide ion produced the aminonitrile 157, the anion of which was alkylated with 1-bromo-3-chloropropane under carefully controlled conditions to give 158 in 94% yield. Replacement of the terminal chloride with cyanide, reduction of the aminonitrile (a masked iminium ion) with sodium borohydride and mild reduction of the terminal nitrile with diisobutylaluminium hydride afforded the aldehyde 159. The construction of the target's bicyclic nucleus was then completed by an intramolecular aldol condensation under acidic conditions, after which stereoselective introduction of the C-5 methyl group into the resulting enone 160 was accomplished with lithium dimethylcuprate. The product, obtained in 91% yield and a diastereomeric excess of better than 99%, contains three of the target's four stereogenic centres; NMR spectroscopy and analysis of NOE effects revealed the desired cis-fused octahydroquinolinone structure, and indicated the preferential adoption of the conformation shown in 161. After defunctionalisation of the ketone by Shapiro reaction of the corresponding tosylhydrazone and catalytic hydrogenation of the resulting alkene , a second electrochemical oxidation of the decahydroquinoline 162 produced the aminonitrile 163 as a mixture of two diastereomers . Alkylation of its anion with 1-bromopropane gave 164 as a single isomer, the structure of which was established by single crystal X-ray analysis. That reduction of the aminonitrile with excess sodium borohydride in ethanol occurred with retention of configuration was confirmed by X-ray crystallography of the product 165. Finally, the N-phenyl protecting group was removed under Birch conditions by reduction with lithium in liquid ammonia with ethanol as the proton source followed by acidic hydrolysis of the intermediate enamine products to give (±)-pumiliotoxin C154 in 15 steps and an overall yield of 5% from 155.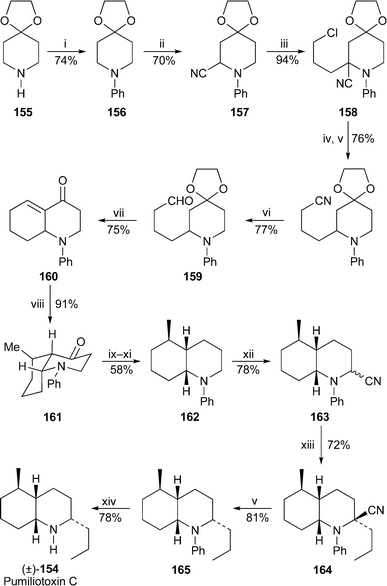 | ||
| Scheme 8 Reagents and conditions: i, PhBr, NaNH2–NaOBut, THF, 50 °C, overnight; ii, 2 e– (graphite felt anode, flow cell, flow rate 5 ml min–1, iox 253 mA), NaCN (6 equiv.), LiOAc in MeOH; iii, LDA, THF, –40 °C, 1 h, then Br(CH2)3Cl, –20 °C, 12 h; iv, NaCN, Bu4NI (10 mol%), DMSO, rt, 48 h; v, NaBH4 (4 equiv.), EtOH, rt, overnight; vi, DIBAL, CH2Cl2, –60 °C, then –20 °C, overnight, then aq. HCl (5%); vii, aq. H2SO4 (1.5 M), THF, reflux, 5 h; viii, Me2CuLi, Et2O, –30 °C, then –15 °C, 30 min; ix, p-TolSO2NHNH2, EtOH, reflux, overnight; x, n-BuLi, THF, –60 °C, 1.5 h, then rt, 2 h, then H2O; xi, H2 (1 atm), 10% Pd/C, MeOH, 10 h; xii, 2 e– (divided cell, vitreous C electrode, +0.80 V, 660 Coulomb ), NaCN (6 equiv.), LiOAc in MeOH; xiii, LDA, THF, –60 °C, 1 h, then n-PrBr, –20 °C, overnight; xiv, Li (10 equiv.), NH3–THF–EtOH (0.5 : 5.0 : 4.5), –40 °C, 1 h, evaporate, then H2SO4–H2O–EtOH (0.5 : 5.0 : 4.5), 60 °C, 10 min. | ||
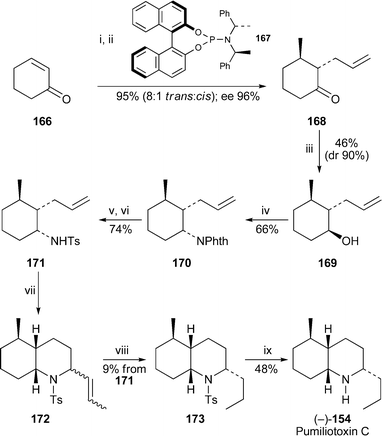
An asymmetric synthesis of (+)-pumiliotoxin C by Feringa and co-workers74 began with the conjugate addition of dimethylzinc to cyclohexenone 166 catalysed by copper(II) triflate and the chiral phosphoramidite 167, followed by allylation with allyl acetate in the presence of tetrakis(triphenylphosphine)palladium(0) (Scheme 9). This tandem bisalkylation reaction produced the ketone 168 as an 8 : 1 mixture of trans and cis isomers in a combined yield of 95% yield. HPLC analysis on a chiral column showed that the enantiomeric excess of the major isomer was 96.2%. Although reduction of the isomeric mixture of ketones with lithium aluminium hydride was virtually quantitative, purification of the resulting mixture of diastereomers by flash chromatography gave the alcohol 169 in only 46% yield and 90% diastereomeric purity. Replacing the alcohol with phthalimide under Mitsunobu conditions afforded the phthalimide derivative 170 with the expected inversion of configuration. Hydrazinolysis and tosylation then produced the crystalline p-toluenesulfonamide 171, the stereochemistry of which was confirmed by single crystal X-ray analysis. Although a tandem Heck reaction–cyclisation of 171 with 1-bromopropene and palladium(II) acetate produced a mixture of several 2-(1-propenyl)decahydroquinoline isomers 172 in modest yield, catalytic hydrogenation of the crude mixture gave mainly two isomeric 2-propyltetrahydroquinolines, which could be separated only by preparative HPLC . The overall yield of the desired product 173 from 171 was a disappointing 9%. Finally, deprotection of 173 with sodium and naphthalene completed the synthesis of (–)-pumiliotoxin C154.
 | ||
| Scheme 9
Reagents and conditions: i, Cu(OTf)2 (0.5 mol%), ligand 167 (1 mol%), PhMe, rt, 1 h, then 166, –30 °C, 10 min, then Me2Zn, –30 °C, 3 h; ii, add H2CCHCH2OAc, Ph(PPh3)4 (2 mol%), 0 °C, overnight; iii, LiAlH4 (2 equiv.), THF, –80 °C, 3 h; iv, phthalimide, PPh3, DEAD, EtNPri2, THF, 30 °C, 3.5 h; v, H2NNH2·H2O, 1-hexene, EtOH, reflux, overnight; vi, p-TsCl, NEt3, CH2Cl2, rt, 60 h; vii, BrCHCHMe, (o-Tol)3P (0.6 equiv.), Pd(OAc)2 (0.3 equiv.), Na2CO3 (3 equiv.), Bu4NCl (2 equiv.), MeCN, 86 °C (sealed tube), 68 h; viii, H2 (45 bar), 10% Pd/C, EtOH, rt, overnight, then separation by preparative HPLC ; ix, Na, naphthalene, MeOCH2CH2OMe, 0 °C, 1 h. | ||
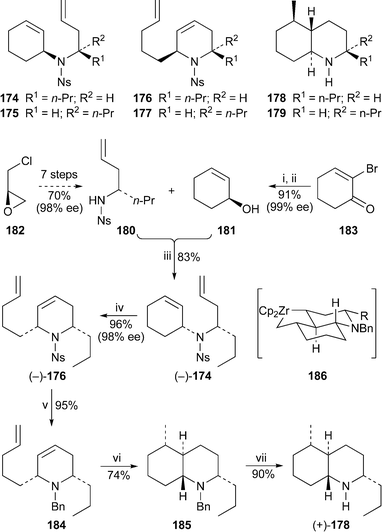
Neidhöfer and Blechert recently reported a synthetic approach to decahydroquinoline alkaloids based on the ring opening–ring closing metathesis (ring rearrangement metathesis) of racemic precursors 174 and 175 to the tetrahydropyridines 176 and 177, followed by diastereoselective zirconium-mediated cyclisation to give, after suitable functional group manipulation, racemic trans-decahydroquinoline 195A (±)-178, another known amphibian metabolite , and its 2-epimer (±)-179 (Scheme 10).75 This work was followed up by an enantioselective synthesis of (+)-trans-195A, also shown in the Scheme.76 The preparation of the metathesis precursor (–)-174 entailed Mitsunobu reaction between two chiral moieties (S)-180 and (S)-181, which were themselves made in several steps from (R)-epichlorohydrin182 and 2-bromocyclohex-2-enone183, respectively. The latter transformation involved enantioselective reduction with the (R)-methoxy Corey–Bakshi–Shibata (CBS) reagent followed by debromination by lithium–halogen exhange. Ring rearrangement metathesis of (–)-174 was effected with the Grubbs first-generation catalyst under an atmosphere of ethylene, and afforded (–)-176 in 96% yield and 98% ee. Although the final ring closure, a diastereoselective zirconium-mediated Negishi alkene coupling with n-butyllithium and zirconocene dichloride, failed with (–)-176 itself, reaction of the N-benzyl analogue (–)-184 gave the bicyclic product (+)-185 in 74% yield. The reaction is thought to proceed through a zirconacycle such as 186. Hydrogenolytic removal of the protecting group completed the synthesis of (+)-decahydroquinoline (+)-178, which proved to be the enantiomer of naturally occurring (–)-trans-195A. The absolute configuration of the natural product, previously unknown, is thus (2R,4aS,5R,8aS).
 | ||
| Scheme 10
Reagents and conditions: i, (R)-MeO-CBS, THF, –10 °C, 45 min; ii, tert-BuLi, Et2O, –78 °C, 1 h, then –20 °C, 3 h, then aq. NaHCO3, rt, 1 h; iii, PriO2CNNCO2Pri, PPh3, THF, 0 °C, 30 min, then rt, 72 h; iv, (PCy3)2Cl2RuCHPh (5 mol%), H2CCH2, CH2Cl2, 50 °C (sealed tube), 24 h; v, PhSH, K2CO3, DMF, 80 °C, 2 h, then Bu4NI, BnBr, 1 h; vi, Cp2ZrCl2, n-BuLi, THF, –78 °C, 45 min, then add 184, THF, rt, 5 h, then aq. HCl (1 M), rt, 30 min; vii, H2 (1 atm), 10% Pd/C, MeOH, overnight. | ||
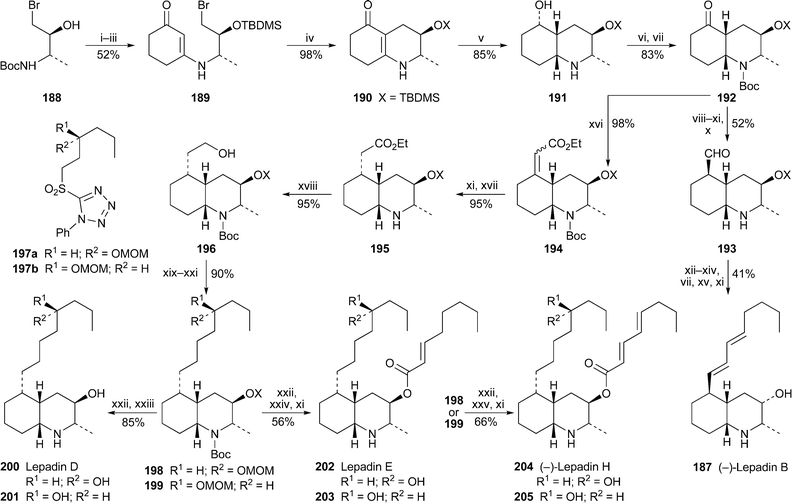
The lepadins are a growing family of biologically active decahydroquinoline alkaloids isolated from a variety of ascidians (sea squirts). For example, (–)-lepadin B187 has been found to act as a reversible blocker of α4β2 and α7 neuronal nicotinic acetylcholine receptors at micromolar concentrations.77 Pu and Ma have made an important contribution to the chemistry of the lepadins by devising an efficient and general approach to their synthesis (Scheme 11), in the process also clarifying the absolute stereochemistry of three partially characterised members of the group.78 The protected amino alcohol 188, prepared from N-Boc-L-alanine, was converted in three steps into the vinylogous amide 189, efficient cyclisation of which produced 190, which contains the alkaloids ' bicyclic core. The desired (4aR,8aR) configuration of the decahydroquinoline ring system was introduced by diastereofacially selective catalytic hydrogenation of the double bond between the fused rings, after which reoxidation of the alcohol in product 191 and protection of the amine gave ketone 192, the pivotal intermediate from which all of the target alkaloids were derived. For lepadin B, the β-orientated side chain at C-5 was installed by Wittig reaction of 192 with methoxymethylenetriphenylphosphorane followed by unmasking of the aldehyde, removal of the N-Boc protecting group and base-induced epimerisation to give 193 as a single isomer. The side chain was elaborated by Horner–Wadsworth–Emmons reaction, giving the (E,E)-diene with greater than 95% selectivity. Finally, the stereochemistry of the oxygen substituent at C-3 was inverted in two steps by oxidation of the free alcohol with Dess–Martin periodinane and stereocontrolled reduction with sodium borohydride. Lepadin B 187, characterised as its trifluoroacetate salt, was thereby obtained in 5.3% overall yield over 20 linear steps from N-Boc-L-alanine. For the remaining targets, the α-orientated side chain at C-5 was introduced by Peterson reaction of 192 with ethyl trimethylsilylacetate, the acrylate product 194 subsequently giving the saturated diastereomer 195 almost exclusively (97 : 3) after N-deprotection and catalytic hydrogenation. The stereochemistry of the product was confirmed by single crystal X-ray analysis of the N-tosyl derivative. Re-protection of 195 followed by reduction with diisobutylaluminium hydride produced alcohol 196, the precursor of all the remaining alkaloids , which share a common side chain at C-5. The strategy for elaborating this chain was by Julia reaction of the corresponding aldehyde. However, since the absolute configuration of the alcohol substituent at C-5′ in the side chain was not determined when the alkaloids were first reported, the present workers chose to carry out the reaction with both enantiomers of the sulfone 197, which they prepared by a sequence entailing Sharpless asymmetric epoxidation and reductive ring opening. Thus Swern oxidation of 196, Julia reaction with 197a or 197b and catalytic hydrogenation of the resulting alkene afforded 198 and 199 in 90% yield. Simple N- and O-deprotection then yielded the two epimeric products 200 and 201. For the final two alkaloids , esterification of the OH substituent at C-3 with appropriate acids produced the epimeric product pairs 202/203 and 204/205. In the latter case, the NMR spectra of 204, but not 205, and the optical rotation of its hydrochloride salt matched those reported for (–)-lepadin H. Although the remaining pairs of products gave very similar spectra and optical rotations, 200 and 202 were assumed to correspond to lepadins D and E, respectively, by analogy with 204. The conclusion is that all three alkaloids share the (2S,3R,4aS,5S,8aR,5′R) absolute configuration .
 | ||
| Scheme 11
Reagents and conditions: i, TBDMSCl, imidazole, DMF; ii, HCO2H, CH2Cl2, then aq. NaHCO3; iii, 1,3-cyclohexanedione, C6H6, reflux; iv, NEt3, NaI, DMF, 110 °C; v, H2 (80 atm), Pt/C, dry AcOH, 50 °C; vi, (Boc)2O, C6H6; vii, Dess–Martin oxidation; viii, Ph3PCHOMe, –40 °C to rt; ix, Cl3CCO2H, H2O (trace), CH2Cl2; x, K2CO3, MeOH; xi, TFA, CH2Cl2; xii, diethyl (2E)-heptenylphosphonate, KHMDS, THF; xiii, (Boc)2O, C6H6, reflux; xiv, Bu4NF, THF; xv, NaBH4, MeOH, –78 °C; xvi, Me3SiCH2CO2Et, LDA, –78 °C to rt; xvii, H2 (1 atm), Pt/C, dry AcOH, 50 °C; xviii, (Boc)2O, DMAP, then DIBAL, –40 °C; xix, Swern oxidation; xx, 197a or 197b + NaHMDS, THF; xxi, H2, Pd/C; xxii, Bu4NF, THF, rt; xxiii, HCl, MeOH–PriOH; xxiv, (2E)-octenoic acid, 2,4,6-Cl3C6H2COCl, EtNPri2, DMAP; xxv, (2E,4E)-octadienoic acid, EDCI, DMAP, CH2Cl2. | ||
2 Quinazoline alkaloids
2.1 Occurrence, characterisation and biological activity
Alkaloids of the vasicine (peganine) class are well-known constituents of the medicinal plants Adhatoda vasica and Peganum harmala. The simultaneous detection and quantification of vasicine206 and vasicinone207 in the former source has now been achieved quite simply by means of high-performance thin-layer chromatography .79 These two alkaloids and the related compounds deoxyvasicine (deoxypeganine) 208 and deoxyvasicinone (pegenone) 209 were found to be the principal alkaloids in shoots, flowers and shoot cultures of the latter source, whereas β-carbolines were dominant in fruits, roots and root cultures.80 Some clues as to how these pharmacologically important alkaloids are metabolised have come from experiments in which the anticholinergic alkaloid deoxyvasicine208 was incubated under aerobic conditions with NADPH and a homogenate from rabbit liver as the enzyme source; both deoxyvasicinone209 and the ring-hydroxylated products vasicinone207 and isovasicinone210 were isolated in effective yields of 78% and 10–15%, respectively.81 Aerial oxidation of 208 to 209 also took place, although less efficiently, in control samples lacking the coenzyme NADPH or the enzyme source. This ready oxidation of deoxyvasicine to the biologically inactive deoxyvasicinone is thus likely to cause problems in the formulation of pharmaceutical products containing the former for the treatment of disorders such as Alzheimer's disease or alcoholism. Synthetic analogues might provide a way around this difficulty; in this regard, a computational approach to evaluating a library of synthetic vasicinone analogues for therapeutically favourable ADME (absorption , distribution , metabolism and excretion) properties has been described.82 The synthetic vasicine analogues 211 appear to be novel leads for developing antiasthmatic drugs by virtue of their ability to modulate activity at CCR3 receptors.83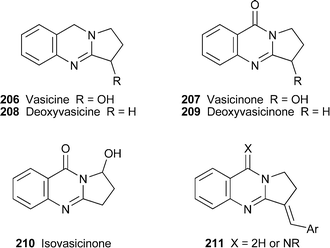
HPLC -based activity profiling of lipophilic extracts from woad (Isatis tinctoria) for 5-lipoxygenase activity pinpointed—perhaps not unexpectedly—the well-known bioactive Isatis constituents tryptanthrin212 and α-linolenic acid as being primarily responsible for the plant's anti-inflammatory activity.84 Interestingly enough, a recent investigation of the constituents of several woad strains showed that tryptanthrin is actually only a minor constituent of freeze-dried plant material; however, its concentration increased substantially in post-harvest treatment, especially when leaves were dried at 40 °C prior to extraction.85 Although details of its formation under these conditions have not yet been elucidated, transformations of other woad constituents such as indoxyl glycosides into various precursors of indigo213 during the drying of leaf samples86 possibly provide a clue to tryptanthrin's origin. The alkaloid's potential as an antitumour agent has received further support from the finding that it strongly inhibited the induction of hepatocytegrowth factor in human dermal fibroblasts.87
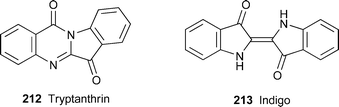
A far more unusual source of tryptanthrin is an aerobic Gram-negative alphaproteobacterial strain, HEL-45T, found in a water sample from the North Sea.88 The organism, which required sodium and 1–7% sea salts for growth, also produced polar lipids , cyclic dipeptides and indole derivatives. Analysis of its gene sequences showed it to belong to a new genus and species, Oceanibulbus indolifex gen. nov., sp. nov.
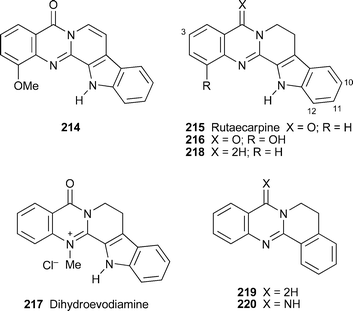
The only new quinazoline alkaloid reported from a higher plant during the review period was 1-methoxy-7,8-dehydrorutaecarpine214, isolated from the root bark of Zanthoxylum integrifoliolum and characterised by means of standard spectroscopic techniques.20 The known alkaloids rutacecarpine215 and 1-hydroxyrutaecarpine216 were also obtained. In in vitro tests, all three alkaloids proved to be reasonably cytotoxic towards the murine leukemia P-388 cell line (ED50 3.88, 10.5 and 3.72 µg ml–1, respectively) and HT-29 cells (ED50 8.54, 33.6 and 7.44 µg ml–1, respectively). Another study of interest89 probed the metabolic fate of rutaecarpine, which is also one of the chief alkaloids of the medicinal herb Evodia rutaecarpa and a proven antithrombotic and vasorelaxant. The oxidation of the alkaloid with liver microsomes from rats previously treated with 3-methylcholanthrene, an inducer of cytochrome P450 -dependent monooxygenase, in the presence of added NADPH was shown to produce predominantly 10-hydroxyrutaecarpine as well as lesser amounts of the 3-, 11- and 12-hydroxy isomers. The structures of the metabolic products were confirmed by HPLC , mass and 1H NMR spectroscopic comparison with authentic synthetic standards. Rutaecarpine has been found to protect rats against ulceration of gastric mucosa by acetylsalicylic acid (aspirin),90 while the related alkaloid dihydroevodiamine217 and various synthetic derivatives of rutaecarpine, notably the analogues 218, 219 and 220, were effective inhibitors of acetylcholinesterase and butyrylcholinesterase and thus possibly useful in antiamnesiac therapy.91
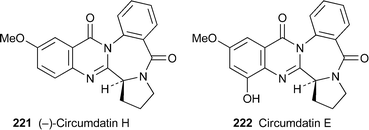
All of the remaining new quinazoline alkaloids in this year's crop were isolated from fungal sources. (–)-Circumdatin H221, the newest member of the growing family of circumdatins, was isolated from the culture broth of the fungus Aspergillus ochraceus and characterised by spectroscopic means.92 The absolute stereochemistry at the sole stereogenic centre, which is probably derived from L-proline, was tentatively assigned by comparing the sign of its specific rotation with that of related alkaloids such as circumdatin E222. Both 221 and 222 were able to inhibit the mitochondrial respiratory chain in submitochondrial particles from beef heart, presumably by interfering with NADH oxidase activity (IC50 1.5 and 2.5 µM, respectively).
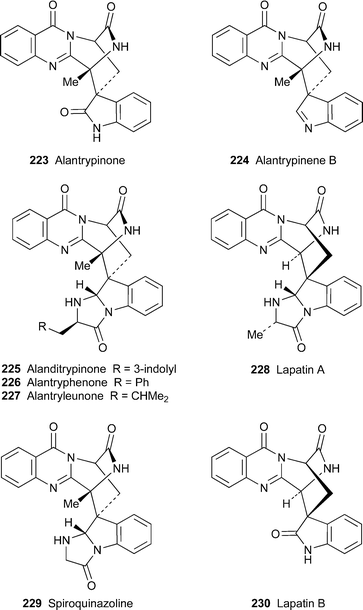
Four new spiroquinazoline alkaloids isolated from an endophytic fungus Eupenicillium sp. found in healthy leaves of the rutaceous plant Murraya paniculata proved to be analogues of the known fungal metabolite alantrypinone223.93 They include the indoline variant (–)-alantrypinene B224 and the three related alkaloids alanditrypinone225, (–)-alantryphenone226 and (–)-alantryleunone227, which appear to be condensation products of alantrypinone with tryptophan, phenylalanine and leucine, respectively. The structures were elucidated with the aid of extensive spectroscopic studies, including two-dimensional NMR spectroscopic and MS-MS experiments. Since plants of the genus Murraya, but not M. paniculata itself, are in general efficient producers of indole and carbazole alkaloids , the authors speculate that the endophytic fungus in the host plant may suppress the biosynthesis of alkaloids by lowering the availability of anthranilate.
Another two novel spiroquinazoline alkaloids were tracked in extracts of Penicillium lapatayae by means of ‘X-hitting’, a mathematical algorithm for the automatic comparison and dereplication of UV spectra of complex mixtures of natural products.94 By comparing the UV spectrum of the reference compound alantrypinone223 with the spectra of suspected quinazolines detected by HPLC in the fungal extract , two components having UV spectra similar to that of 223 were identified as candidates for isolation by preparative HPLC and structural elucidation. Detailed spectroscopic investigations, including NOE and HMBC correlations, showed that the compounds were indeed analogues of known alkaloids . (+)-Lapatin A228 turned out to be a structural isomer of spiroquinazoline229, the difference lying in the placement of a methyl substituent, while (–)-lapatin B230 was found to be a demethylated alantrypinone, possibly reflecting its biogenesis from glycine rather than alanine. However, both new alkaloids gave CD spectra with Cotton effects that were the opposite of those in alantrypinone. Since the absolute configuration of alantrypinone has been established by X-ray crystallography, the implication is that the relative positioning of the quinazolinone and tryptophan-derived chromophores must be reversed in the new alkaloids . Their absolute configurations are thus assumed to be as illustrated in 228 and 230.
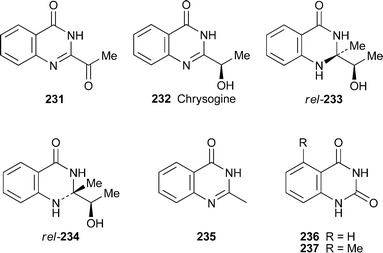
The known alkaloids 2-acetylquinazolin-4(3H)-one231 and chrysogine232 have been isolated from a new Penicillium species, P. persicinum sp. nov., obtained from a soil sample collected in Qinghai Province, China,95 and detected in various strains of the ascomycete Fusarium langsethiae and one strain of F. sporotrichioides.96 The production of chrysogine in Penicillium notatum strain KL1 could be boosted by culturing in the presence of the antibiotic strobilurin A.97 The two new alkaloids 233 and 234, isolated from the fermentation broth of Streptomyces strain GW23/1540,98 are structurally similar to chrysogine but bear an additional methyl substituent at C-2; as cyclic aminals , they are presumably diastereomeric at C-2 rather than in the side chain. Although both showed optical activity, attempts to determine their absolute configurations by analysis of Mosher esters were unsuccessful. Also obtained from the same source were the known alkaloids 2-methylquinazolin-4(3H)-one235 and quinazoline-2,4-dione236. The former has previously been isolated from other bacteria, while the latter has hitherto been found only in plants. A different Streptomyces strain, coded as GW2/577, produced a methylated quinazoline-2,4-dione; but since it was not possible to distinguish between the 5-methyl and 7-methyl isomers on the basis of available spectroscopic data, a sample of the latter was prepared by condensing 3-methylanthranilic acid with urea. The synthetic compound did not correlate with the natural product, which was thus assumed to be the 5-methyl isomer 237.
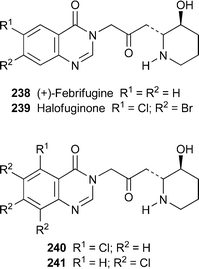
Although the potent antimalarial alkaloid febrifugine238, first reported in the late 1940s, has never been used clinically because it causes severe gastrointestinal irritation and induces emesis, renewed interest in the alkaloid has led to a search for synthetic variants with comparable efficacy but diminished toxicity. A suite of analogues first prepared at a military research laboratory in the 1960s, briefly tested at the time and then put into storage has recently been re-examined with the aid of more sophisticated testing methods.99 Out of ten analogues—all bearing halogen or methylenedioxy substituents on the aromatic ring—that displayed impressive in vitro antimalarial activity in tests with chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum, the clear winner proved to be the known coccidiostat halofuginone239 (IC50 0.12 and 0.145 ng ml–1, respectively; cf. 0.34 and 0.53 ng ml–1 for febrifugine, and less than 30 ng ml–1 for all but one of the remaining analogues). Furthermore, the host macrophages (J744 cells) for the parasites were 50 to 100 times less sensitive to the analogues than the parasites were, while the mammalian neuronal cell line NG108 was even more robust, all of which bode well for establishing a good therapeutic index for the compounds. The four most active compounds 238–241 were also tested in vivo in mice infected with the murine parasite P. berghei, and succeeded in reducing parasitaemia to undetectable levels, or apparently even curing some animals, when administered either orally or by subcutaneous injection at finely-balanced sub-lethal dosages. Intriguingly, gastrointestinal irritation was not observed when the compounds were injected, which suggests the approach to follow for future clinical testing of febrifugine analogues. However, parasite infestation recurred after cessation of treatment; and although most of the infected animals died, those that had apparently been cured succeeded in eliminating the parasites after several days and recovered completely. While the authors refreshingly admit that they are ‘absolutely clueless' about how the mice survived the recrudescence of the infestation, the observation is quite encouraging. The recommendation is that halofuginone be subjected to intensive testing as a potential new antimalarial drug, especially in view of the current interest in the compound for a variety of other conditions, including the control of diseases involved in excessive collagen synthesis, scleroderma and cancer therapy (cf.ref. 23f). On this score, several recent articles on the antifibrotic effects of halofuginone should be noted,100–103 as well as a study on its ability to suppress angiogenesis and metastasis in rat brain tumours.104
2.2 Synthesis and other chemical studies
The Eguchi protocol—a synthetic procedure involving tandem Staudinger and intramolecular aza-Wittig reactions on precursors containing aryl azide and amide functionalities—has become a powerful tool for the synthesis of quinazolin-4(3H)-one alkaloids and other cyclic imines. Eguchi himself has now comprehensively reviewed the process, the versatility of which is demonstrated by the inclusion of many examples illustrating applications in the total synthesis of natural products and various bioactive heterocycles.105 The procedure, traditionally carried out in solution, has recently been modified to proceed on solid supports, as shown in the synthesis of deoxyvasicinone209 (= 242, R1, R2 = H; n = 1) and two homologues by Gil and Bräse106 (Scheme 12). The polymer-bound anthranilate 243, which contains a triazene linker, was converted in situ into the acid chloride, which was then coupled with lactams 244 to give the imides 245. The resins were cleaved with trifluoroacetic acid, after which treatment with trimethylsilyl azide produced the liberated intermediates 246 in modest yield. Treating these aryl azides with phosphine-bound polystyrene at elevated temperature gave the target compounds 242 (R1, R2 = H; n = 1–3) in 99% yield. A more conventional solution-phase Eguchi protocol, also shown in Scheme 12, was used for making several substituted deoxyvasicinones from the 2-azidobenzoyl chlorides 247, the ring closure in this case being accomplished with sodium iodide in acetic acid at reflux rather than with the customary phosphines.107Deoxyvasicinone itself has been acylated with N,N-dimethylformamide under Vilsmeier–Haack conditions to give the enol 248, which could be alkylated under basic conditions with concomitant loss of the formyl group to afford the α-methyl or α-benzyl products 249.108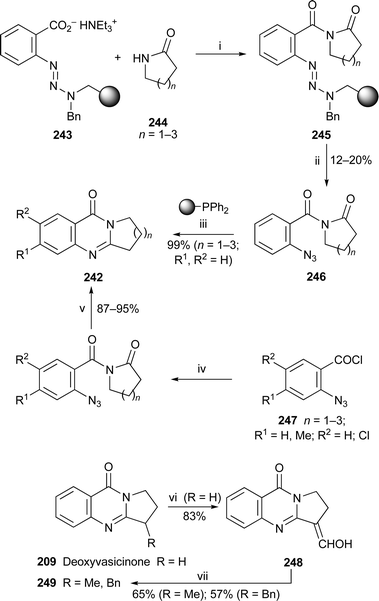 | ||
| Scheme 12 Reagents and conditions: i, resin 243 (1 equiv.), PPh3 (3 equiv.), lactam 244 (2 equiv.), EtNPri2 (4 equiv.), CCl4 (3 equiv.), THF, reflux, 5 h; ii, TFA in CH2Cl2 (5%), rt, 5–10 min, then Me3SiN3; iii, polystyrene–phosphine resin (4 equiv.), toluene, rt, 1 h, then 100 °C, 132 h; iv, lactam 244, NEt3, THF, rt; v, NaI, AcOH, reflux, 3 h; vi, DMF, POCl3, rt, overnight, then 95 °C, 2 h; vii, EtOH, NaOH, 30 min, then MeI or BnCl, 75–80 °C, 6 h. | ||
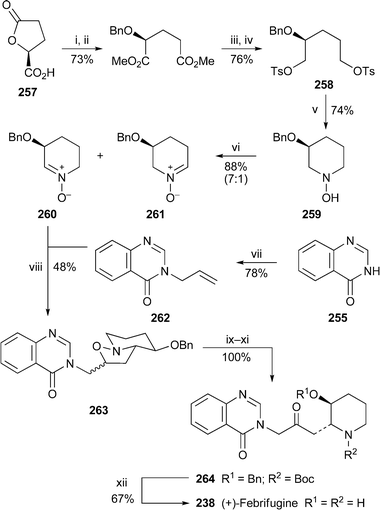
Two total syntheses of (+)-febrifugine238 were reported during the period under review. That by Honda and co-workers109 began with 250, a derivative of commercially available (4R)-hydroxy-L-proline, which was converted into the substituted pyrrolidine 251 in six steps (Scheme 13). The stereochemistry of intermediate 252 was confirmed by single crystal X-ray analysis of its N-deprotected derivative. However, the novelty in the route lies in the reductive deamination of 251 with samarium diiodide in the presence of methanol as a proton source, the intermediate amino ester then recyclising to produce the piperidinone 253 in high yield. In the final stages of the synthesis, the silyl enol ether prepared in situ from the methyl ketone 254 was brominated with N-bromosuccinimide, and the resulting α-bromoketone was coupled with quinazolin-4(3H)-one 255 to give the protected febrifugine derivative 256. Deprotection with aqueous hydrochloric acid then afforded (+)-febrifugine238. The short convergent route by Ashoorzadeh and Caprio110 (Scheme 14) began with the chiral lactone 257, derived from L-glutamic acid, from which the bis(tosylate) 258 was prepared in a further four steps. Reaction with hydroxylamine produced the N-hydroxypiperidine 259, oxidation of which with manganese dioxide gave the stable, readily separated chiral nitrones 260 and 261. The former underwent 1,3-dipolar cycloaddition with 3-allylquinazolin-4-one262 to give adduct 263 as the major product. Cleavage of the oxazolidine ring with zinc and acetic acid, protection of the free secondary amine and oxidation of the alcohol to the ketone quantitatively gave the febrifugine derivative 264, which was converted into the target alkaloid by removal of the protecting groups with aqueous hydrochloric acid.
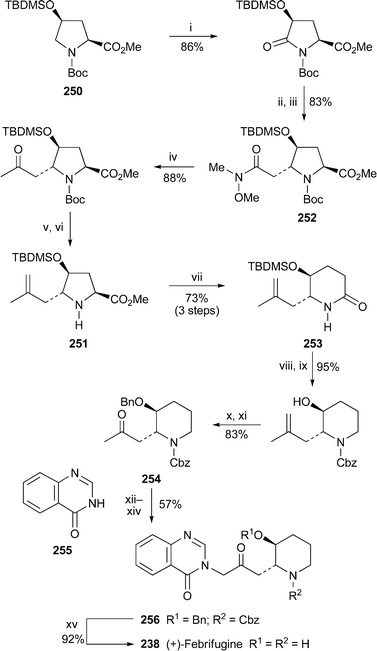 | ||
| Scheme 13 Reagents and conditions: i, RuO2 (cat.), NaIO4, EtOAc–H2O, rt; ii, LiEt3BH, THF, –78 °C; iii, (EtO)2P(O)CH2CON(Me)OMe, NaH, THF, rt; iv, MeMgBr, THF, 0 °C; v, Tebbe reagent, THF, –40 °C to rt; vi, ZnBr2, CH2Cl2, rt; vii, SmI2, THF–HMPA, MeOH, 0 °C to rt; viii, LiAlH4, THF, 65 °C; ix, ClCO2Bn, NEt3, DMAP, CH2Cl2, rt; x, BnBr, NaH, DMF, 0 °C; xi, O3, MeOH, –78 °C, then Me2S; xii, TMSOTf, EtNPri2, CH2Cl2; xiii, NBS, rt; xiv, 255, KH, DMF, 70 °C; xv, aq. HCl (6 M), reflux. | ||
 | ||
| Scheme 14
Reagents and conditions: i, conc. HCl, MeOH, reflux, 12 h; ii, BnBr, Ag2O, EtOAc, rt, 48 h; iii, LiAlH4, Et2O, 24 h; iv, p-TsCl, NEt3, DMAP, CH2Cl2, rt, 12 h; v, NH2OH·HCl, NEt3, reflux, 4 h; vi, MnO2, CH2Cl2, 0 °C, 12 h; vii, H2CCHCH2Br, NaOMe, MeOH, rt, 12 h; viii, PhMe, reflux, 24 h; ix, Zn, AcOH, reflux; x, (Boc)2O, NEt3, CH2Cl2, xi, Dess–Martin periodinane, py, CH2Cl2; xii, aq. HCl (6 M), reflux. | ||
The remarkable cytotoxicity displayed by luotonin A265 towards murine leukemia P-388 cells even at low concentrations has prompted several groups to devise new synthetic approaches to the alkaloid and its analogues. Recognising that a strategy in which the central five-membered ring is assembled at a late stage might permit access to three different luotonins from a common intermediate, Chavan and Sivappa devised the short synthesis shown in Scheme 15.111 Condensing anthranilamide 266 with the known quinoline-2-aldehyde 267 under mild conditions produced the dihydroquinazolinone 268, oxidation of which with potassium permanganate gave key intermediate 269 in an overall yield of 76%. The same compound could also be obtained directly if the condensation was performed under more vigorous conditions. Hydrolysis of the acetal in 269 and reduction of the resulting aldehyde then gave the alcohol 270 which could be cyclised to luotonin A either under Mitsunobu conditions or, more conveniently, by treatment with sulfuric acid in ethanol. Alternatively, treating 265 with aqueous acid completed a synthesis of luotonin B271, while acidic hydrolysis in methanol afforded luotonin E272.
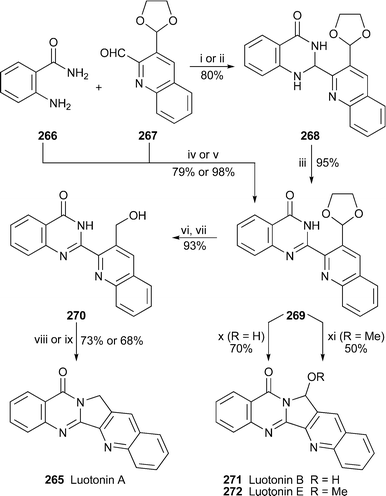 | ||
| Scheme 15 Reagents and conditions: i, aq. NaOH (15%), EtOH, reflux, 1 h; ii, MeCONMe2, 80 °C, 3 h; iii, KMnO4, Me2CO, reflux; iv, aq. KOH (20%), EtOH, reflux; v, NaHSO3, MeCONMe2, 150 °C, 2 h; vi, aq. HCl (10%)–THF (1 : 1), rt, 2 h; vii, NaBH4, MeOH, rt, 1 h; viii, H2SO4 in EtOH (60%), 115 °C, 3 h; ix, PPh3, DEAD, THF, rt, 2 h; x, aq. HCl (60%), rt, 1 h, then reflux, 1 h; xi, conc. HCl, MeOH, rt, 1 h, then reflux, 2 h. | ||
Two short syntheses of luotonin A265, although not especially efficient, included steps in which the alkaloid's pentacyclic core was constructed by interesting new procedures (Scheme 16). In the approach by Bowman and co-workers,112 cyclisation of the vinyl iodide 273, made in two steps from ethyl anthranilate, with various radical initiators yielded luotonin A, presumably via the vinyl and iminyl radicals 274 and 275, as well as deiodinated products. In the approach by Twin and Batey, the 2-formylquinazolinone 276, prepared in six steps from isatoic anhydride, was converted directly into luotonin A in 51% yield when condensed with aniline in the presence of dysprosium(III) triflate.113 The reaction presumably proceeds by way of Lewis acid-catalysed intramolecular hetero Diels–Alder (Povarov) reaction of the imine intermediate 277.
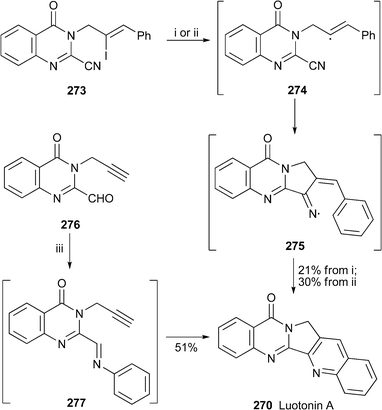 | ||
| Scheme 16 Reagents and conditions: i, (Me3Sn)2 (14 equiv.), PhBut, sun lamp irradiation (300 W), 150 °C, 46 h; ii, (Me3Sn)2 (3.8 equiv.), (ButO)2 (2.0 equiv.), PhBut, 120 °C, 24 h; iii, Dy(OTf)3 (10 mol%), PhNH2, MeCN, rt, 24 h. | ||
Several previously communicated syntheses of the luotonins have recently been reported either with full experimental details or with extensions that include the preparation of substituted analogues. Harayama's route114 (cf.ref. 23g), entailing the intramolecular palladium-assisted biaryl coupling of 278, has been optimised;115luotonin A was obtained in 86% yield when using precisely specified quantities of palladium(II) acetate (0.1 equivalents), tricyclohexylphosphine and potassium acetate (2 equivalents each) in N,N-dimethylformamide at reflux. A similar reaction with the indole-containing precursor 279 gave rutaecarpine215 in 24% yield at best; but with the N-acetyl precursor 280, the yield of rutaecarpine was improved to 89%. Experimental details for the oxidation of luotonin A265 to luotonin B271 were also reported. The action of luotonin A as a topoisomerase poison was first demonstrated by Hecht and co-workers,116 who also synthesised several ring E-modified analogues in modest yield by condensing the tricyclic lactam 281 with methyl anthranilates in the presence of phosphorus oxychloride in dichloromethane117 (cf.ref. 23h); the new follow-up publication includes full synthetic details for a larger range of novel synthetic luotonin analogues 282 and 283, as well as cytotoxicity and topoisomerase inhibition studies for these compounds.118 Among the new products were the interesting isoluotonins 284, which were the major products when the syntheses were performed in tetrahydrofuran. The most active analogues for stabilising the topoisomerase I–DNA covalent binary complex were 282 (R = 8-NH2, F, CH2OH), the thiophene 283 and its isoluotonin analogue, and isoluotonin A284 (R = H) itself; while luotonin A, isoluotonin A and the amino derivative 282 (R = 8-NH2) showed notable topoisomerase I-dependent cytotoxicity towards Saccharomyces cerevisiae. Dallavalle's preparation of luotonin A from the same tricyclic lactam 281119 (cf.ref. 23i) has been extended to include various substituted anthranilic acids or isatoic anhydrides as reaction partners, thereby giving a range of ring E-substituted analogues 285 for cytotoxicity studies.120 In addition, two ring A-substituted derivatives 286 were prepared by Friedländer condensation of 287 with 2-aminobenzaldehydes. All of the products showed cytotoxicity towards the human non-small lung carcinoma cell line H460, a feature related to their ability to stimulate topoisomerase I-mediated cleavage of DNA.
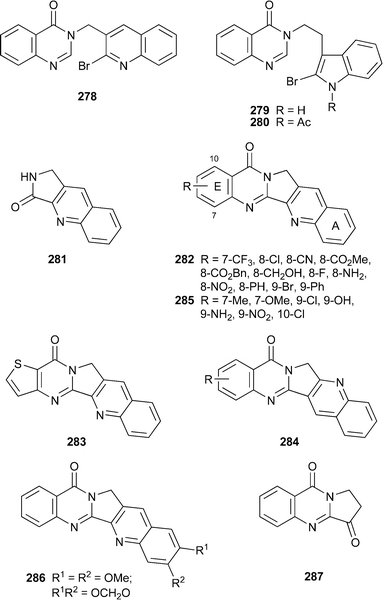
Miscellaneous publications of interest include a short synthesis of the two simple quinazoline-2,4-dione alkaloids 288 (R = H, OMe), originally isolated from seed husks of Zanthoxylum arborescens, from isatoic anhydride and the corresponding 2-phenylethylamines.121 The stereochemistry of C-1 alkylation of dilithiated azaenolates derived from the tricyclic precursors 289 has been studied as part of an ongoing investigation into the synthesis of various pyrazino[2,1-b]quinazoline alkaloids .122 A microwave-assisted three-component coupling of tryptanthrin212 with diethyl acetylenedicarboxylate and cyclohexyl isocyanide on montmorillonite K10 as solid support gave the spiroiminolactones 290 in moderate yield (64–68%) within a matter of minutes.123 Arylimines prepared from tryptanthrin have also been shown to undergo 1,3-dipolar cycloaddition with nitrile oxides to give spirocyclic oxadiazoles 291 in yields of 76–89%.124
3 Acridone alkaloids
Known acridone alkaloids reported from novel sources during the period covered by this review include 1-hydroxy-3-methoxy-N-methylacridone292 from Almeidea rubra4 and Glycosmis arborea,10 and normelicopicine293 from Sarcomelicope follicularis.14Alkaloid 292 showed modest trypanocidal activity against Trypanosoma cruzi;4 while the related alkaloid arborinine294 was an effective inhibitor of the induction of Epstein–Barr virus early antigen (EBV-EA) in Raji cells, and thus potentially useful as an antitumour promoter.10Two new acridone alkaloids isolated from the roots of the Japanese shrub Boenninghausenia japonica were identified by means of spectroscopic methods as 1,3-dihydroxy-4-[(Z)-3-hydroxy-3-methylbut-1-enyl]-N-methylacridone295 and (–)-1,3-dihydroxy-4-(2-hydroxy-3-hydroxymethyl-3,4-epoxybutyl)-N-methylacridone296.125 The unusual (Z)-alkene side chain in the former was deduced from the size of the coupling constant (J 10 Hz) for the vinyl hydrogen atoms, while HMBC correlations were needed to elucidate the structure of the side chain in the latter, although neither the relative nor the absolute configuration could be determined. Alkaloid 296 showed high antiproliferative activity against human gastric carcinoma (MK-1), human uterus carcinoma (HeLa) and murine melanoma (B16F10) cells (GI50 0.056, 0.056 and 1.76 µM, respectively).
The chemotherapeutic properties of acridone alkaloids and their synthetic analogues continue to attract interest. The antitumour alkaloid glyfoline297 was found to induce apoptotic changes and arrest cell cycle progression at the G2/M phase in nasopharyngeal carcinoma by assisting the leakage of cytochrome C from mitochondria into the cytosol.126 Benzo[b]fused analogues of the antitumour alkaloid acronycine298 have been extensively studied by Tillequin and co-workers in recent years; some of their results have now been summarised in short reviews that also highlight the impressive antitumour spectrum of the cis-diacetoxy derivative 299 and the putative mode of action of related esters.127–129 The covalent binding of 299 and related compounds to double-stranded DNA has been found to induce helix opening and the formation of single-stranded DNA—apparently a novel DNA-bonding mechanism.130 Structure–activity relationships for some of these benzannelated acronycine derivatives and related acridones have been evaluated with the aid of a computational model, which showed that electrostatic descriptors provided the best correlations for the observed biological activity.131
In a new departure in the chemistry and biology of acridone alkaloid derivatives, several acronycine-inspired thioacridones 300 were synthesised for evaluation as antitumour agents.132 All showed in vitro activity against HL-60 cells (IC50 2.3–22 µg ml–1), with compound 301 being the most active. In addition, the C-1 amines were found to bind to DNA, probably by intercalation, with most binding constants approximately 102 M–1 and that for 301 being substantially greater (2.6 × 104 M–1). The thioacridones were also tested for antimalarial activity against a chloroquine-sensitive strain of the malaria parasite Plasmodium falciparum; all compounds showed useful antiplasmodial activity (IC50 0.4–27 µg ml–1), with compound 301—clearly a promising lead for future drug design —not only being the most potent once again, but also being effective against a chloroquine-resistant strain of P. falciparum.133
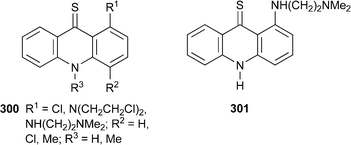
The unique azaacridone alkaloid 302, prosaically named azaacridone A, has been synthesised by Scopton and Kelly by the route shown in Scheme 17.134 Annulation of phloroglucinol303 with dimethylacrylic acid304 followed by selective protection of the non-hydrogen bonded phenol as the methoxymethyl ether afforded the chromanone 305, which was further elaborated to the chromene 306 by methylation of the remaining phenol and conversion of the ketone into an alkene . Directed ortho-lithiation of this intermediate at the desired C-6 position proved troublesome. With n-butyllithium in diethyl ether, metallation occurred mainly at C-8 rather than at C-6 (49% vs. 17%, as determined after quenching with deuterium oxide); in the presence of HMPA, however, the intermediate 307 was generated predominantly (70% vs. 17%, again after analysis of deuteriated products). Formylation of 307, accomplished with iron pentacarbonyl, gave the aldehyde 308 in 53% yield together with a quantity of the C-8 isomer (18%). Addition of the bis-lithiated pyridine 309 followed by oxidation of the resulting alcohol produced the diaryl ketone 310, the N-methyl derivative of which was then treated with trifluoroacetic acid. Under these conditions, the acid-labile protecting groups were removed, and cyclisation (possibly via the enone tautomer of the phenol) gave azaacridone A302 directly. The spectroscopic data were in agreement with those reported for the natural product, thus confirming the original structural assignment of this unusual alkaloid . It is of interest that when the methoxymethyl and methyl protecting groups on the cyclisation precursor were interchanged, intermediate 311 cyclised regiospecifically to 312, the linear isomer of azaacridone A.
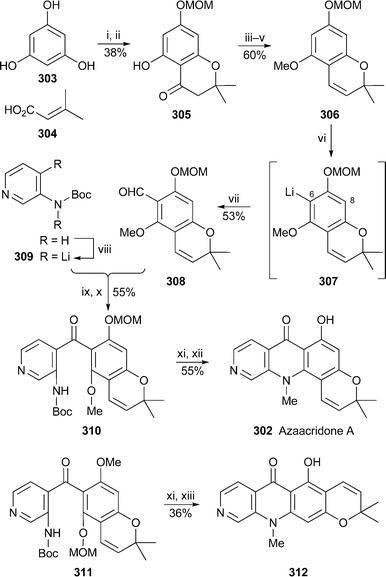 | ||
| Scheme 17 Reagents and conditions: i, 303 + 304, ZnCl2, POCl3, 50 °C, 4 h; ii, MOMCl, K2CO3, DMF, rt, 12 h; iii, MeI, K2CO3, DMF, 80 °C, 2.5 h; iv, NaBH4, MeOH, rt, 3 h; v, aq. HCl (1 M), THF, rt, 5 h; vi, n-BuLi, HMPA (2 equiv.), THF, 0 °C, 1.5 h; vii, Fe(CO)5, 0 °C, then rt, 2.5 h, then AcOH, rt, 15 min; viii, ButLi, THF, –78 °C, 2 h; ix, 308 + 309, –78 °C, 0.5 h, then rt, 1.5 h; x, IBX, DMSO, rt, 2 h; xi, MeI, NaH, DMF, 0 °C, 10 min, then rt, 3 h; xii, TFA, ClCH2CH2Cl, rt, 1 h, then reflux, 12 h; xiii, TFA, CH2Cl2, rt, overnight. | ||
4 Acknowledgements
This material is based upon work supported by the National Research Foundation under grant number 2053652. Financial support from the University of the Witwatersrand is also gratefully acknowledged.5 References
- K. Günaydin and S. Savci, Nat. Prod. Res., 2005, 19, 203 CrossRef.
- I.-S. Chen and C.-M. Teng, Front. Nat. Prod. Chem., 2005, 1, 201 Search PubMed.
- L. G. Rocha, J. R. G. S. Almeida, R. O. Macêdo and J. M. Barbosa-Filho, Phytomedicine, 2005, 12, 514 Search PubMed.
- A. R. P. Ambrozin, J. Mafezoli, P. C. Vieira, J. B. Fernandes, M. F. das G. F. da Silva, J. A. Ellena and S. de Albuquerque, J. Braz. Chem. Soc., 2005, 16, 434 CAS.
- A. N. García-Argáez, N. M. González-Lugo, H. Parra-Delgado and M. Martínez-Vázquez, Biochem. Syst. Ecol., 2005, 33, 441 CrossRef CAS.
- K.-W. Nam, K.-H. Je, Y.-J. Shin, S. S. Kang and W. Mar, Arch. Pharm. Res., 2005, 28, 675 Search PubMed.
- B. A. Barros-Filho, F. M. Nunes, M. C. F. de Oliveira, J. Mafezoli, M. Andrade-Neto, E. R. Silveira and J. R. Pirani, Biochem. Syst. Ecol., 2004, 32, 817 CrossRef CAS.
- F. M. Nunes, B. A. Barros-Filho, M. C. F. de Oliveira, J. Mafezoli, M. Andrade-Neto, M. C. de Mattos, E. R. Silveira and J. R. Pirani, Magn. Reson. Chem., 2005, 43, 180 CrossRef CAS.
- Y. Diao, Y. Gao and X. Peng, Zhongcaoyao, 2004, 35, 1098 ( Chem. Abstr. , 2006 , 144 , 134822 ) Search PubMed.
- C. Ito, M. Itoigawa, A. Sato, C. M. Hasan, M. A. Rashid, H. Tokuda, T. Mukainaka, H. Nishino and H. Furukawa, J. Nat. Prod., 2004, 67, 1488 CrossRef CAS.
- V. I. Akhmedzhanova, K. A. Rasulova, I. A. Bessonova, A. S. Shashkov, N. D. Abdullaev and L. Angenot, Chem. Nat. Compd., 2005, 41, 60 CrossRef CAS.
- X. Feng, Y. Dong, M. Wang and Y. Zhao, Zhongcaoyao, 2004, 35, 1336 ( Chem. Abstr. , 2006 , 144 , 11245 ) Search PubMed.
- C. Ito, M. Itoigawa, A. Furukawa, T. Hirano, T. Murata, N. Kaneda, Y. Hisada, K. Okuda and H. Furukawa, J. Nat. Prod., 2004, 67, 1800 CrossRef CAS.
- E. Chosson, A.-E. Hay, A. Chiaroni, A. Favel, F. Tillequin, M. Litaudon and E. Seguin, Heterocycles, 2004, 63, 2043 CrossRef CAS.
- N. Sultana, N. Afza and Atta-ur-Rahman, Sci. Int. (Lahore, Pak.), 2004, 16, 271 ( Chem. Abstr. , 2006 , 144 , 57041 ) Search PubMed.
- P. K. Tarus, P. H. Coombes, N. R. Crouch, D. A. Mulholland and B. Moodley, Phytochemistry, 2005, 66, 703 CrossRef CAS.
- M.-J. Cheng, C.-C. Wu, I.-L. Tsai and I.-S. Chen, J. Chin. Chem. Soc., 2004, 51, 1065 CAS.
- M. M. Rahman, M. A. Islam, P. Khondkar and A. I. Gray, Biochem. Syst. Ecol., 2005, 33, 91 CrossRef.
- V. A. Facundo, A. S. Pinto da Silveira, R. Braz Filho, A. C. Pinto and C. M. Rezende, Quim. Nova, 2004, 28, 224 ( Chem. Abstr. , 2005 , 143 , 129965 ).
- J.-J. Chen, H.-Y. Fang, C.-Y. Duh and I.-S. Chen, Planta Med., 2005, 71, 470 CrossRef CAS.
- D.-X. Li and Z.-D. Min, Zhongguo Tianran Yaowu, 2004, 2, 285 ( Chem. Abstr. , 2005 , 143 , 23012 ) Search PubMed.
- A. J. El-Rehaily, M. S. Ahmad, I. Muhammad, A. A. Al-Thukair and H. P. Perzanowski, Phytochemistry, 2003, 64, 1405 CrossRef.
- (a) J. P. Michael, Nat. Prod. Rep., 2005, 22, 627 RSC; (b) J. P. Michael, Nat. Prod. Rep., 1991, 8, 61 Search PubMed; (c) J. P. Michael, Nat. Prod. Rep., 2005, 22, 632 Search PubMed; (d) J. P. Michael, Nat. Prod. Rep., 2004, 21, 657 Search PubMed; (e) J. P. Michael, Nat. Prod. Rep., 1998, 15, 599 Search PubMed; (f) J. P. Michael, Nat. Prod. Rep., 2005, 22, 637 Search PubMed; (g) J. P. Michael, Nat. Prod. Rep., 2004, 21, 663 Search PubMed; (h) J. P. Michael, Nat. Prod. Rep., 2005, 22, 638 Search PubMed; (i) J. P. Michael, Nat. Prod. Rep., 2003, 20, 487 Search PubMed.
- J. L. Gaston and M. F. Grundon, J. Chem. Soc., Perkin Trans. 1, 1989, 905 RSC.
- R. Liu, X. Chu, A. Sun and L. Kong, J. Chromatogr., A, 2005, 1074, 139 CrossRef CAS.
- X. Luo, B. Chen and S. Yao, Talanta, 2005, 66, 103 CrossRef CAS.
- M. Adams, O. Kunert, E. Haslinger and R. Bauer, Planta Med., 2004, 70, 904 CrossRef CAS.
- H. Ohmiya, H. Yorimitsu and K. Oshima, Chem. Lett., 2004, 33, 1240 CrossRef CAS.
- R. Martínez, G. J. Brand, D. J. Ramón and M. Yus, Tetrahedron Lett., 2005, 46, 3683 CrossRef CAS.
- K. Motokura, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2004, 45, 6029 CrossRef CAS.
- D. Shi, L. Rong, C. Shi, Q. Zhuang, X. Wang, S. Tu and H. Hu, Synthesis, 2005, 717 CrossRef CAS.
- T. Demaude, L. Knerr and P. Pasau, J. Comb. Chem., 2004, 6, 768 CrossRef CAS.
- K. H. Kumar, D. Muralidharan and P. T. Perumal, Tetrahedron Lett., 2004, 45, 7903 CrossRef CAS.
- X.-F. Lin, Y. Li and D.-W. Ma, Chin. J. Chem., 2004, 22, 932 CAS.
- C. Theeraladanon, M. Arisawa, M. Nakagawa and A. Nishida, Tetrahedron: Asymmetry, 2005, 16, 827 CrossRef CAS.
- L. Xu, K. H. Lam, J. Ji, J. Wu, Q.-H. Fan, W.-H. Lo and A. S. C. Chan, Chem. Commun., 2005, 1390 RSC.
- S.-M. Lu, X.-W. Han and Y.-G. Zhou, Adv. Synth. Catal., 2004, 346, 909 CrossRef CAS.
- T. Suresh, T. Dhanabal, R. N. Kumar and P. S. Mohan, Heterocycl. Commun., 2005, 11, 79 CAS.
- B. Lal, N. B. Bhise, R. M. Gidwani, A. D. Lakdawala, K. Joshi and S. Patvardhan, ARKIVOC, 2005, ii, 77.
- U. Bhoga, R. S. Mali and S. R. Adapa, Tetrahedron Lett., 2004, 45, 9483 CrossRef CAS.
- H. M. R. Hoffmann and J. Frackenpohl, Eur. J. Org. Chem., 2004, 4293 CrossRef CAS.
- T. S. Kaufman and E. A. Rúveda, Angew. Chem., Int. Ed., 2005, 44, 854 CrossRef CAS.
- J. G. Díaz, J. G. Sazatornil, M. L. Rodríguez, L. R. Mesía and G. V. Arana, J. Nat. Prod., 2004, 67, 1667 CrossRef CAS.
- L. Ruiz-Mesía, W. Ruiz-Mesía, M. Reina, R. Martínez-Diaz, C. de Inés, A. Guadaño and A. González-Coloma, J. Agric. Food Chem., 2005, 53, 1921 CrossRef CAS.
- P. V. V. S. Sarma, D. Han, J. R. Deschamps and J. M. Cook, J. Nat. Prod., 2005, 68, 942 CrossRef CAS.
- Y.-F. Su, Y. Luo, C.-Y. Guo and D.-A. Guo, J. Asian Nat. Prod. Res., 2004, 6, 223 ( Chem. Abstr. , 2004 , 141 , 377086 ) Search PubMed.
- D. de N. M. Machado, S. F. Palmeria Júnior, L. M. Conserva and R. P. de Lyra Lemos, Biochem. Syst. Ecol., 2005, 33, 555 CrossRef CAS.
- S. C. S. M. Hoelzel, E. R. Vieira, S. R. Giacomelli, I. I. Dalcol, N. Zanatta and A. F. Morel, Phytochemistry, 2005, 66, 1163 CrossRef CAS.
- G. J. Kapadia, B. D. Paul, J. V. Silverton, H. M. Fales and E. A. Sokoloski, J. Am. Chem. Soc., 1975, 79, 6814 CrossRef.
- X. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752 RSC.
- H. Morita, Y. Hirasawa, T. Shinzato and J. Kobayashi, Tetrahedron, 2004, 60, 7015 CrossRef CAS.
- Y. Hirasawa, H. Morita and J. Kobayashi, Heterocycles, 2004, 64, 515 CrossRef CAS.
- M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685 CrossRef CAS.
- (a) K. C. Nicolaou, B. S. Safina, M. Zak, A. A. Estrada and S. H. Lee, Angew. Chem., Int. Ed., 2004, 43, 5087 CrossRef CAS; (b) K. C. Nicolaou, M. Zak, B. S. Safina, S. H. Lee and A. A. Estrada, Angew. Chem., Int. Ed., 2004, 43, 5092 CrossRef CAS.
- F. Bredenbruch, M. Nimtz, V. Wray, M. Morr, R. Müller and S. Hässler, J. Bacteriol., 2005, 187, 3630 CrossRef CAS.
- A. du Moulinet d'Hardemare, G. Serratrice and J.-L. Pierre, BioMetals, 2004, 17, 691 Search PubMed.
- S. Sonjak, J. C. Frisvad and N. Gunde-Cimerman, FEMS Microbiol. Ecol., 2005, 53, 51 CrossRef CAS.
- G. Schmeda-Hirschmann, E. Hormazabal, L. Astudillo, J. Rodriguez and C. Theoduloz, World J. Microbiol. Biotechnol., 2005, 21, 27 CrossRef CAS.
- M. Abe, T. Imai, N. Ishii, M. Usui, T. Okuda and T. Oki, Biosci., Biotechnol., Biochem., 2005, 69, 1202 CrossRef CAS.
- W.-G. Kim, N.-K. Song and I.-D. Yoo, J. Antibiot., 2001, 54, 831 CAS.
- X. Zhang, W. Jiang and Z. Sui, J. Org. Chem., 2003, 68, 4523 CrossRef CAS.
- S.-J. Park, K.-N. Cho, W.-G. Kim and K.-I. Lee, Tetrahedron Lett., 2004, 45, 8793 CrossRef CAS.
- T. G. Back and J. E. Wulff, Angew. Chem., Int. Ed., 2004, 43, 6493 CrossRef CAS.
- Y. Morimoto and H. Shirahama, Tetrahedron, 1996, 52, 10631 CrossRef CAS.
- M. Ori, N. Toda, K. Takami, K. Tago and H. Kogen, Angew. Chem., Int. Ed., 2003, 42, 2540 CrossRef CAS.
- M. Ori, N. Toda, K. Takami, K. Tago and H. Kogen, Tetrahedron, 2005, 61, 2075 CrossRef CAS.
- C. L. Francis, N. M. Williamson and A. D. Ward, Synthesis, 2004, 2685 CAS ; also see: N. M. Williamson and A. D. Ward, Tetrahedron, 2005, 61, 155 Search PubMed.
- H. Beiderbeck, K. Taraz, H. Budzikiewicz and A. E. Walsby, Z. Naturforsch., C, 2000, 55c, 681.
- Y. Itou, S. Okada and M. Murakami, Tetrahedron, 2001, 57, 9093 CrossRef CAS.
- Y. Ito, K. Ishida, S. Okada and M. Murakami, Tetrahedron, 2004, 60, 9075 CrossRef CAS.
- K. Gademann and Y. Bethuel, Angew. Chem., Int. Ed., 2004, 43, 3327 CrossRef CAS.
- K. Gademann and Y. Bethuel, Org. Lett., 2004, 6, 4707 CrossRef CAS.
- N. Girard, J.-P. Hurvois, C. Moinet and L. Toupet, Eur. J. Org. Chem., 2005, 2269 CrossRef CAS.
- E. W. Dijk, L. Panella, P. Pinho, R. Naasz, A. Meetsma, A. J. Minnaard and B. L. Feringa, Tetrahedron, 2004, 60, 9687 CrossRef CAS.
- J. Neidhöfer and S. Blechert, Synthesis, 2004, 3047.
- N. Holub, J. Neidhöfer and S. Blechert, Org. Lett., 2005, 7, 1227 CrossRef CAS.
- H. Tsuneki, Y. You, N. Toyooka, T. Sasaoka, H. Nemoto, J. A. Dani and I. Kimura, Biol. Pharm. Bull., 2005, 28, 611 CrossRef CAS.
- X. Pu and D. Ma, Angew. Chem., Int. Ed., 2004, 43, 4222 CrossRef CAS.
- C. Das, R. Poi and A. Chowdhury, Phytochem. Anal., 2005, 16, 90 CrossRef CAS.
- R. Zayed and M. Wink, Z. Naturforsch., C, 2005, 60c, 451.
- S. Illgner and R. Matusch, Arch. Pharm. Chem. Life Sci., 2005, 338, 49 Search PubMed.
- D. S. Samiulla, V. V. Vaidyanathan, P. C. Arun, G. Balan, M. Blaze, S. Bondre, G. Chandrasekhar, A. Gadakh, R. Kumar, G. Kharvi, H.-O. Kim, S. Kumar, J. A. Malikayil, M. Moger, M. K. Mone, P. Nagarjuna, C. Ogbu, D. Pendhalkar, A. V. S. Raja Rao, G. Venkateshwar Rao, V. K. Sarma, S. Shaik, G. V. R. Sharma, S. Singh, C. Sreedhar, R. Sonawane, U. Timmanna and L. W. Hardy, Mol. Diversity, 2005, 9, 131 Search PubMed.
- R. Anderskewitz, R. Bauer, G. Bodenbach, D. Gester, B. Gramlich, G. Morschhäuser and F. W. Birke, Bioorg. Med. Chem. Lett., 2005, 15, 669 CrossRef CAS.
- C. Oberthür, R. Jäggi and M. Hamburger, Fitoterapia, 2005, 76, 324 CrossRef CAS.
- C. Oberthür and M. Hamburger, Planta Med., 2004, 70, 642 CrossRef.
- C. Oberthür, H. Graf and M. Hamburger, Phytochemistry, 2004, 65, 3261 CrossRef.
- T. Motoki, Y. Takami, Y. Yagi, A. Tai, I. Yamamoto and E. Gohda, Biol. Pharm. Bull., 2005, 28, 260 CrossRef CAS.
- I. Wagner-Döbler, H. Rheims, A. Felske, A. El-Ghezal, D. Flade-Schröder, H. Laatsch, S. Lang, R. Pukall and B. J. Tindall, Int. J. Syst. Evol. Microbiol., 2004, 54, 1177 Search PubMed.
- Y.-F. Ueng, H.-J. Yu, C.-H. Lee, C. Peng, W.-C. Jan, L.-K. Ho, C. F. Chen and M.-J. Don, J. Chromatogr., A, 2005, 1076, 103 CrossRef CAS.
- L. Wang, C.-P. Hu, P.-Y. Deng, S.-S. Shen, H.-Q. Zhu, J.-S. Ding, G.-S. Tan and Y.-J. Li, Planta Med., 2005, 71, 416 CrossRef CAS.
- M. Decker, Eur. J. Med. Chem., 2005, 40, 305 CrossRef CAS.
- M. P. López-Gresa, M. C. González, J. Primo, P. Moya, V. Romero and E. Estornell, J. Antibiot., 2005, 58, 416 CrossRef CAS.
- F. A. Proença Barros and E. Rodrigues-Filho, Biochem. Syst. Ecol., 2005, 33, 257 CrossRef CAS.
- T. O. Larsen, B. O. Petersen, J. Ø. Duus, D. Sørensen, J. C. Frisvad and M. E. Hansen, J. Nat. Prod., 2005, 68, 871 CrossRef CAS ; also see: M. E. Hansen, J. Smedsgaard and T. O. Larsen, Anal. Chem., 2005, 77, 6805 Search PubMed.
- L. Wang, H.-B. Zhou, J. C. Frisvad and R. A. Samson, Antonie van Leeuwenhoek, 2004, 86, 173 CrossRef CAS.
- U. Thrane, A. Adler, P.-E. Clasen, F. Galvano, W. Langseth, H. Lew, A. Logrieco, K. F. Nielsen and A. Ritieni, Int. J. Food Microbiol., 2004, 95, 257 CrossRef CAS.
- M. Kettering, O. Sterner and T. Anke, Z. Naturforsch., C, 2004, 59c, 816.
- R. P. Maskey, M. Shaaban, I. Grün-Wollny and H. Laatsch, J. Nat. Prod., 2004, 67, 1131 CrossRef CAS.
- S. Jiang, Q. Zeng, M. Gettayacamin, A. Tungtaeng, S. Wannaying, A. Lim, P. Hansukjariya, C. O. Okunji, S. Zhu and D. Fang, Antimicrob. Agents Chemother., 2005, 49, 1169 CrossRef CAS.
- R. A. Nowak, Drug Discovery Today, 2004, 1, 237 CAS.
- S. L. Young, A. Al-Hendy and J. A. Copland, Semin. Reprod. Med., 2004, 22, 121 CrossRef CAS.
- M. Van de Casteele, T. Roskams, I. Van der Elst, J. F. van Pelt, J. Fevery and F. Nevens, Liver Int., 2004, 24, 502 Search PubMed.
- D. C. Marchion, P. A. Weber, L. Lomnitski, C. M. G. Duran and D. T. Cheung, Tissue Eng., 2004, 10, 1076 CAS.
- R. Abramovitch, A. Itzik, H. Harel, A. Nagler, I. Vlodavsky and T. Siegal, Neoplasia, 2004, 6, 480 CrossRef CAS.
- S. Eguchi, ARKIVOC, 2005, ii, 98.
- C. Gil and S. Bräse, Chem.–Eur. J., 2005, 11, 2680 CrossRef.
- A. Kamal, A. V. Ramana, K. S. Reddy, K. V. Ramana, A. H. Babu and B. R. Prasad, Tetrahedron Lett., 2004, 45, 8187 CrossRef CAS.
- B. Z. Elmuradov and K. M. Shakhidoyatov, Chem. Nat. Compd., 2004, 40, 496 CrossRef CAS.
- M. Katoh, R. Matsune, H. Nagase and T. Honda, Tetrahedron Lett., 2004, 45, 6221 CrossRef CAS.
- A. Ashoorzadeh and V. Caprio, Synlett, 2005, 346 CAS.
- S. P. Chavan and R. Sivappa, Tetrahedron, 2004, 60, 9931 CrossRef CAS.
- W. R. Bowman, M. O. Cloonan, A. J. Fletcher and T. Stein, Org. Biomol. Chem., 2005, 3, 1460 RSC.
- H. Twin and R. A. Batey, Org. Lett., 2004, 6, 4913 CrossRef CAS.
- T. Harayama, Y. Morikami, Y. Shigeta, H. Abe and Y. Takeuchi, Synlett, 2003, 847 CrossRef CAS.
- T. Harayama, A. Hori, G. Serban, Y. Morikami, T. Matsumoto, H. Abe and Y. Takeuchi, Tetrahedron, 2004, 60, 10645 CrossRef CAS.
- A. Cagir, S. H. Jones, R. Gao, B. M. Eisenhauer and S. M. Hecht, J. Am. Chem. Soc., 2003, 125, 13628 CrossRef CAS.
- A. Cagir, S. H. Jones, B. M. Eisenhauer, R. Gao and S. M. Hecht, Bioorg. Med. Chem. Lett., 2004, 14, 2051 CrossRef CAS.
- A. Cagir, B. M. Eisenhauer, R. Gao, S. J. Thomas and S. M. Hecht, Bioorg. Med. Chem., 2004, 12, 6287 CrossRef CAS.
- S. Dallavalle and L. Merlini, Tetrahedron Lett., 2002, 43, 1835 CrossRef CAS.
- S. Dallavalle, L. Merlini, G. L. Beretta, S. Tinelli and F. Zunino, Bioorg. Med. Chem. Lett., 2004, 14, 5757 CrossRef CAS.
- I. A. Rivero, K. Espinoza and R. Somanathan, Molecules, 2004, 9, 609 Search PubMed.
- F. Hernández, V. Morales, F. L. Buenadicha, M. Söllhuber and C. Avendaño, Tetrahedron: Asymmetry, 2004, 15, 3045 CrossRef CAS.
- J. Azizian, A. R. Karimi, A. A. Mohammadi and M. R. Mohammadizadeh, Heterocycles, 2004, 63, 2225 CrossRef CAS.
- J. Azizian, M. Madani and S. Souzangarzadeh, Synth. Commun., 2005, 35, 765 CrossRef CAS.
- N. Chaya, K. Terauchi, Y. Yamagata, J. Kinjo and H. Okabe, Biol. Pharm. Bull., 2004, 27, 1312 CrossRef CAS.
- T.-L. Su, H.-M. Huang, C.-K. Wang, H.-C. Wu and C. T. Lin, Planta Med., 2005, 71, 28 CrossRef CAS.
- S. Michel, T. Gaslonde and F. Tillequin, Eur. J. Med. Chem., 2004, 39, 649 CrossRef CAS.
- F. Tillequin and M. Koch, Ann. Pharm. Fr., 2005, 63, 35 ( Chem. Abstr. , 2005 , 142 , 366592 ) Search PubMed.
- E. Seguin and F. Tillequin, Ann. Pharm. Fr., 2005, 63, 44 ( Chem. Abstr. , 2005 , 142 , 366593 ) Search PubMed.
- M.-H. David-Cordonnier, W. Laine, A. Lansiaux, F. Rosu, P. Colson, E. de Pauw, S. Michel, F. Tillequin, M. Koch, J. A. Hickman, A. Pierré and C. Bailly, Mol. Cancer Ther., 2005, 4, 71 Search PubMed.
- M. R. F. Neto, J. B. N. DaCosta, C. M. R. Sant'Anna and J. W. M. Carneiro, Arzneim.-Forsch., 2005, 55, 282 CAS.
- J. P. Dheyongera, W. J. Geldenhuys, T. G. Dekker and C. J. Van der Schyf, Bioorg. Med. Chem., 2005, 13, 689 CrossRef CAS.
- J. P. Dheyongera, W. J. Geldenhuys, T. G. Dekker, M. G. Matsabisa and C. J. Van der Schyf, Bioorg. Med. Chem., 2005, 13, 1653 CrossRef CAS.
- A. Scopton and T. R. Kelly, Org. Lett., 2004, 6, 3869 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |