Indolizidine and quinolizidine alkaloids
Joseph P. Michael*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Wits 2050, South Africa. E-mail: jmichael@chem.wits.ac.za
First published on 3rd January 2007
Abstract
Covering: July 2004 to June 2005
This review covers the isolation, structure determination, synthesis, chemical transformations and biological activity of indolizidine and quinolizidine alkaloids. Included in the review are the hydroxylated indolizidines lentiginosine, swainsonine, castanospermine and their analogues; alkaloids from animal sources, including ants, amphibians and beetles; indolizidine alkaloids from the genera Polygonatum, Prosopis and Elaeocarpus; indolizinium and phenanthroindolizidine alkaloids; alkylquinolizidine alkaloids, including myrtine, epimyrtine, plumerinine and Lycopodium metabolites; Lythraceae and Nuphar alkaloids; lupine alkaloids; and alkaloids from marine sources; 150 references are cited.
1 Hydroxylated indolizidine alkaloids
A review describing recent advances in the chemistry of azapyranose sugars pays brief attention to the glycosidase inhibitors lentiginosine, swainsonine and castanospermine and their naturally occurring diastereomers, and highlights aspects of their toxicity and therapeutic applications.1 Polyhydroxylated indolizidine alkaloids are also included in a review of various chemical approaches to the synthesis of naturally occurring iminosugars.21.1 Lentiginosine
The allylic alcohol 1, obtained from D-tartaric acid by a previously reported procedure,3 was the starting material in a new synthesis of (+)-lentiginosine 2 by Ichikawa et al.4 (Scheme 1). The addition of diethylzinc to the corresponding aldehyde 3 was performed in the presence of the chiral ligand (S)-DPMPM 4 to give the allylic alcohol 5 in 91% yield and high diastereoselectivity (93 : 7). In situ dehydration of the carbamate derivative 6, still as a mixture of diastereomers, was followed by a spontaneous [3,3]-sigmatropic rearrangement of the allyl cyanate 7 with transfer of chirality to produce the isocyanate 8 (major isomers shown), which contains all three of the target alkaloid's stereogenic centres. This intermediate was trapped with 2,2,2-trichloroethanol as the carbamate 9. Subsequent deprotection with zinc in acetic acid and sulfonylation of the free amine with 2-nitrobenzenesulfonyl chloride gave a product 10 that could be separated from the minor diastereomer by recrystallisation. After Mitsunobu alkylation of 10 with but-3-en-1-ol, ring-closing metathesis of the diene 11 in the presence of the Grubbs first-generation catalyst (PCy3)2Cl2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh afforded the tetrahydropyridine 12 in 92% yield. The synthesis of (+)-lentiginosine 2 was completed as illustrated by a sequence of conventional steps.
CHPh afforded the tetrahydropyridine 12 in 92% yield. The synthesis of (+)-lentiginosine 2 was completed as illustrated by a sequence of conventional steps.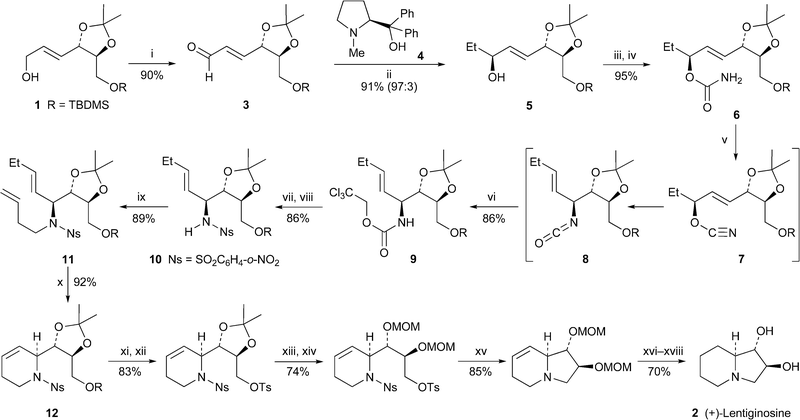 | ||
| Scheme 1 Reagents and conditions: i, IBX, DMSO, 0 °C, then rt, 4 h; ii, (S)-(+)-DPMPM 4 (6 mol%), cyclohexane, reflux, 30 min, then Et2Zn, 0 °C, 7 h; iii, Cl3CCONCO, CH2Cl2, 0 °C, 20 min; iv, K2CO3, MeOH–H2O, rt, 7 h; v, CBr4, PPh3, NEt3, CH2Cl2, −20 °C to 0 °C, 20 min; vi, Cl3CCH2OH, 0 °C, 1 h; vii, Zn, AcOH, THF, rt, 4 h; viii, ClSO2C6H4-2-NO2, aq. NaHCO3, CH2Cl2, 0 °C, 2 h; ix, H2CCHCH2CH2OH, DEAD, PPh3, C6H6, 0 °C, then rt, 7 h; x, (PCy3)2Cl2RuCHPh (7 mol%), C6H6, −78 °C, then 60 °C, 1.5 h; xi, Bu4NF, MeCN, rt, 6 h; xii, p-TsCl, NEt3, CH2Cl2, rt, 4 h, then Me2N(CH2)3NH2; xiii, aq. HCl (3 M), THF, 50 °C, 5 h; xiv, (MeO)2CH2, P2O5, rt, 1.5 h; xv, PhSH, Cs2CO3, MeCN, rt, 1.5 h; xvi, H2 (1 atm), Pt/C, EtOH, 2 h; xvii, aq. HCl (3 M), MeOH, 55 °C, 5 h; xviii, ion exchange (IRA-410). | ||
1.2 Swainsonine and related compounds
An extensive phytochemical study of 129 plant species belonging to 29 genera of the Convolvulaceae and including plants from all continents has revealed the presence of calystegines (polyhydroxylated tropanes) in no fewer than 62 species and 22 genera.5 However, neither swainsonine 13 nor castanospermine (vide infra) was detected in any of the specimens despite previous reports of the former alkaloid in several species of Ipomoea, one of the largest genera of the Convolvulaceae.North American locoweeds of the genera Astragalus and Oxytropis, which cause neurological disorders in animals that graze on them, are another important source of swainsonine. It has previously been suggested that an endophytic fungus, possibly Embellisia sp., may be the actual source of the toxic alkaloid6 (cf.ref. 7a). Tests have now shown that rats fed either locoweed or Embellisia fungi displayed indistinguishable symptoms and morphological changes in renal, pancreatic and hepatic tissue, consistent with swainsonine poisoning.8
The many novel synthetic approaches to (−)-swainsonine 13 published since 1999 have been summarised in a useful review by Pyne,9 whose own contributions in this regard are summarised in a more personal overview on the asymmetric synthesis of polyfunctionalised pyrrolidine alkaloids and related compounds.10 Disappointed by a poor yield in the Sharpless asymmetric hydroxylation of intermediate 14 in their previous synthesis of (−)-swainsonine11 (cf.ref. 7b), Lindsay and Pyne have now devised an alternative route to the alkaloid in which dihydroxylation of a less basic intermediate, the oxazolidinone 15, was expected to be a more efficient process12 (Scheme 2). Proceeding (with minor experimental improvements) through the same initial intermediates 16–19 that had featured in their earlier synthesis, they cyclised the latter by treatment with sodium hydride to give compound 15. The same product could be prepared in a shorter but less efficient manner from 17 by sequential treatment with triphosgene followed by ring-closing metathesis. When 15 was dihydroxylated with the Sharpless AD-mix-β reagent, the reaction proved to be highly diastereoselective in favour of product 20 (20 : 1 with exo-diol), but the reaction was very slow, giving a yield of only 46% in six days; fortunately, unreacted 15 could be recovered (45%). The isomers could be separated after benzylation of the hydroxy groups. After hydrolysis of the benzylated oxazolidinone to the amino alcohol 21, straightforward protection–deprotection protocols produced 22, which was cyclised in situvia the corresponding bromide to give the indolizidine 23 in 93% yield. Removal of the protecting groups and purification of the product by ion-exchange chromatography completed the synthesis of (−)-swainsonine 13.
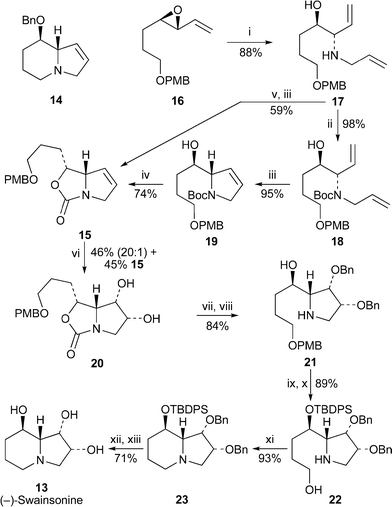 | ||
| Scheme 2 Reagents and conditions: i, H2CCHCH2NH2, LiOTf, microwave, 110 °C; ii, Boc2O, NEt3, THF, rt, 24 h; iii, (PCy3)2Cl2RuCHPh (4 mol%), CH2Cl2, reflux, 18 h; iv, NaH, PhMe, 50 °C, 24 h; v, triphosgene, NEt3, CH2Cl2, 0 °C, 2 h; vi, AD-mix-β, (DHQD)2PHAL, MeSO2NH2, ButOH–H2O, rt, 6 d; vii, BnBr, NaH, Bu4NI, THF, rt, 2 d; viii, NaOH, MeOH, H2O, microwave, 110 °C, 2 h; ix, TBDPSCl, imidazole, 65 °C, 3 d; x, CAN, MeCN, H2O, rt, 3 h; xi, CBr4, PPh3, NEt3, CH2Cl2, 0 °C, 16 h; xii, Bu4NF, THF, rt, 5 d; xiii, H2 (1 atm), PdCl2, MeOH, rt, 2 h, then ion exchange. | ||
Mariano and co-workers previously demonstrated the photochemical conversion of pyridinium salts into stereochemically defined 4-aminocyclopentene-3,5-diol derivatives such as the acetate 24, and the subsequent enzymatic desymmetrisation to produce the useful building block 25.13 Such chiral precursors have now been employed by Mariano's team in a novel synthesis of (−)-swainsonine14 (Scheme 3). An immediately striking feature of this route is the regiospecific tandem ring opening–ring closing metathesis of the N-allyl derivative 26, catalysed by the Grubbs second-generation catalyst, to give the tetrahydropyridine 27 in 90% yield. Regioselective dihydroxylation of the side chain in preference to the ring double bond required conversion into the acetate 28; even so, reaction with osmium tetroxide and N-methylmorpholine N-oxide afforded a mixture of diol diastereomers 29 and 30 in a ratio of 4.4 : 1 (81% yield). The unseparated mixture was converted into the indolizidines 31 and 32 by sequential hydrolysis, cyclisation with diethyl azodicarboxylate and triphenylphosphine, and re-acetylation. The two isomers were isolated in yields of 47% and 11%, respectively, but their rather disappointing enantiomeric excesses (79% and 78% ee, by chiral HPLC) reflect the efficiency of the earlier enzymatic hydrolysis (i.e., 24 → 25; 80% ee). The synthesis of (−)-swainsonine 13 from 31 was completed by hydrogenation of the double bond, hydrogenolysis of the benzyl protecting group and acidic hydrolysis of the acetates in a yield of 80% over the three steps. Similarly, 32 was transformed in 63% yield into (−)-2-epi-swainsonine 33.
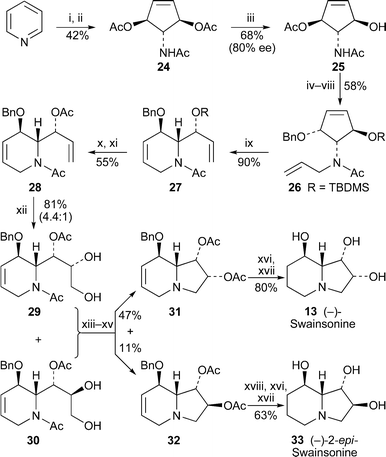 | ||
| Scheme 3 Reagents and conditions: i, aq. HClO4 (0.7%), hν (254 nm), 20 h; ii, Ac2O, DMAP, py, 25 °C, 24 h; iii, electric eel acetylcholinesterase, NaH2PO4 buffer (pH 6.9), 15–20 °C, 5 h; iv, TBDMSCl, imidazole, CH2Cl2, 25 °C, 12 h; v, NaOMe, MeOH, 25 °C, 10 h; vi, Burgess salt, THF, 70 °C, 3 h, then aq. NaH2PO4, 25 °C, 12 h; vii, NaH, DMF, 0 °C, 20 min, then BnBr, 25 °C, 2 h; viii, NaH, DMF, 0 °C, 20 min, then H2CCHCH2Br, 0 °C, 1 h; ix, Grubbs II catalyst (10 mol%), H2CCH2, CH2Cl2, reflux, 16 h; x, Bu4NF, THF, 25 °C, 2 h; xi, AcCl, NEt3, THF, 25 °C, 12 h; xii, OsO4 (cat.), NMO, H2O–Me2CO, 25 °C, 3 h, then aq. Na2S2O3, 1 h; xiii, HCl (6 M), THF, 70 °C, 4 h; xiv, DEAD, PPh3, 4 Å molecular sieves, py, 0 °C, 3 h; xv, Ac2O, DMAP, py, CH2Cl2, 25 °C, 12 h; xvi, H2 (1 atm), PdCl2, MeOH, 1 h; xvii, HCl (6 M), THF, 25 °C, 24–48 h, then ion exchange (Dowex 1-X8, OH− form, 100–200 mesh); xviii, H2 (1 atm), 10% Pd/C, EtOH, 25 °C, 2 h. | ||
Ring-closing metathesis also featured in a formal synthesis of (−)-swainsonine by Riera and co-workers15 (Scheme 4). In this case, the alkene-bearing oxazolidinone (+)-34, prepared in two steps from the chiral epoxide (−)-35, was converted into the bicyclic oxazolidinone (+)-36 in good yield with the Grubbs first-generation catalyst. After the uneventful conversion of 36 into the aldehyde 37, olefination with triethyl phosphonoacetate in the presence of DBU and lithium chloride afforded predominantly (14 : 1) the (E)-unsaturated ester 38 in 94% yield. With methyl bis(trifluoroethoxy)phosphonoacetate, however, the (Z)-isomer 39 was the major product (5 : 1). The ensuing dihydroxylation of both geometric isomers was diastereofacially selective; 38 yielded diol 40 as the sole product (72%), while the mixture containing predominantly 39 gave the expected 5 : 1 mixture of 41 and 40. Hydrolysis and cyclisation of this mixture followed by protection of the diol as the acetonide produced 42 as a single diastereomer after chromatographic purification. Finally, reduction of the lactam yielded 43, thereby completing a formal synthesis16 of (−)-swainsonine 13. A similar sequence of reactions on diol 40 did not require acetonide formation, and yielded the benzyl-protected 2-epi-swainsonine 44.
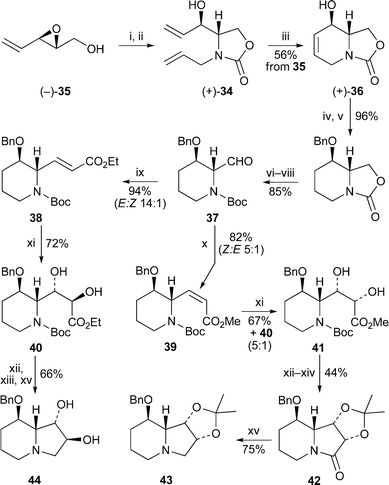 | ||
| Scheme 4 Reagents and conditions: i, NEt3, Et2O, rt, 30 min, then H2CCHCH2NCO, 65 °C, 4 h; ii, NaHMDS, THF, rt, 30 min; iii, (PCy3)2Cl2RuCHPh (5 mol%), CH2Cl2, rt, 1 h; iv, H2 (1 atm), Pd/C, EtOAc, rt, 1 h; v, NaH, DMF, −20 °C, 30 min, then BnBr, rt, 14 h; vi, aq. NaOH (6 M), MeOH–H2O (9 : 1), 100 °C, 14 h; vii, Boc2O, aq. NaHCO3, EtOAc, rt, 14 h; viii, Dess–Martin periodinane, CH2Cl2, rt, 1 h; ix, (EtO)2POCH2CO2Et, LiCl, DBU, MeCN, rt, 2 h; x, KHMDS, (CF3CH2O)2POCH2CO2Me, 18-crown-6, THF, −78 °C, 4 h; xi, OsO4 in ButOH (cat.), NMO, H2O–Me2CO (1 : 10), rt, 48 h; xii, HCl (1 M in Et2O), rt, 4 h; xiii, EtNPri2, THF, reflux, 14 h; xiv, (MeO)2CMe2, p-TsOH (cat.), Me2CO, rt, 3 h; xv, BH3·SMe2, THF, rt, 14 h. | ||
Asymmetric dihydroxylation of the unsaturated pyridine N-oxide 45 with the Sharpless AD-mix-β reagent was the key stereodifferentiating step in a formal synthesis of (−)-swainsonine by Reiser and co-workers17 (Scheme 5). The product 46, obtained in 65% yield and 98% ee, was hydrogenated over platinum dioxide to give the indolizidinone 47 as a separable mixture of epimers (3 : 2) at the bridgehead site—of no consequence, since the bridgehead stereocentre was eventually to be destroyed. The stereocentre at C-1 in the unseparated mixture was inverted via the derivative 48 by intramolecular SN2 displacement of the corresponding C-1 trifluoromethanesulfonate by the neighbouring benzoate group to afford compound 49, again as a mixture of epimers at C-8a. Hydrolysis then afforded the diol 50, from which the major isomer 8aβ-H-50 could be obtained by recrystallisation. Its structure was confirmed by X-ray crystallography. Once again, however, the synthesis was continued with the diastereomeric mixture 50, which was protected as the acetonide 51. Interestingly, regioselective bridgehead hydroxylation of this mixture with ruthenium tetroxide indicated that the two epimers did not react at the same rate; after workup with acetic acid, the unconverted epimer 8aα-H-51 was isolated in 29% yield together with the desired eliminated product 52 (50%). By contrast, isomer 8aβ-H-51, which has the bridgehead hydrogen on the more accessible convex face of the molecule, was readily oxidised to 53 with retention of configuration. Acid-induced elimination then furnished 52 in 79% yield over the two steps. Since racemic 52 has previously been converted into (±)-swainsonine rac-13,18 this route in effect completes a formal synthesis of the alkaloid's (−)-enantiomer. The authors also applied their methodology to the transformation of diol 54via indolizidinone 55 into 2,8a-di-epi-swainsonine 56.
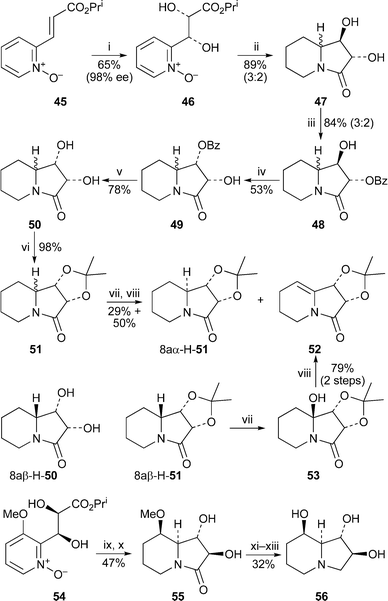 | ||
| Scheme 5 Reagents and conditions: i, AD-mix-β, MeSO2NH2, ButOH–H2O (1 : 1), rt, 24 h; ii, H2 (1 atm), PtO2 (cat.), MeOH, rt, 14 d; iii, PhCOCl (syringe pump), DMAP, py, −30 °C, 1 h; iv, (CF3SO2)2O, py, −30 °C to rt, then 1 h; v, NaOMe, MeOH, rt, 3 h; vi, (MeO)2CMe2, p-TsOH (cat.), CH2Cl2, rt, 2 h; vii, aq. NaOCl (12%), RuO2·xH2O (cat.), EtOAc, 0 °C, 9 h; viii, AcOH, CHCl3, rt, 1 h; ix, H2 (1 atm), 5% Pt/C, AcOH, rt, 7 d; x, NEt3, CHCl3, rt, 24 h; xi, aq. HBr (48%), 140 °C, 30 min; xii, BH3·SMe2, THF, rt, 32 h; xiii, ion exchange (Dowex 1-X8, OH− form, 100–200 mesh). | ||
The transformation of the enantiomerically pure epoxysulfone 57 into the unstable pyrrolidine-2-carbaldehyde 58 by treatment with benzylamine19 represents a formal synthesis of (−)-swainsonine 13 in the light of methodology previously reported by Ikota and Hanaki.20 New but exotic synthetic analogues of swainsonine reported during the review period include the benzo-fused trihydroxy compounds 59 and 60, prepared in several steps from N-benzylpyroglutamic acid, and the corresponding dihydroxy compounds 61 and 62, which can also be viewed as analogues of lentiginosine.21 Also of interest is the pyrroloazepine 63, which was found to be a good inhibitor of yeast α-glucosidase (90% at 1 mM).22
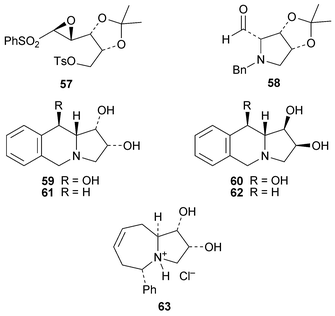
1.3 Castanospermine and related compounds
The antiviral effects of castanospermine 64, a potent inhibitor of endoplasmic reticulum α-glucosidase I, have been ascribed to its ability to disrupt the folding of certain viral proteins by suppressing the removal of terminal glucose residues from N-linked glycans. Recent in vitro studies with various flaviviruses, which are assembled at the endoplasmic reticulum, showed that the alkaloid was able to inhibit infection by four different serotypes of dengue virus at the level of secretion and infectivity of viral particles.23 However, yellow fever virus was partially resistant to the effects of the drug; and West Nile virus was almost completely unaffected. Most significantly, a 90% survival rate was recorded for mice infected with dengue fever after treatment with the alkaloid at a daily dosage of 50 mg kg−1, whereas untreated mice died. Since dengue fever is a globally prevalent disease for which no vaccine has yet been approved, further exploration of castanospermine as a possible treatment for human infections seems warranted.A new synthesis of (+)-castanospermine by Cronin and Murphy24 commenced with the known glucose derivative 65, which was prepared in five steps from methyl α-D-glucopyranoside 66 (Scheme 6). The corresponding benzyl analogue 67 was epoxidised with trifluoroacetone and oxone to give an epimeric mixture of epoxides 68 in a ratio of 1.7 : 1. Acid-induced methanolysis of the epoxide, followed by oxidation with tetrapropylammonium perruthenate then afforded the separable aldehydes 69 and 70 in yields of 46% and 12%, respectively. The relative configurations of the two isomers were determined by means of NOE spectroscopy. Condensation of the major isomer 69 with the lithium enolate of ethyl acetate produced the separable β-hydroxy esters 71 and 72 in yields of 16% and 35%, respectively. The key step in the synthesis, induced by hydrogenating 71 over palladium hydroxide, proved to be a novel cascade process involving reduction of the azide, intramolecular reductive amination, lactam formation and debenzylation to give lactam 73 in 62% overall yield. This highly stereoselective reaction also succeeded in producing the correct bridgehead stereochemistry for the target alkaloid (+)-64, which was obtained from 73 by silylation of the free alcohol groups, reduction of the lactam and hydrolytic work-up. A similar sequence of reactions was performed on the hydroxy ester 72 to yield (+)-1-epi-castanospermine 74, an unnatural epimer of 64.
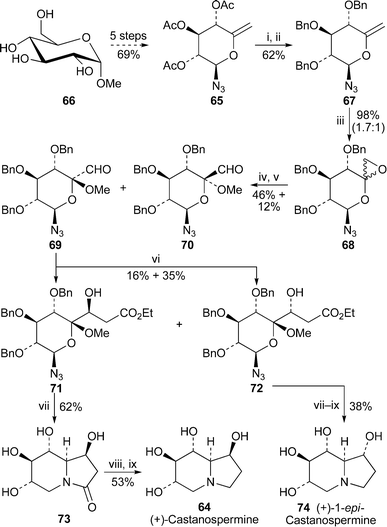 | ||
| Scheme 6 Reagents and conditions: i, NaOMe, MeOH, 1 min; ii, NaH, DMF, 0 °C, 30 min, then BnBr, rt, 3 h; iii, Na-EDTA, F3CCOMe, Na2CO3, oxone, MeCN, 0 °C, 1 h, rt, 30 min; iv, CSA, MeOH, rt, 10 min; v, TPAP, NMO, 4 Å molecular sieves, CH2Cl2, rt, 15 h, then chromatography; vi, LiCH2CO2Et (from LDA + EtOAc), THF, −78 °C to rt, 1.5 h, then chromatography; vii, H2 (500 psi), 5% Pd(OH)2/C, HCO2H, MeOH, 48 h; viii, TMSOTf, 2,6-lutidine, py, CH2Cl2, 0 °C, then rt, 12 h; ix, LiAlH4, THF, rt, 16 h, then H2O workup. | ||
Transformations of (+)-castanospermine into other derivatives continue to be of interest in view of the wide range of biological activities displayed by the alkaloid and its analogues. The direct oxidation of castanospermine tetraacetate 75 with N-bromosuccinimide in aqueous dioxane afforded the crystalline lactam 76 in 19% yield.25 Treatment of 76 with DBU then induced a double elimination to give 77 in 89% yield, while reaction with sodium methoxide in methanol gave the deacetylated tetrol 78 in 37% yield together with the diene 79 (23%). The structure of 78 was confirmed by X-ray crystallography.
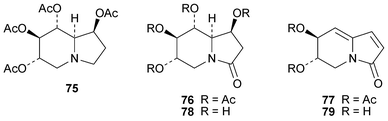
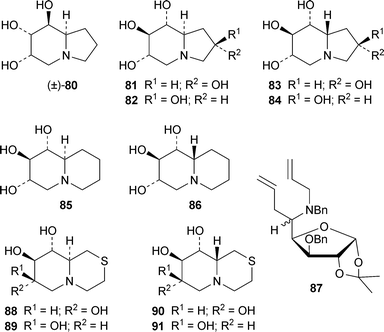
Synthetic analogues of castanospermine reported during the review period include (±)-1-deoxy-6,8a-di-epi-castanospermine 8026 and the four 2-hydroxy-1-deoxycastanospermine epimers (+)-81–83 and (−)-84.27 All four compounds proved to be inhibitors of various glycosidases, displaying activities in the micromolar range. Two quinolizidine analogues of 1-deoxycastanospermine, (−)-85 and (−)-86, were obtained by routes involving ring-closing metathesis of sugar derivatives 87.28 Perhaps the most interesting new compounds are the related thiaquinolizidines 88–91, which represent a novel class of glycosidase inhibitors.29 X-Ray crystal structures revealed that both 88 and 89 adopt conformations with a flattened but trans-fused thiaquinolizidine ring system. Compound 88, which has the D-gluco configuration, was a specific but modest inhibitor of yeast and rice α-glucosidases, but it failed to inhibit β-glucosidase, α- and β-galactosidases and α-mannosidase. Its inhibition constants were in the millimolar range. By contrast, the L-ido derivative 91 inhibited almond β-glucosidase but not the α-glucosidases.
2 Alkaloids from ants and amphibians
2.1 Occurrence and biological activity
Preliminary studies on biologically active substances extracted from 21 genera of frogs and toads from Thailand have turned up surprisingly few alkaloids of relevance for this review.30 Only in extracts from skins of the ranid frog Limnonectes kuhli were minute traces of pumiliotoxins detected by means of GC-MS. The dominant alkaloid, coded as 295D and apparently a new amphibian metabolite, was assigned the partial pumiliotoxin structure 92. The fragmentation pattern suggested that the unknown appendage R corresponds to C5H9O2 or C6H13O, either of which must be able to lose CH2OH or OCH3. The rather uncommon alkaloids pumiliotoxin 265G 93 and homopumiliotoxin 223G 94 were also present, but at 20-fold lower levels. The minuscule levels of the three alkaloids suggest that their presence in the extracts is fortuitous rather than the result of efficient uptake and storage, as occurs with other anurans.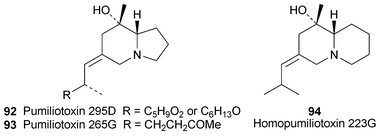
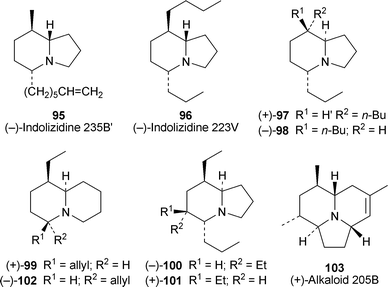
Nicotinic acetylcholine receptors (nAChRs)—ligand-gated ion channels that play a central role in cholinergic transmission in the nervous system—are key molecules in the physiological processes of learning and memory, among others. Several amphibian alkaloids and related compounds previously synthesised by Toyooka and his co-workers (vide infra, Section 2.3) have recently been tested for inhibitory activity towards a range of nAChRs expressed in oocytes obtained from Xenopus laevis frogs.31 At a concentration of 0.3 µM, the 5,8-disubstituted indolizidine (−)-235B′ 95 proved to be a potent but noncompetitive blocker of α4β2 nicotinic receptors (IC50 74 nM), as judged by its ability to suppress acetylcholine-elicited electric currents in appropriately treated oocytes. The alkaloid showed considerable selectivity for this nAChR subtype in comparison to α7, α3β2, α3β4 and α4β4 receptors, and various additional experiments indicated that it probably acts as an open-channel blocker rather than through an indirect effect. In comparison, synthetic (−)-96 (indolizidine 223V; cf. Section 2.3), its diastereomers 97 and 98 and (+)-quinolizidine 207I 99 (the enantiomer of the natural product) were considerably less effective (IC50 4.5–20.1 µM) and less selective blockers for α4β2 than 95; in general they were also almost equally active in inhibiting responses from α7 and α3β4 receptors (IC50 1.8–14.7 µM). While the trisubstituted indolizidine (−)-223A 100 was similar to this group of compounds in its activity and lack of specificity, its synthetic 6-epimer (+)-101 had a negligible effect on acetylcholine-elicited currents in oocytes expressing α4β2 or α7 receptors, but interacted with α3β4 receptors (IC50 15.1 µM). Finally, both synthetic (−)-1-epi-quinolizidine 207I 102 and the tricyclic compound (+)-205B 103—the enantiomer of natural (−)-205B—proved to be selective in blocking α7 receptor-mediated currents (IC50 0.6 and 2.5 µM, respectively). All these results suggest that frog alkaloids may be useful lead compounds in the design of drugs for treating cholinergic disorders of the central nervous system.
2.2 Synthesis of 5-alkylindolizidine alkaloids
A new synthesis of (+)-indolizidine 167B 104 by Kunz and co-workers (Scheme 7) employed the N-(tetra-O-pivaloyl-β-D-galactosyl) substituent as an unusual chiral auxiliary, and entailed the stereoselective desymmetrisation of the 4-pyridone 105 by treatment of the corresponding O-silyl pyridinium salt with Grignard reagents.32 In the example of relevance, addition of n-propylmagnesium chloride gave predominantly (89 : 11) the (S)-adduct 106, which could be separated from the (R)-diastereomer by preparative HPLC. Unfortunately, poor cis-selectivity in attaching alkyl substituents to C-6 by conjugate addition necessitated removal of the auxiliary at this point. Since the enaminone could not withstand acid hydrolysis, it was masked as the phenylselenenyl derivative 107 before replacement of the galactosyl substituent by benzyloxycarbonyl (Cbz) and oxidative elimination of SePh from intermediate 108 to reintroduce the enaminone. Even so, conjugate addition of the cuprate prepared from 3-(1-ethoxyethoxy)propyl bromide to enaminone 109 proceeded with only modest stereoselectivity, giving the 2,6-cis-disubstituted piperidine 110 in 73% yield and a diastereomer ratio of 2 : 1. The diastereomers could be separated after conversion of the alcohol into the chloride 111. Removal of the carbonyl group was achieved by hydrogenation of the corresponding enol triflate, after which the ring-closing N-alkylation afforded the alkaloid (+)-104 in an overall yield of 6.3% from the N-galactosylpyridine 105. This synthesis complements Kunz's previously published route to indolizidine 167B33 (cf.ref. 7c), which he has summarised in a recent review34 that highlights his approach to the synthesis of various alkaloids via carbohydrates as stereodifferentiating auxiliaries.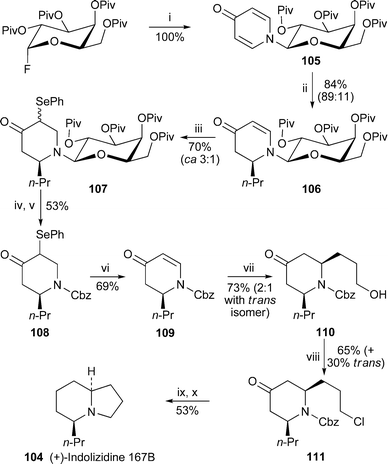 | ||
| Scheme 7 Reagents and conditions: i, 4-(trimethylsilyloxy)pyridine, ClCH2CH2C1, 70 °C, then TiCl4, reflux, 1 h; ii, TIPSOTf, CH2Cl2, rt, 30 min, then 2,6-lutidine, n-PrMgCl, then HPLC separation; iii, L-selectride, THF, −78 °C, 40 min, then PhSeCl, 2 h; iv, HCl (1 M), MeOH, 12 h; v, ClCO2Bn, aq. satd. NaHCO3, 2 h; vi, H2O2–urea complex, CH2Cl2–H2O (20 : 1), 2 h; vii, EtOCH(Me)O(CH2)3MgBr, CuBr·SMe2, BF3·OEt2, THF, −78 °C, 5 h, then aq. HCl (2 M)–THF (1 : 1), rt, 20 min; viii, NCS, PPh3, CH2Cl2, −40 °C, then rt, overnight, then chromatographic separation; ix, LiHMDS, THF, −78 °C, 1 h, then 5-Cl-2-NTf2-pyridine, rt, overnight; x, H2, 10% Pd/C, Li2CO3, EtOAc, 1 h. | ||
Two shorter syntheses of indolizidine 167B are also of interest (Scheme 8). In the first, the optically active hexahydrooxazolo[3,2-a]pyridinium salt 112 was prepared in two steps from the thiolactam 113,35 and then reduced with L-selectride to give a mixture of oxazolidine diastereomers 114 in a ratio of 4 : 1.36 Reaction of this mixture with 2-(1,3-dioxolan-2-yl)ethylmagnesium bromide produced the 2,6-cis-disubstituted piperidine 115 in 88% yield after chromatography. Finally, catalytic hydrogenation in acidic methanol in the presence of palladium on carbon removed both protecting groups and effected intramolecular reductive amination, quantitatively giving the hydrochloride salt of (+)-indolizidine 167B 104. In the second route, the ready cyclodehydration of the glycine-derived pyrrole-aldehyde 116, accomplished merely by heating the reactant in DMSO at 100 °C, afforded the 5,6-dihydroindolizine 117 in 55% yield.37 Chemoselective reduction of the ester to an aldehyde followed by Wittig olefination yielded the trans-alkene 118, which underwent a totally diastereoselective hydrogenation in the presence of 5% rhodium on carbon—apparently never before used for the total reduction of indolizine systems—to complete the synthesis of (±)-indolizidine 167B, rac-104. The 5-ethyl analogue of 104 was also obtained by this method.
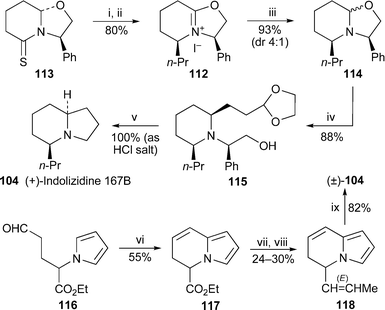 | ||
| Scheme 8 Reagents and conditions: i, n-PrMgCl, THF, −20 °C, 2 h; ii, MeI, THF, 5 °C, 4 h; iii, L-selectride, THF, −10 °C, 10 min, then aq. H2O2 (30%), 0 °C, then aq. NaOH (3 M); iv, 2-(1,3-dioxolan-2-yl)ethylmagnesium bromide, THF, −40 °C, 15 h; v, H2, Pd/C, MeOH, HCl; vi, DMSO, 100 °C, 2 h; vii, DIBAL, THF, −78 °C, 1.5 h; viii, EtPPh3+ Br−, NaNH2, THF, rt, 12 h; ix, H2 (10 atm), 5% Rh/C, 90 min. | ||
The initial steps in a novel synthesis of (−)-indolizidine 209D 119 by Patil et al.38 commenced with chain extension of the (−)-N-Boc-prolinal 120 to the alcohol 121 (Scheme 9). Subsequent treatment of the corresponding iodide with 1-hexynyllithium and removal of the Boc protecting group produced 122, thereby opening the way for what appears to be a new route to indolizidines: intramolecular hydroamination of an alkyne. The reaction, induced by treatment with tetrakis(triphenylphosphine)palladium and benzoic acid, presumably proceeds via a π-allylpalladium intermediate, which undergoes cyclisation by way of a chair-like transition state in which the hydrocarbon chain preferentially takes up an equatorial position to avoid 1,3-diaxial interactions. This appears to account for the high diastereoselectivity of the process, as evinced by the isolation of a single product 123 (74% yield). The synthesis of the target alkaloid (−)-119 was completed by catalytic hydrogenation of the hexenyl substituent.
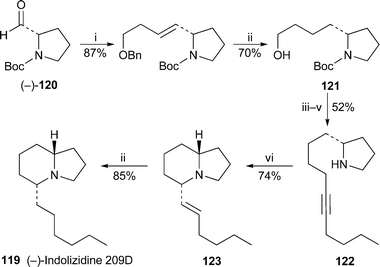 | ||
Scheme 9 Reagents and conditions: i, BnO(CH2)3PPh3+ Br−, NaHMDS, THF, −78 °C, 1 h; ii, H2, 10% Pd/C, MeOH, 24 h; iii, PPh3, I2, imidazole, THF, rt, 12 h; iv, LiC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(CH2)3Me, THF, −78 °C, 8 h; v, TFA–CH2Cl2 (3 : 2), 0 °C, 3 h; vi, Pd(PPh3)4 (5 mol%), C6H5CO2H (10 mol%), NEt3, dioxane, 100 °C, 12 h (in screw-capped vial). C(CH2)3Me, THF, −78 °C, 8 h; v, TFA–CH2Cl2 (3 : 2), 0 °C, 3 h; vi, Pd(PPh3)4 (5 mol%), C6H5CO2H (10 mol%), NEt3, dioxane, 100 °C, 12 h (in screw-capped vial). | ||
A short synthesis of racemic indolizidine 209D (±)-119 by Blechert and co-workers39 made use of cross-metathesis between alkenes 124 and 125, which was efficiently promoted by the ruthenium (Grubbs–Hoveyda) catalyst 126 (Scheme 10). The product 127, obtained in 87% yield, then underwent hydrogenation and reductive amination in acidic methanol to give the target alkaloid in 88% yield. This cross-metathesis–cyclisation strategy was also used to prepare several piperidines and pyrrolizidines, as well as the indolizidines 128 and 129, which are obvious candidates for further elaboration to amphibian alkaloids.
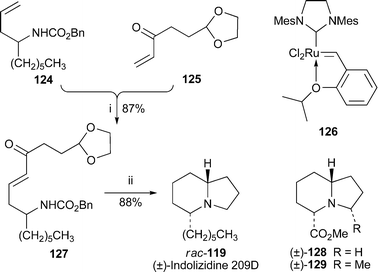 | ||
| Scheme 10 Reagents and conditions: i, catalyst 126 (5 mol%), CH2Cl2, 40 °C, 16 h; ii, H2, Pd/C, MeOH–HCl, 24 h. | ||
2.3 Synthesis of dialkylindolizidine alkaloids
Among a suite of synthetic 5,8-disubstituted indolizidines reported by Toyooka, Daly and co-workers in 199740 (cf.ref. 7d) was compound (−)-96 (see Section 2.1 above). It has recently been shown by GC-MS and GC-FTIR that the synthetic compound is actually identical to indolizidine 223V, a minor amphibian alkaloid.41 In the earlier article, it was also tentatively suggested that another minor alkaloid, indolizidine 223I, might have the structure 130 with the rare (5,8a-E) relative stereochemistry between the attached hydrogen atoms. However, the new report includes enantioselective syntheses of the two 223V diastereomers (5R,8S,9R)-(+)-130 (= 97) and (5R,8R,9R)-(−)-130 (= 98; also see Section 2.1), GC-MS and GC-FTIR analysis of which proved that neither corresponded to indolizidine 223I. The central step in the synthesis of (5R,8S,9R)-(+)-130 was the stereoselective conjugate addition of lithium dibutylcuprate to the bicyclic oxazolidinone 131, which produced 132 as a single diastereomer in 96% yield. The synthesis was completed in 22 steps by a series of chain extensions, functional group manipulations and a final cyclisation of the late intermediate 133via the corresponding bromide. Isomer (5R,8R,9R)-(−)-130 was obtained by another lengthy route in which enzymatic desymmetrisation of diol 134 served as the prelude to preparing the lactam 135 (16 steps), the enol triflate of which underwent palladium-catalysed coupling with allyltributyltin to give the 2,3,6-trisubstituted piperidine 136 (4 steps from 135). Completion of the synthesis via137 then required a further 9 steps. Thus, whatever the structure of natural alkaloid 223I may be, it clearly does not possess the (5,8a-E) relative stereochemistry. The authors suspect that it may not even be a 5,8-dialkylindolizidine!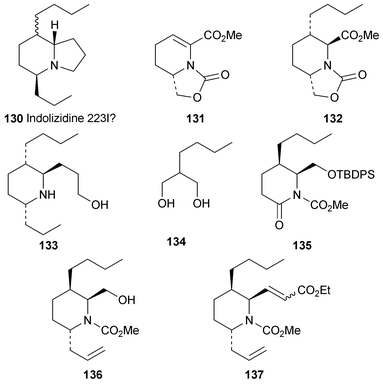
By modifying strategies previously devised for the enantioselective total synthesis of 5,8-disubstituted indolizidine alkaloids, Toyooka and co-workers have been able to establish the relative and absolute stereochemistry of alkaloid 237D, a partially characterised 5,8-disubstituted indolizidine alkaloid detected as a minor component in extracts from Dendrobates pumilio and D. speciosus42,43 (Scheme 11). Chain extension at C-2 and C-6 of the trisubstituted piperidine 138, previously used by these authors in syntheses of other amphibian alkaloids,44,45 yielded the advanced intermediate 139. Reduction of the unsaturated side chain, removal of the protecting groups and ring closure then afforded the (−)-indolizidine 140, which co-chromatographed on a non-chiral GC column with natural alkaloid 237D. The mass and infra-red spectra of the natural and synthetic products were also identical, thereby substantiating the assignment of the alkaloid's relative stereochemistry. In order to establish the absolute configuration, (−)-indolizidine 235B′ 95 (cf. Section 2.1) and (+)-indolizidine 235B″ 141, two unambiguously characterised naturally occurring analogues of 237D with unsaturated side chains, were hydrogenated to give (−)- and (+)-indolizidine 237D, respectively. These proved to be separable on a chiral GC column, and natural indolizidine 237D was found to co-chromatograph with the (−)-enantiomer. Synthetic (−)-140 and reduced (−)-indolizidine 235B′ also eluted simultaneously and gave identical GC-EIMS and GC-FTIR spectra. The absolute configuration of (−)-indolizidine 237D thus appears to be (5R,8R,9S). The authors also adapted the present route to prepare samples of (−)-indolizidine 207A 142 and the homologues (−)-143 and (−)-144 for evaluation as nAChR inhibitors.43 It should be noted that in the two articles cited, the authors have inadvertently drawn the wrong enantiomers of all the structures; one of the articles has since been corrected, and a second corrigendum will be published in due course.46
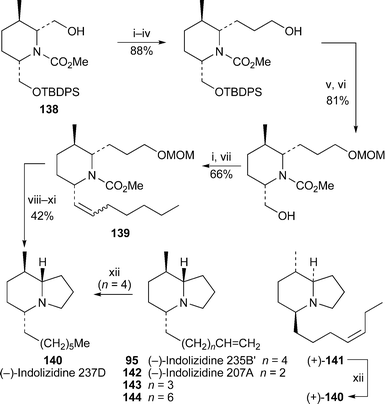 | ||
| Scheme 11 Reagents and conditions: i, Swern oxidation; ii, (EtO)2POCH2CO2Et, NaH, THF, 0 °C, 30 min, then aldehyde, rt, 20 h; iii, H2 (4 atm), 10% Pd/C, EtOAc, rt, 40 h; iv, Super-Hydride (1 M in THF), THF, 0 °C, 1.5 h; v, MOMCl, EtNPri2, CH2Cl2, rt, 45 h; vi, Bu4NF, THF, rt, 2 h; vii, Ph3PCH(CH2)4Me, THF, rt, 14 h; viii, H2 (1 atm), 10% Pd/C, EtOAc, rt, 48 h; ix, n-PrSLi, HMPA, THF, rt, 54 h; x, conc. HCl, MeOH, reflux, 1 h; xi, CBr4, PPh3, CH2Cl2, 0 °C, 1 h, then NEt3, rt, 30 min; xii, H2, 10% Pd/C. | ||
The trifluoromethyl analogue of monomorine 145, the trail pheromone of the Pharaoh ant, has been synthesised as shown in Scheme 12.47 Compound 146, formed by condensation between the keto acid 147 and (S)-phenylglycinol, was converted into the enol triflate 148, which was coupled with hept-1-yn-3-ol to give the enyne 149. Diastereofacially selective hydrogenation of 149 then afforded 150 as a 1 : 1 mixture of alcohol epimers. Oxidation of this mixture to the ketone 151 was followed by one-pot hydrogenolysis of the N-benzyl substituent and intramolecular reductive amination to give the target 152. Alternatively, the two epimers of 150 could be separated and converted into their mesylates 153 and 154. Hydrogenolysis and cyclisation of the former gave the expected indolizidine 152, whereas mesylate 154, which failed to cyclise, yielded the 2,6-cis-disubstituted piperidine 155 instead.
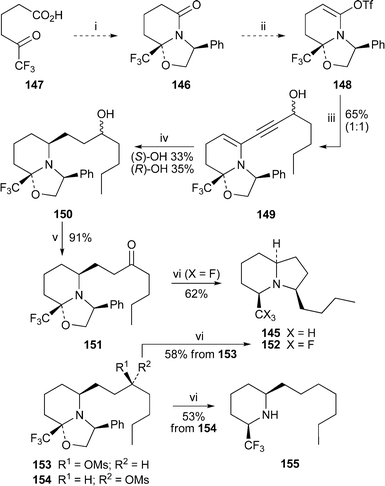 | ||
| Scheme 12 Reagents and conditions: i, (S)-phenylglycinol, condensation; ii, 5-Cl-2-NTf2-pyridine; iii, hept-1-yn-3-ol, PdCl2(PPh3)2, CuI, Pri2NH, THF, rt; iv, H2 (50 psi), PtO2, PhMe; v, Dess–Martin periodinane, CH2Cl2; vi, H2, Pd(OH)2, EtOH, rt. | ||
2.4 Synthesis of trialkylindolizidine alkaloids
The structure of indolizidine 223A was originally thought to be 101 (cf. Section 2.1) until Toyooka et al. proved by synthesis that it was actually the 5,6-cis-isomer 10048 (cf.ref. 7e). The results revealed in this earlier communication have now been reported in a full paper that contains complete experimental details for the synthesis of both (−)-indolizidine 223A and (+)-6-epi-indolizidine 223A, as well as rationalisations for the steps that proceed with high diastereoselectivity.45 Also included in the article are full experimental procedures for the synthesis of the tricyclic alkaloid (+)-205B 103, which had also been outlined in a prior communication44 (cf.ref. 7f).A concise synthesis of (−)-indolizidine 223A 100 by Davis and Yang49 provides a further demonstration of their use of chiral N-sulfinylimines as building blocks for the enantioselective synthesis of alkaloids (Scheme 13). In this case, the (E)-enolate of heptan-4-one 156, generated by treating the ketone with lithium hexamethyldisilazide in diethyl ether, reacted at the si face of the (R)-(−)-sulfinylimine 157 under carefully controlled conditions to give (−)-158 as the major adduct (78%) together with the diastereomer (−)-159 (8%). The major isomer is probably formed through a cyclic transition state such as 160 in which the N-sulfinyl group dictates the orientation of approach. After removal of the N-sulfinyl group from 158, treatment with crotonaldehyde produced imine 161, acid-mediated intramolecular Mannich reaction of which afforded the two (+)-piperidinones 162 and 163 in isolated yields of 18% and 58%, respectively with no observable epimerisation of the ethyl group adjacent to the ketone. The relative stereochemistry of both products was confirmed by appropriate NOE experiments. For comparison, two further diastereomers of these piperidinones (not illustrated) were similarly prepared from the minor condensation product 159. To complete the synthesis, N-allylation of 163 yielded (+)-164, ring-closing metathesis of which was performed with the Grubbs first-generation catalyst to give, after hydrogenation, the indolizidinone (+)-165 in 72% yield. However, defunctionalisation of the ketone proved to be problematic. In the end, reduction to the alcohol (+)-166 with sodium borohydride (90%) was followed by free-radical deoxygenation of the corresponding phenylthionocarbonate with tributyltin hydride to yield (−)-indolizidine 223A 100 in 9.3% overall yield from the N-sulfinylimine (−)-157.
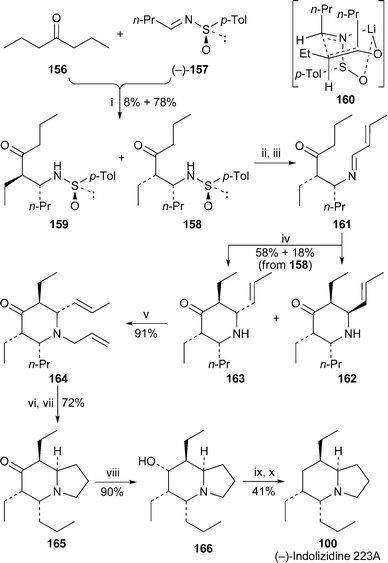 | ||
| Scheme 13 Reagents and conditions: i, 156 + LiHDMS, Et2O, −78 °C, 1 h, add over 30 min to 157, Et2O, −78 °C, then 1 h; ii, TFA, MeOH, rt, 2 h; iii, MeCHCHCHO, MgSO4, CH2Cl2, rt, 2 h; iv, p-TsOH·H2O, C6H6, rt, 40 h; v, H2CCHCH2Br, Na2CO3, EtOH, reflux, 6 h; vi, (PCy3)2Cl2RuCHPh (5 mol%), CH2Cl2, reflux, 2 h; vii, H2 (1 atm), 10% Pd/C, rt, 4 h; viii, NaBH4, MeOH, −78 °C, 2 h; ix, PhOC(S)Cl, py, CH2Cl2, 0 °C to rt, 24 h; x, Bu3SnH, AIBN, C6H6, reflux, 4 h, then KF, CH2Cl2. | ||
Ma and co-workers have improved on their original synthesis of (−)-indolizidine 223A50 (cf.ref. 7g) by the short, efficient route shown in Scheme 14.51 Although the new synthesis began with the same initial transformations of the unsaturated ester 167 into the amino alcohol 168, it diverged at this point by first replacing the alcohol by chloride before hydrogenolytic removal of the N-benzyl substituents. When the resulting ammonium salt 169 was heated with the substituted propiolic ester 170 in the presence of potassium carbonate, a reaction cascade involving sequential displacement of iodide, intramolecular conjugate addition to the alkynoate and a final cyclisation by displacement of chloride took place, giving the bicyclic vinylogous urethane 171 in 80% yield. Diastereofacially selective cis-hydrogenation of the double bond followed by base-catalysed epimerisation of the ester to the more stable equatorial position produced 172 as a single isomer in 75% yield. Finally, reduction of the ester to the alcohol 173, chain extension by Swern oxidation and Wittig methylenation, and catalytic reduction of the resulting terminal alkene afforded the target alkaloid (−)-100 in 11 linear steps and 14.5% overall yield from the unsaturated ester 167. This appears to be the most efficient route to the alkaloid to date.
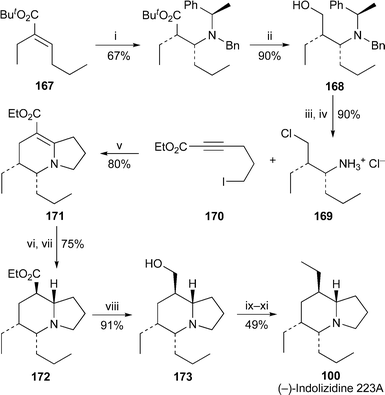 | ||
| Scheme 14 Reagents and conditions: i, (R)-PhCH(Me)NBnLi, THF, −78 °C, 1 h, −40 °C, 1 h, then quench with 2,6-di-tert-butyl-4-methylphenol, THF, −78 °C to rt; ii, LiAlH4, THF, reflux, 12 h; iii, SOCl2, CHCl3, 0 °C, then reflux, 1 h; iv, H2 (50 atm), 20% Pd(OH)2/C, MeOH, 40 °C, 48 h; v, K2CO3, 4 Å molecular sieves, MeCN, reflux; vi, H2 (1 atm), PtO2, AcOH, rt, 2 h; vii, NaOEt (cat.), EtOH, reflux, 5 h; viii, LiAlH4, THF, rt, 1 h; ix, (COCl)2, DMSO, NEt3, CH2Cl2, −78 °C, 15 min, then warm ro rt; x, Ph3PCH2, THF, −78 °C, 1 h, then warm to 0 °C; xi, H2 (1 atm), 10% Pd/C, EtOAc, rt, 2 h. | ||
Before the structure of indolizidine 223A had been corrected, RajanBabu and co-workers had embarked on a strategy for preparing trisubstituted indolizidines with 5,6-trans-stereochemistry by palladium-mediated cyclisation of allene-aldehyde substrates in the presence of a trialkylstannylsilane.52 In a highly relevant model study (Scheme 15), the racemic precursor 174, used as a mixture of four diastereomers, was rapidly cyclised with trimethyl(tri-n-butylstannyl)silane and allylpalladium chloride dimer to give a mixture of the four indolizidinones 175–178, bearing five contiguous stereogenic centres, in a combined yield of 82%. The relative configurations of the products were established by a combination of two-dimensional NMR experiments and X-ray crystallographic analyses. Manipulation of these isomers provided further proof of concept. For example, desilylation of the major isomer 175 gave 179, the structure of which was confirmed by single-crystal X-ray analysis. Hydrogenation followed by free-radical deoxygenation of the alcohol via the phenylthionocarbonate produced the lactam 180, borane reduction of which gave (±)-5,8-di-epi-indolizidine 223A 181 in 32% overall yield from 175. Desilylation of the second most abundant product, 176, afforded 182, but the dehydration of the axially-orientated alcohol produced mainly the unconjugated diene 183 instead of the expected conjugated diene, thereby destroying the stereochemistry at C-8 and preserving that at C-6—precisely the opposite effect to what is required for indolizidine 223A. Catalytic hydrogenation at the more exposed face of 183 followed by borane reduction of the lactam gave mainly (±)-6,7-di-epi-indolizidine 223A 184. Finally, desilylation of the minor isomer 177 yielded alcohol 185, which was converted by two different methods into the alkene 186. A final catalytic hydrogenation produced the lactam 187. Although the borane reduction of this lactam was not performed, it would presumably give 8-epi-indolizidine 223A.
![Reagents and conditions: i, Me3SiSnBu3, [(allyl)PdCl]2 (5 mol%), THF, rt, 10 min; ii, Bu4NF, THF–DMSO (2 : 1), 75 °C, 2.5–3 h; iii, H2 (60 psi), 5% Pd/C, EtOH, rt, 4 h; iv, PhCO(S)Cl, DMAP, CH2Cl2, rt, 3 d; v, Bu3SnH, AIBN, PhMe, 100 °C, 2.5 h; vi, BH3·THF, THF, rt, 24 h; vii, C6F5OC(S)Cl, DMAP, CH2Cl2, reflux, 18 h; viii, H2 (60 psi), 5% PtO2/C, EtOH, rt; ix, POCl3, py, rt, 6 h; x, H2 (200 psi), [Ir(COD)py(PCy3)]+ PF6− (5 mol%), CH2Cl2, rt, 12 h.](/image/article/2007/NP/b509525p/b509525p-s15.gif) | ||
| Scheme 15 Reagents and conditions: i, Me3SiSnBu3, [(allyl)PdCl]2 (5 mol%), THF, rt, 10 min; ii, Bu4NF, THF–DMSO (2 : 1), 75 °C, 2.5–3 h; iii, H2 (60 psi), 5% Pd/C, EtOH, rt, 4 h; iv, PhCO(S)Cl, DMAP, CH2Cl2, rt, 3 d; v, Bu3SnH, AIBN, PhMe, 100 °C, 2.5 h; vi, BH3·THF, THF, rt, 24 h; vii, C6F5OC(S)Cl, DMAP, CH2Cl2, reflux, 18 h; viii, H2 (60 psi), 5% PtO2/C, EtOH, rt; ix, POCl3, py, rt, 6 h; x, H2 (200 psi), [Ir(COD)py(PCy3)]+ PF6− (5 mol%), CH2Cl2, rt, 12 h. | ||
Some results from a failed attempt at a synthesis of (−)-indolizidine 223A are of interest.53 The two enantiomerically pure thiolactams 188 and 189 underwent Eschenmoser sulfide contraction with ethyl or benzyl bromoacetate to give the vinylogous urethanes 190 and 191 in yields of 97% and 96% (R = Et) or 90% and 81% (R = Bn), respectively (Scheme 16). Hydrolysis of the tert-butyl ester of 190 with trifluoroacetic acid followed by treatment with acetic anhydride and potassium carbonate afforded the (5R,6R)-(+)-indolizidinones 192 in 85% (R = Et) and 80% (R = Bn) yields. However, when the same cyclisation protocol was applied to 191, a 3 : 1 mixture of indolizidinones 192 and 193 was isolated in about 52% yield, epimerisation at the enolisable C-6 site having occurred under the reaction conditions. Chemoselective reduction of the CC double bond of 192 was accomplished with lithium aluminium hydride in THF at −78 °C to give mixtures of 194 and 195 in good yield. Hydride reduction of the minor isomer 193 (R = Et) also gave a mixture of isomers 194 and 195 in which the former dominated (4 : 1), showing that epimerisation at C-6 is a persistent problem in this series of compounds. Unfortunately, all attempts to prepare suitable derivatives of 194 for defunctionalisation of the ketone at C-7 failed; with hindsight, the reduction–radical deoxygenation approach adopted by Davis (vide supra, Scheme 13) would probably have been a better alternative. The mixture of β-keto esters 194 and 195 (R = Et) could, however, be ethylated to give diastereomers of 196 in moderate yield, but attempted hydrolysis and decarboxylation of the ester failed, although there was no difficulty in hydrolysing 192 (R = Et) itself to produce enaminone 197. Attempts to alkylate this nucleophilic intermediate exclusively on the enamine carbon atom (C-8) were inconclusive.
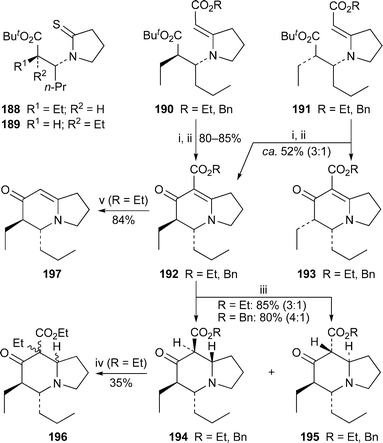 | ||
| Scheme 16 Reagents and conditions: i, TFA, rt, 3 h; ii, Ac2O, K2CO3, MeCN, rt, 18 h, then reflux, 3 h (R = Et), or 50 °C, 36 h (R = Bn); iii, LiAlH4, THF, −78 °C, 2.5 h, then warm to rt; iv, NaH, THF, 0 °C, 1.5 h, then EtI, rt, 7 d; v, aq. NaOH (1 M), reflux, 1.5 h, then conc. HCl, reflux. 30 min. | ||
2.5 Synthesis of homopumiliotoxins
A formal synthesis of (±)-homopumiliotoxin 223G 94 by Chen et al. commenced with base-induced cyclocondensation between ethyl 2-methoxyacrylate 198 and the sulfonylacetamide 199 to give the substituted glutarimide 200 in 73% yield54 (Scheme 17). Regioselective reduction and reaction with allyltrimethylsilane via an acyliminium ion yielded 201 as a mixture of three diastereomers—of no consequence, since later steps entailed convergence to a single product. The second ring was constructed by metathesis of 201 with Grubbs catalyst, which produced the quinolizidinone 202 in 71% yield. Hydrogenation, cleavage of the methyl ether and reductive removal of the sulfone substituent afforded the alcohol 203, which was then oxidised with Jones reagent. Addition of methylmagnesium bromide to the resulting ketone 204 was stereoselective, giving 205 as the sole product in 76% yield, and thereby completing the formal synthesis55 of 94.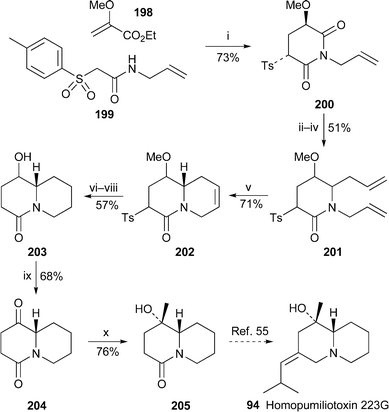 | ||
| Scheme 17 Reagents and conditions: i, NaH, THF, rt, 7 h; ii, LiAlH4, THF, −10 °C, 3 h; iii, BF3·Et2O, MeOH, rt, 15 h; iv, H2CCHCH2SiMe3, BF3·Et2O, CH2Cl2, 0 °C to rt; v, (PCy3)2Cl2RuCHPh (4 mol%), CH2Cl2, rt, 12 h; vi, H2, 10% Pd/C, MeOH, rt, 3 h; vii, Me3SiCl, NaI, MeCN, reflux, 15 h; viii, Na/Hg (6%), Na3PO4, MeOH, rt, 2 h; ix, Jones reagent, Me2CO, 0 °C, 10 min; x, MeMgBr, THF, rt, 10 min. | ||
3 Polygonatum alkaloids
In 1997, the isolation of the 5,6,7,8-tetrahydroindolizinone 206 from extracts of the rhizomes of Polygonatum sibiricum (Liliaceae) was reported in the Chinese literature.56 This compound, unnamed at the time, possesses a curious ethoxymethyl substituent; it is also unusual in its occurrence in a plant family from which indolizine derivatives (and, indeed, pyrrole alkaloids) had never previously been isolated. The same workers have now reported their results in a more accessible journal,57 but do not cite their original publication. Compound 206, to which the name polygonatine B has now been given, was accompanied by the parent alcohol polygonatine A 207, which was not reported in the earlier article. The structures of both alkaloids were elucidated with the aid of IR, UV, MS and one-and two-dimensional NMR spectroscopic data. However, since ethanol was used in the extraction of the alkaloids, one is tempted to repeat the speculation (cf.ref. 7h) that polygonatine B may be an artifact of the isolation procedure, as it is known that 2-hydroxymethylpyrroles and their esters are susceptible to nucleophilic displacement of the oxygen substituent. Interestingly enough, compound 207 has previously been reported as the unexpected major product (65% yield) of acyl radical cyclisation from the reaction between Se-phenyl 3-(2-formyl-1H-pyrrol-1-yl)propaneselenoate and tributyltin hydride.58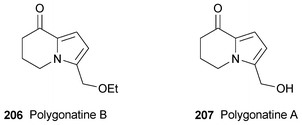
4 Prosopis alkaloids
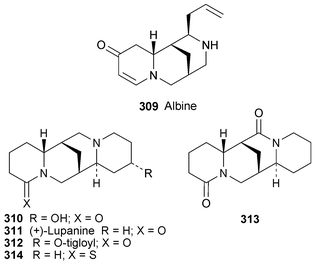
Six Prosopis alkaloids 208–213, the last four of which were recently reported as new metabolites of Prosopis juliflora59 (cf.ref. 7h), have been evaluated as plant growth inhibitors.60 With the exception of juliprosopine 208, the alkaloids were more effective in inhibiting the growth of roots rather than shoots; and all showed some activity towards the growth of both dicotyledons and monocotyledons. The most active compound was juliprosine 209, followed by a mixture of the oxo analogues 211 and 212 and juliprosopine 208, while secojuliprosopinal 213 was only weakly active.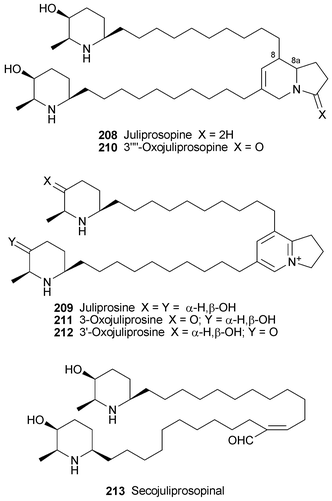
The first syntheses of juliprosopine and juliprosine, by Snider and Neubert, have as their central feature the construction of the indolizine nucleus by a biomimetic Chichibabin pyridine synthesis61 (Scheme 18). The aldehyde component 214 was prepared from the bromopyridinol 215 in six steps, which included a Kumada coupling between the protected pyridinol 216 and the Grignard reagent 217. When two equivalents of the aldehyde 214 were condensed with one equivalent of 1-pyrroline 218 in acetic acid at ambient temperature, the bis(Troc) derivative 219 was isolated in 49% yield. Deprotection with zinc and hydrochloric acid then afforded the bis(hydrochloride) salt of juliprosine 209 in 72% yield. Neutralisation with aqueous ammonium hydroxide yielded the parent alkaloid with chloride as the counter-ion. Although reduction of juliprosine itself to give juliprosopine was unsuccessful, the derivative 219 could be reduced with sodium borohydride to provide a separable mixture of Troc-protected indolizidines 220 and 221 (39% each). Deprotection of these isomers was accomplished by alkaline hydrolysis to give 208 and 222, both in 94% yield. Comparison of spectroscopic data showed that the trans isomer 208 was identical to natural juliprosopine—a significant result, since the alkaloid's 8,8a relative stereochemistry has never been established previously, even though the natural product was first isolated more than a quarter of a century ago.
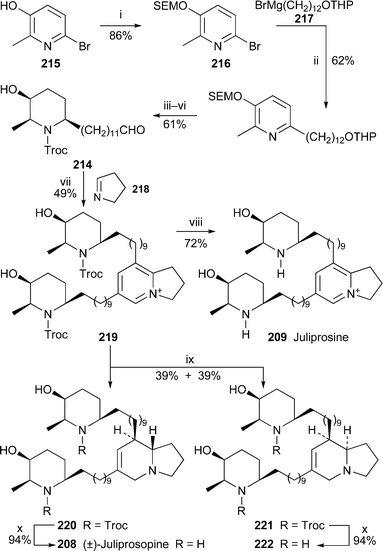 | ||
| Scheme 18 Reagents and conditions: i, SEMCl, Pri2NEt; ii, 217, Ni(dppp)Cl2, THF, 50 °C, 18 h; iii, H2 (50 psi), 5% Rh/Al2O3, MeOH, 24 h; iv, TrocCl, CH2Cl2–py (20 : 1), 25 °C, 24 h; v, aq. H2SO4 (2 M), MeOH, 65 °C, 24 h; vi, TEMPO, aq. KBr (0.5 M), CH2Cl2, 0 °C, then aq. NaOCl (0.35 M), 3 h; vii, 214 (2 equiv.), 218 (1 equiv.), AcOH, 25 °C, 24 h; viii, Zn, conc. HCl, MeOH, 65 °C, 3 h; ix, NaBH4, EtOH, 25 °C, 30 min, then reflux, 30 min; x, KOH, PriOH, H2O, 100 °C (sealed tube), 2 d. | ||
5 Elaeocarpus alkaloids
Extracts from the leaves of the Australian rainforest tree Elaeocarpus grandis, selected for further investigation after high-throughput screening indicated interaction with the human δ-opioid receptor, yielded the known metabolite (−)-isoelaeocarpiline 223 and the two new alkaloids (+)-grandisine A 224 and (+)-grandisine B 225.62 These novel compounds, the structures and relative configurations of which were elucidated by means of one- and two-dimensional NMR spectroscopic techniques, have unprecedented skeletons. Grandisine B, in particular, is the only known alkaloid to contain both indolizidine and isoquinuclidone moieties. Interestingly, the isoquinuclidone unit 226 is the known alkaloid mearsine, which was isolated from Peripentadenia mearsii, another member of the Elaeocarpaceae, some decades ago. Despite the apparent structural dissimilarity of 223–225, the authors propose a plausible mutual biogenesis from ornithine and six acetate units via227, intramolecular aldol reaction of which would give the common precursor 228 (Scheme 19). Reduction to 229 followed by enolisation and intramolecular conjugate addition would then yield isoelaeocarpiline 223. Alternatively, conjugate addition of an ammonia equivalent to 229 would give 230, from which grandisine B 225 could be derived by intramolecular imine formation. Finally, retro-aldol reaction of 228 to produce 231 followed by selective reduction of the methyl ketones would give 232, cyclisation and dehydration of which would lead to grandisine A 224. Since the absolute stereochemistry of isoelaeocarpiline is known to be as illustrated, the biogenetic scheme also implies that the absolute configurations of 224 and 225 might be as shown. The purified alkaloids 223–225 were found to inhibit the binding of [125I]-deltorphin II to human δ-opioid receptors (IC50 11, 2.7 and 52.2 µM, respectively).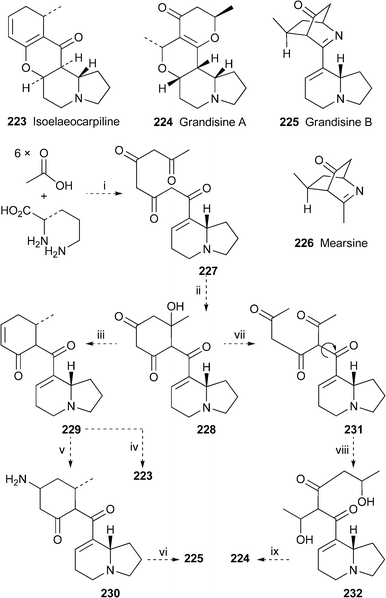 | ||
| Scheme 19 Possible biogenetic processes: i, condensation; ii, aldol reaction; iii, reduction; iv, enolisation and cyclisation; v, amination; vi, cyclisation and dehydration (imine formation); vii, retroaldol reaction; viii, reduction; ix, cyclisation, dehydration. | ||
6 Indolizinium alkaloids
The antimicrobial activity demonstrated by extracts from the wood of the South American tropical tree Aniba panurensis (Lauraceae) appears to be due to the novel indolizinium alkaloid 233, the trifluoroacetate salt of which was characterised by mass spectrometry and a comprehensive range of NMR spectroscopic techniques.63 The (Z)-configuration of both unsaturated chains was deduced from the observed coupling constants of 11.4 and 11.3 Hz. Compound 233 appears to be the first reported example of a naturally occurring tetrasubstituted indolizinium alkaloid. In a series of screening tests for antimicrobial activity, it proved to be a powerful inhibitor of an azole-resistant strain of Candida albicans (MIC50ca. 0.25 µg ml−1), and similar antifungal activity was found towards several other Candida species and towards Cryptococcus neoformans. It also showed promising but unselective activity in tests with a panel of human tumour cell lines (LC50ca. 10−5 M), but its potent hemolytic activity makes it unsuitable for development as an anticancer or antifungal agent.Snider and Neubert have reported a short preparation of the indolizinium alkaloid ficuseptine 234 by a route in which the key step was an intramolecular Chichibabin pyridine synthesis similar to the one they used in making juliprosine61 (see Section 4, Scheme 18). In this case, condensation of two equivalents of 4-methoxyphenylacetaldehyde 235 with 4-aminobutanal dimethyl acetal 236 in acetic acid at 95 °C followed by work-up with sodium chloride and purification on an ion exchange resin gave the chloride salt of ficuseptine directly in 52% yield.
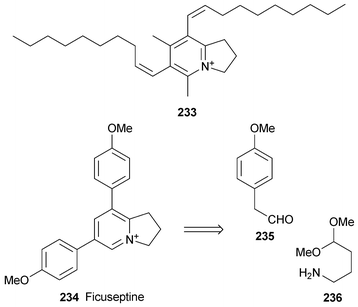
7 Phenanthroindolizidine alkaloids
Condensation of veratraldehyde 237 with homoveratric acid 238 in the presence of acetic anhydride and triethylamine followed by methylation of the resulting carboxylic acid constituted the initial steps in a new synthesis of (S)-(+)-tylophorine 239 by Jin et al.64 (Scheme 20). The product 240, an 88 : 12 mixture of (Z)- and (E)-isomers, underwent coupling of the aromatic rings with vanadyl chloride to produce the phenanthrene 241. A further two steps yielded the chloride 242, reaction of which with the anion of tert-butyl L-pyroglutamate afforded 243. After in situ preparation of the corresponding acid chloride, intramolecular Friedel–Crafts acylation catalysed by tin tetrachloride brought about the construction of the phenanthroindolizidine skeleton, giving 244 in a yield of 92% over the two steps. Deoxygenation of 244 by reduction with lithium aluminium hydride followed by triethylsilane in acidic medium completed this efficient synthesis of (+)-239 in ten linear steps and an overall yield of 60% based on veratraldehyde.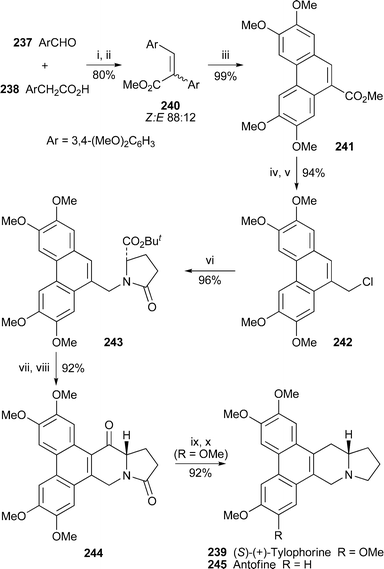 | ||
| Scheme 20 Reagents and conditions: i, Ac2O, NEt3, reflux; ii, CH2N2, Et2O, rt; iii, VOCl3, CH2Cl2, −78 °C to rt; iv, LiAlH4, THF; v, CCl4, PPh3, CHCl3; vi, NaH, tert-butyl pyroglutamate, DMSO, rt; vii, TFA, CH2Cl2; viii, (COCl)2, DMF, CH2Cl2, then SnCl4, reflux; ix, LiAlH4, THF, reflux; x, Et3SiH, TFA. | ||
Tylophora indica, one of the principal sources of tylophorine, is an important medicinal plant in India. In attempts to increase the production of biologically active alkaloids by the plant, intact shoots as well as leaf and stem explants were inoculated with suspensions of Agrobacterium rhizogenes strain A4 in order to induce the growth of genetically transformed roots at the wounded sites.65 When these roots were excised and cloned in culture under a variety of conditions, most grew faster and produced much higher levels of tylophorine than untransformed controls, in some cases releasing the alkaloid into the culture medium. The technique thus shows potential for producing useful levels of a metabolite with proven immunosuppressive, antitumour and anti-inflammatory properties.
The related alkaloid antofine 245 also shows antitumour activity and pronounced cytotoxicity. Fluorescence spectroscopy has recently been used to show that the alkaloid (referred to in the article by the long-superseded name ‘tylophorine B’, and also incorrectly termed ‘autofine’) is able to bind to various synthetic oligodeoxyribonucleotides at submicromolar concentrations, the interaction with bulged DNA being particularly strong (Kd 0.018 µM).66 Once bound, 245 also appears to stabilise the bulged hairpin oligonucleotide, as demonstrated by thermal melting experiments. The findings may shed light on the mode of action of phenanthroindolizidine alkaloids in biological systems, and also assist the rational design of analogues with sequence-specific DNA binding ability.
8 Myrtine and epimyrtine
A short synthesis of (±)-myrtine 246 by Back and co-workers commenced with the conjugate addition of the homopipecolic ester (±)-247 to the alkynylsulfone 248, followed by base-induced cyclisation of the product 249 to give the bicyclic enaminone 250 in 42% overall yield67 (Scheme 21). Reduction with sodium borohydride produced a saturated alcohol as a single diastereomer of undetermined stereochemistry, Swern oxidation of which not only re-oxidised the ketone, but also effected α-chlorination at C-3. X-Ray crystallography of the product 251 revealed the relative stereochemistry of the axial methyl group at C-4 and its cis-relationship to the hydrogen atom at C-10. Among several other standard oxidation methods attempted on the intermediate alcohol, Moffatt oxidation proved most successful, giving ketone 252 in a moderate yield of 44%. The synthesis of (±)-myrtine rac-246 was completed by reductive desulfonation with lithium 4,4′-di-tert-butylbiphenylide (LiDBB).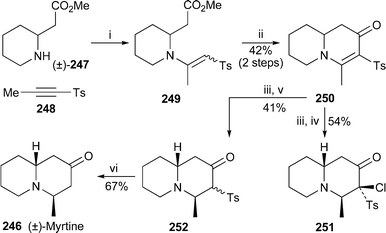 | ||
| Scheme 21 Reagents and conditions: i, MeOH, rt, 20 h; ii, LDA, THF, −78 °C, 5 min; iii, NaBH4, MeOH, rt, 30 min; iv, (COCl)2, DMSO, CH2Cl2, −78 °C, 30 min, then NEt3, rt, 18 h; v, DCC, DMSO, py, C6H6, 0 °C, then TFA, rt, 18 h; vi, LiDBB, THF, 0 °C, 2 min. | ||
A cascade of iminium ion cyclisations initiated by reaction between the monoprotected dialdehyde 253 and the optically active amine-substituted allylsilane 254 and terminated by the addition of cyanide ion produced a 90% yield of the quinolizidines 255 as a mixture of diastereomers at C-1068 (Scheme 22). After reducing this compound—in effect a masked iminium ion—with sodium cyanoborohydride to give a 95 : 5 mixture of products 256, Martin and co-workers simply ozonolysed the exo-methylene substituent to produce an inseparable mixture (95 : 5) of (−)-epimyrtine 257 and (+)-myrtine 246. As the authors comment, this is an extraordinarily concise route to the target alkaloids.
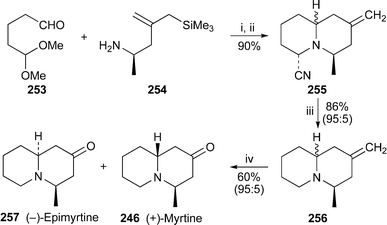 | ||
| Scheme 22 Reagents and conditions: i, 4 Å molecular sieves, MeCN, 0 °C, 2 h and rt, 12 h, then TFA, 0 °C, 2 h; ii, NaCN, H2O, 0 °C, then rt, 4 h; iii, NaBH3CN, MeCN; iv, TFA, then O3, CH2Cl2–MeOH, −78 °C, 5 min, then Me2S, rt, 12 h. | ||
9 Plumerinine
The quinolizidinol structure 258 proposed for plumerinine when its isolation from Plumeria rubra was reported in 198969 was discredited in 2002 after Comins et al. showed that the spectra of their synthetic product did not agree with those recorded on the natural product70 (cf.ref. 7i). Maldaner and Pilli have now prepared another two diastereomers of 258 as shown in Scheme 23.71 Addition of the silyl enol ether 259 to the acyliminium ion generated from 260 in the presence of trimethylsilyl triflate yielded the 2,3,6-trisubstituted piperidine 261, the relative stereochemistry of which was established by means of NOE experiments. Once the Boc protecting group had been removed, it proved difficult to induce cyclisation by intramolecular conjugate addition; the ring closure was eventually achieved in modest yield by extended heating with aqueous ammonia solution. However, the product isolated was not the expected quinolizidinone 262 but the isomer 263 in which the C-2/C-3 relative stereochemistry of the precursor had not been conserved. The orientations of the substituents and conformation of this product were established by NOE studies and corroborated by molecular mechanics methods, which showed that the compound adopted the cis-fused bicyclic structure 263′. The scrambling of the stereochemistry can be rationalised by invoking equilibria involving retro-Mannich or retro-Michael processes from the expected products 262 in order to attain the thermodynamically most stable structure. Reduction of 263 with lithium aluminium hydride produced a mixture of diastereomers 264 and 265 in a ratio of 1.2 : 1, whereas product 264, which has an axial alcohol substituent, was obtained exclusively with lithium tris(sec-butyl) borohydride as the reductant. Unfortunately, neither product gave spectra that matched those of natural plumerinine. The structure of the alkaloid thus remains a mystery.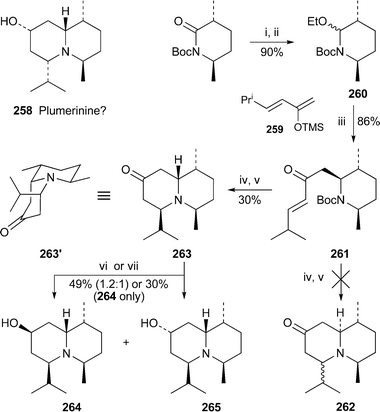 | ||
| Scheme 23 Reagents and conditions: i, LiBHEt3, THF, −78 °C; ii, HCl, EtOH (pH 3); iii, 259, TMSOTf (0.4 equiv.), CH2Cl2, −78 °C; iv, TFA, CH2Cl2, rt; v, aq. NH3, MeOH, 60 °C (sealed tube), 24 h; vi, LiAlH4, THF; vii, Li(s-Bu3)BH, THF, −78 °C. | ||
10 Lycopodium alkaloids
A suite of novel alkaloids isolated from methanolic extracts of the club moss Lycopodium cernuum included the simple (+)-2,4-dimethylquinolizidine 266, given the name (+)-cermizine C, and a new C16N2-type alkaloid, (−)-cermizine D 267.72 Chloroform extracts of a different club moss, L. chinense, contained another two new alkaloids, (−)-senepodine G 268 and the related iminium species (−)-senepodine H 269. A full array of NMR and other spectroscopic methods were used to characterise the new alkaloids, the relative stereochemistries of which were determined by means of selected NOESY correlations and corroborated by Monte Carlo simulations. In addition, the absolute configuration of senepodine H, which has a secondary alcohol substituent, was ascertained by NMR spectroscopic analysis of its (R)- and (S)-Mosher's esters. Cermizine D and senepodines G and H exhibited cytotoxicity against murine lymphoma L1210 cells in vitro (IC50 7.5, 7.8 and 8.2 µg ml−1, respectively). The more complex alkaloid (−)-senepodine F 270 isolated from L. chinense was also characterised spectroscopically, although broadening of NMR spectroscopic signals due to slowly interconverting amide rotamers made complete assignment difficult.73 However, hydrolysis of 270 with aqueous hydrochloric acid produced the deacetyl derivative 271, which gave well-resolved signals that facilitated a full assignment of the alkaloid's structure and relative stereochemistry.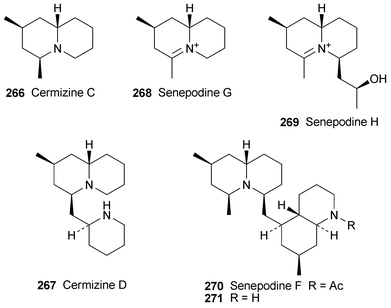
11 Lythraceae alkaloids
A concise synthesis of (−)-lasubine II 272 by Back and Hamilton, reported in a communication in 200274 (cf.ref. 7j), has now been published with amplifying discussion.67 The route is similar to that shown for (±)-myrtine (see Section 8, Scheme 21).Blechert's ring opening–ring closing–cross metathesis strategy for constructing alkaloid skeletons has been effectively applied to a novel synthesis of (−)-lasubine II from the chiral monoacetylated cyclopentenediol 27375 (Scheme 24). After the high-yielding (93%) metathetical rearrangement of the cyclopentene 274via its silyl ether to the tetrahydropyridine 275, cross metathesis with 3,4-dimethoxystyrene in the presence of the Grubbs–Hoveyda catalyst 126 (cf.Scheme 10) followed by oxidation of the alcohol gave the unsaturated ketone 276 in 85% yield. Alternatively, 274 could be oxidised to the cyclopentenone 277 initially, after which a one-pot metathesis cascade with the Grubbs second-generation catalyst in tandem with 3,4-dimethoxystyrene produced 276 directly, although in a poorer overall yield of 48%. Removal of the N-Boc group and treatment with base induced the desired intramolecular conjugate addition to give a 2 : 3 mixture of the two diastereomeric ketones 278 and 279 in a combined yield of 77%. Compound 279, in which the aryl substituent is axial, could be epimerised completely to 278 by prolonged heating with methanolic sodium hydroxide, presumably through a retro-Michael–recyclisation equilibrium. The synthesis of (−)-lasubine II 272 was completed by reducing the alkene and carbonyl groups of 278, the latter task being accomplished stereoselectively with L-Selectride at low temperature.
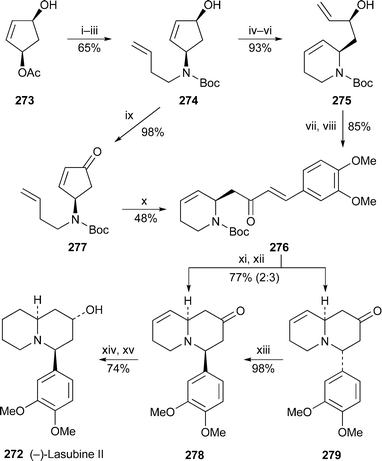 | ||
| Scheme 24 Reagents and conditions: i, H2CCHCH2CH2NH2, NEt3, THF, rt, 1 h; ii, add Pd(PPh3)4 (5 mol%), THF, 40 °C, 15 h; iii, Boc2O, NEt3, MeOH, 70 °C 4 h; iv, TBDMSCl, imidazole, DMF, rt, 48 h; v, (PCy3)2Cl2RuCHPh (5 mol%), H2CCH2, CH2Cl2, rt, 57 h; vi, Bu4NF, THF, rt, 1 h; vii, Dess–Martin periodinane, CH2Cl2, rt, 30 min; viii, Grubbs–Hoveyda catalyst 126 (4 mol%), 3,4-dimethoxystyrene, CH2Cl2, reflux, 14 h; ix, MnO2, Me2CO, rt, 15 h; x, Grubbs II catalyst (5 mol%), 3,4-dimethoxystyrene, CH2Cl2, reflux 12 h; xi, TFA, CH2Cl2, 0 °C, 1 h; xii, DBU, CH2Cl2, rt, 24 h; xiii, aq. NaOH (2 M), MeOH, rt, 48 h; xiv, H2, 10% Pd/C, EtOAc, rt, 12 h; xv, L-Selectride, THF, −78 °C, 12 h, then rt, 3 h. | ||
The opening gambit in a new asymmetric approach to 4-arylquinolizidine alkaloids by Kibayashi and co-workers was the diastereofacially selective addition of allyllithium to the chiral oxime ether 28076 (Scheme 25). Removal of the chiral auxiliary from the adduct 281, isolated as a 4 : 1 mixture of diastereomers, by cleavage with zinc and acetic acid, produced the chiral homoallylic amine 282, which was protected as the phthalimide derivative before Wacker oxidation of the terminal vinyl group to give 283. The liberated amine, protected as the ketal 284, participated in a Mannich-type cyclisation with aldehyde 285 to give the 2,6-cis-disubstituted piperidine 286 in 88% yield. A standard sequence of transformations was then employed to furnish the quinolizidin-2-ol 287, which is actually the silylated derivative of another known lythraceous alkaloid. In this case, however, 287 was acylated with the protected feruloic anhydride 288, after which removal of the protecting groups yielded (+)-abresoline 289. This is the first reported synthesis of the alkaloid in optically active form. Oddly enough, neither the optical rotation nor the absolute configuration of the natural product have been reported, although the absolute configuration shown in 289 seems plausible by analogy with (−)-lasubine II 272.
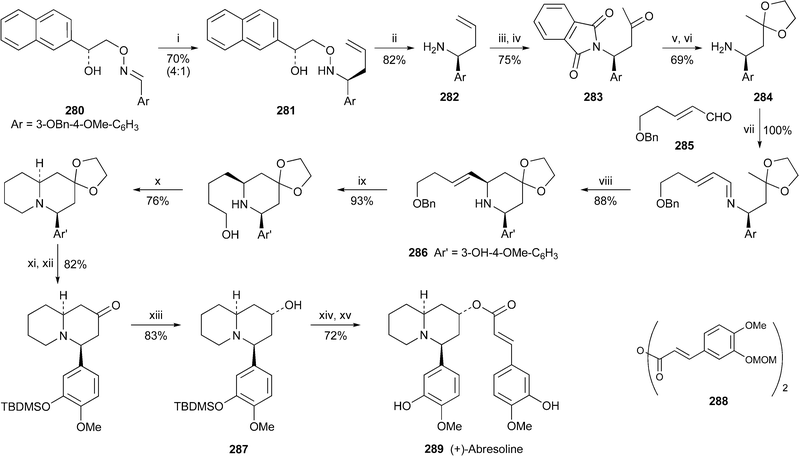 | ||
| Scheme 25 Reagents and conditions: i, H2CCHCH2Li, PhMe–Et2O (1 : 1), 45 °C, 20 min; ii, Zn, AcOH, THF–H2O, 60 °C; iii, phthalic anhydride, PhMe, reflux; iv, PdCl2, CuCl2, O2, DMF–H2O, 85 °C; v, HOCH2CH2OH, p-TsOH, C6H6, reflux; vi, H2NNH2·H2O, EtOH, reflux; vii, 285, CHCl3, rt; viii, TiCl4, CH2Cl2, 0 °C; ix, H2, Pd/C, THF–MeOH; x, CBr4, PPh3, NEt3, CH2Cl2, rt; xi, HCl, THF, reflux; xii, TBDMSOTf, py, 0 °C; xiii, LS-Selectride, THF, −78 °C; xiv, 288, DMAP, py, reflux; xv, BF3·Et2O, MeCN, 0 °C. | ||
12 Nuphar alkaloids
The recently communicated formal synthesis of (−)-deoxynupharidine 290 by Harrity and-co-workers77 (cf.ref. 7k) has now been published in full with some modifications and extensions.78 In the improved final stages (Scheme 26), the advanced intermediate 291 was treated with 3-furyllithium, after which in situ reduction of the resulting carbinolamine with diisobutylaluminium hydride produced 292 as a single diastereomer in 76% yield. Hydrogenation of this intermediate over rhodium on alumina then afforded the target alkaloid (−)-290 in a reasonable yield of 60%. However, a disappointingly unselective hydroboration of 292 with borane–dimethyl sulfide complex followed by oxidative work-up produced a 1 : 1 mixture of (−)-nupharolutine 293 and (−)-castoramine 294 in 52% yield. Alternatively, diastereofacially selective dihydroxylation of 292 yielded diol 295, the primary alcohol substituent of which could be removed by tosylation and reduction to give (−)-nupharolutine 293 as the sole product.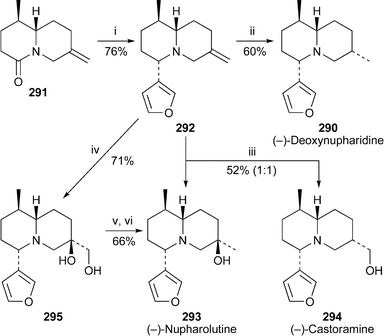 | ||
| Scheme 26 Reagents and conditions: i, 3-furyllithium, Et2O, −78 °C to rt, 1.5 h, then DIBAL, CH2Cl2, rt, 1 h; ii, H2 (1 atm), 5% Rh/Al2O3, EtOAc, rt, 3 h; iii, BH3·Me2S, THF, 0 °C to rt, 1 h; then aq. NaOH (1 M), aq. H2O2 (30%), 1 h; iv, OsO4 (cat.), NMO, Me2CO–H2O (4 : 1), rt, 1 h; v, p-TsCl, NEt3, DMAP, CH2Cl2, rt, 48 h; vi, LiAlH4, THF, 0 °C, then reflux, 3 h. | ||
13 Alkaloids of the lupinine–cytisine–sparteine–matrine–Ormosia group
13.1 Occurrence, analysis and chemical ecology
For the first time in years, no new compounds appear in the list of lupin alkaloids isolated from, or detected in, new plant sources during the review period79–85 (Table 1). However, some of the new results are chemotaxonomically interesting. For example, the isolation of typical Ormosia alkaloids from fruits and leaves of the Vietnamese plant Connarus paniculatus, which belongs to the family Connaraceae, is surprising, since such alkaloids have previously been found only in members of the Fabaceae (Leguminosae). Detailed NMR spectroscopic studies revealed both the structures and the relative conformations of the six alkaloids 18-epipiptanthine 296, homoormosanine 297, homopodopetaline 298, ormosanine 299, piptanthine 300 and podopetaline 301, the last-named alkaloid having been isolated as its monohydrochloride salt.79 The relatively rigid, all-chair conformations 296′ and 300′ for 18-epipiptanthine and piptanthine were deduced largely from NOESY effects and analysis of coupling constants, with intramolecular hydrogen bonding between N-12 and N-23 assisting the attainment of the observed conformations. Chemical exchange between N-12⋯HN-23 and N-12H⋯N-23 was inferred from the line broadening of several adjacent protons. For ormosanine, a trans ring junction between rings C and D appears to correlate with a preferred boat conformation for ring C in solution, as shown in 299′. A similar boat conformation for ring C in homoormosanine and homopodopetaline is stabilised by the additional ring F. However, the spectroscopic data indicate a preferred cis C/D ring junction and ring C-chair conformation for podopetaline.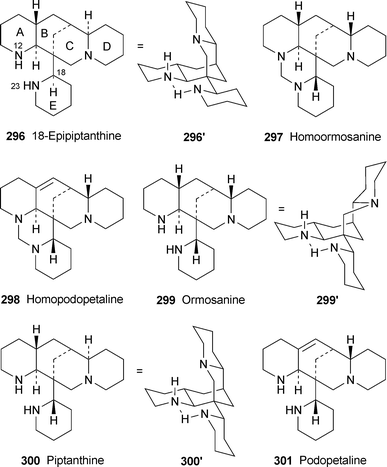
| Speciesa | Alkaloidb | Ref. |
|---|---|---|
| a Unless otherwise specified, all of the listed species are members of the Fabaceae (Leguminosae).b Only new records for a given species are listed in the Table; previously reported alkaloids from the species are not listed. Structures of known alkaloids, if not specifically numbered, may be found in previous reviews in this series.c L. montanus.d L. madrensis.e L. rotundiflorus.f L. exaltatus.g L. mexicanus.h L. elegans. | ||
| Connarus paniculatus (Connaraceae) | 18-Epipiptanthine 296 | 79 |
| Homoormosanine 297 | ||
| Homopodopetaline 298 | ||
| Ormosanine 299 | ||
| Piptanthine 300 | ||
| Podopetaline 301 (as monohydrochloride) | ||
| Crotalaria emarginella, C. fascicularis, C. phillipsiae, C. spinosa | Tashiromine 302 | 80 |
| Cyclolobium brasiliense | α-Isosparteine 303 | 81 |
| β-Isosparteine 304 | ||
| N-Methylcytisine 308 | ||
| 17-Oxo-β-isosparteine 305 | ||
| 17-Oxosparteine 306 | ||
| Sparteine 307 | ||
| Houttuynia cordata | Matrine N-oxide 320 | 82 |
| Lupinus angustifolius, L. campestris | 5,6-Dehydrolupanine | 83 |
| Multiflorine | ||
| 11,12-Seco-12,13-didehydromultiflorine | ||
| Lupinus elegans, L. exaltatus, L.madrensis, L. mexicanus, L. montanus, L. rotundiflorus | 13α-Angeloyloxylupaninec | 84 |
| Angustifolinec,d | ||
| Aphyllidinee | ||
| Aphyllinee,f,g,h | ||
| 5,6-Dehydro-α-isolupaninee,f | ||
| 5,6-Dehydrolupaninee | ||
| 11,12-Dehydrolupaninee,f,g | ||
| 11,12-Dehydrooxosparteinee,f,g | ||
| 3β,13α-Dihydroxylupanined | ||
| Epiaphyllidinef | ||
| Epiaphyllinef,g | ||
| 3β-Hydroxylupaninec–g | ||
| 13α-Hydroxylupaninec,d310 | ||
| α-Isolupaninee,f,g | ||
| Lupaninec–h311 | ||
| Multiflorinec,d,f,g | ||
| 17-Oxolupaninee,f,g313 | ||
| 17-Oxosparteinec | ||
| Sparteinec | ||
| Tetrahydrorhombifolinec | ||
| 4β-Tigloyloxylupanineef | ||
| 13α-Tigloyloxylupaninec,e312 | ||
| Sophora flavescens | (−)-5,6-Dehydrolupanine | 85 |
| (−)-14β-Hydroxymatrine | ||
| (−)-12β-Hydroxysophocarpine | ||
| (−)-14β-Hydroxysophoridine | ||
The genus Crotalaria (Fabaceae) is an important source of pyrrolizidine alkaloids, the ingestion of which is responsible for hepatotoxicity and other adverse reactions in both livestock and humans. GC-MS analysis of extracts from 12 Ethiopian Crotalaria species has now revealed for the first time the occurrence of an indolizidine alkaloid, the simple compound tashiromine 302, in the genus.80 This alkaloid, a minor metabolite in leaf and twig extracts of C. fascicularis, proved to be the major component in similar extracts of C. emarginella and C. phillipsiae and in the pods of C. spinosa, which also contained several unidentified minor alkaloids that are suspected to be esters of tashiromine.
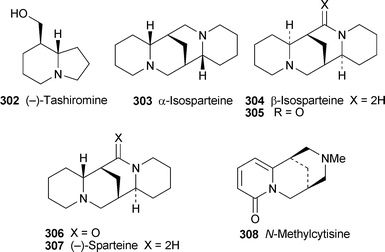
The debated position of the taxonomically isolated genus Cyclolobium within the Leguminosae has been clarified by chemotaxonomic investigation of its secondary metabolites.81 The detection of several typical quinolizidine alkaloids (α-isosparteine 303, β-isosparteine 304, 17-oxo-β-isosparteine 305, 17-oxosparteine 306 and sparteine 307, but no α-pyridones of the cytisine class other than N-methylcytisine 308) in leaf and fruit extracts of C. brasiliense, the only indisputable member of the genus, supports its removal from the non-alkaloid producing tribe Millettieae, and suggests an affinity with the Brongniartieae and other genistoid tribes.
The nutritional value of lupin seeds, widely used as animal feeds and for making lupin flour, has to be balanced against the presence of bitter and frequently toxic quinolizidine alkaloids. A study of the variation in quinolizidine alkaloids during germination of three Lupinus species has shown that the transformation of certain alkaloids into more toxic derivatives such as esters takes place within a matter of days.83 In L. albus, for instance, levels of albine 309 and 13-hydroxylupanine 310 decreased substantially as lupanine 311 and especially 13-tigloyloxylupanine 312 increased, the latter alkaloid also increasing markedly during germination of L. angustifolius. In the American species L. campestris, hydroxyaphylline and hydroxyaphyllidine (unspecified isomers) increased at the expense of epihydroxyaphylline and dehydroepihydroxyaphylline. It appears that an optimal germination period of three days is desirable in order to minimise the presence of antinutritive factors and avoid the formation of quinolizidine esters.
Most of the alkaloids detected in six species of Mexican wild lupins belong to the lupanine class.84 The total alkaloidal extract from seeds of Lupinus exaltatus was found to stimulate the growth of paprika plants and increase the yield of fruits when applied to the soil in which the plants were grown, thus supporting previous reports of lupin alkaloids as growth stimulants. A number of these alkaloids, including 13a-hydroxylupanine 310, lupanine 311, 17-oxolupanine 313 and in particular the synthetic thiono analogue of lupanine 314 (thionosparteine), were found to enhance glucose-induced insulin secretion in isolated rat pancreatic islets, which makes them potentially useful in the treatment of type 2 diabetes.86
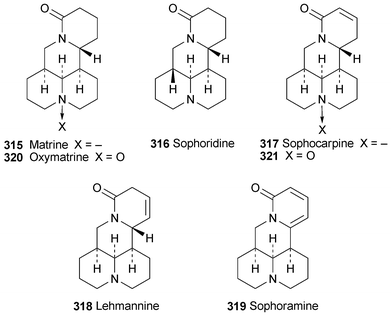
Preparations made from plants of the genus Sophora have numerous applications in Oriental medicine, their efficacy arising from the presence of alkaloids of the matrine group. A review on the extraction and identification of the active principles in the roots of S. flavescens provides an overview of the use of HPLC, GC and capillary electrophoresis in the separation and quantitation of the principal alkaloidal constituents, which include matrine 315 itself as well as sophoridine 316, sophocarpine 317, lehmannine 318, sophoramine 319, oxymatrine 320 and oxysophocarpine 321.85 Specific conditions for the determination of these and related alkaloids in S. alopecuroides, S. flavescens and S. tonkinensis by capillary electrophoresis have been described,87 while a method for the HPLC determination of matrine, sophoridine and sophocarpine in S. flavescens samples relied on the detection of electrogenerated chemiluminescence from the reaction between tris(2,2′-bipyridyl)ruthenium(II) and the tertiary amine site on the alkaloids.88 Matrine, known to have antinociceptive (pain relief) properties, apparently acts through multiple mechanisms such as increasing cholinergic activation in the central nervous system rather than by direct interaction with opioid receptors.89
13.2 Structural, spectroscopic and computational studies
Solid-state IR and Raman spectra recorded on a commercial polycrystalline sample of cytisine 322 have shown the presence of only the conformation 322′, in which the system is stabilised by intermolecular CO⋯H–N hydrogen bonds.90 Two different environments for the associated molecules were detected in the sample. The results were supported by semi-empirical and ab initio calculations performed for the cytisine monomer and hydrogen-bonded dimeric, trimeric and tetrameric aggregates. Ab initio calculations have also been used to explore the preferred sites of protonation of cytisine.91 Surprisingly, the carbonyl oxygen of the pyridone ring was found to be the primary proton acceptor in the gas phase, whereas in water the nitrogen atom in ring C was the most basic site, as expected.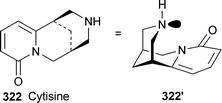
The 1H and 13C NMR spectra of lupanine 311 have been completely assigned.92 IR and NMR spectroscopic studies on the synthetic 17-ethyl and 17-butyl derivatives of lupanine and their monoperchlorate salts have shown that the alkyl substituent is equatorially orientated, while ring C adopts a boat conformation similar to that found in the parent alkaloid.93 Spectroscopic and crystallographic investigations of the related compounds (+)-2-thiono-17-oxosparteine 323 and (+)-2,17-dithionosparteine 324 reveal that they are conformationally rigid, with rings A and C adopting distorted sofa conformations due to the essentially flat geometry around the carbonyl and thiocarbonyl groups.94
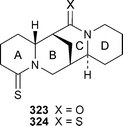
It is now well known that the preferred chair–chair–boat–chair conformation 325 of rings A–D in sparteine changes to a folded all-chair arrangement when the alkaloid forms 1 : 1 complexes with metal ions. The observation has been confirmed for the copper(II), cobalt(II), nickel(II) and zinc(II) chloride complexes 326 of sparteine and 2-methylsparteine in an investigation in which the absorption spectra of the solids were studied by IR, near-IR and diffuse-reflectance UV-VIS spectroscopy.95 The structure of the zinc complex was also confirmed by X-ray crystallography and NMR spectroscopy. Similar results have been found for the symmetrical α-isosparteine–zinc(II) complexes 327 with chloride, bromide and cyanide as counter-ions.96 Another crystallographic investigation of the 1 : 1 complex of sparteine with copper(II) bromide revealed two polymorphs in which the individual structural units were basically identical, although the packing modes differed considerably.97 The weak antiferromagnetic interaction in one of the polymorphs was ascribed to the presence of Cu–Br⋯Br–Cu close contacts. A similar reason has been advanced for the weak antiferromagnetism in sparteinium tetrabromocuprate monohydrate, the crystal structure of which indicates that the rings adopt the conventional chair–chair–boat–chair arrangement because the diprotonated alkaloid does not complex with the discrete CuBr42− anion, which is extensively hydrogen-bonded to the water molecule.98 Interestingly, although the crystal structure of the corresponding tetrachlorocuprate monohydrate complex proved to be almost identical, the substance is paramagnetic. The X-ray crystal structures of the 1 : 1 and 1 : 2 complexes of sparteine with copper(I) chloride showed that the former was a discrete dimer having the structure [Cu2Cl2spa2] 328; while the latter, which has the formula [Cu4Cl4spa2], possesses an unusual ladder-like structure 329.99 The structures are reminiscent of those previously found for the 1 : 1 and 1 : 2 complexes of sparteine with methyllithium and phenyllithium, respectively100 (cf.ref. 7l). Coincidentally, complexes of O'Brien's (+)-sparteine surrogate 330 with methyllithium and phenyllithium have recently been prepared and studied by X-ray crystallography.101 The former proved to be a 2 : 2 dimer 331 akin in structure to 328, while the ladder-like 4 : 2 structure of the latter parallels that of 329. The article also contains improved crystallographic data for the (−)-sparteine–methyllithium complex.
The preferential C–D⋯N bridged conformation 332 adopted by the monodeuteriated N-methyl-α-isosparteinium cation, previously demonstrated by variable temperature NMR spectroscopy and backed up by quantum chemical calculations102 (cf.ref. 7l), has received further support from additional DFT calculations.103 Furthermore, tritium NMR spectroscopy on the cationic species prepared by alkylating (−)-α-isosparteine 303 with enantiomerically enriched isotopically labelled methylamine bis(p-toluenesulfonamide) Ts2NCHDT having an (R)–(S) ratio of about 83 : 17 showed that the 3H chemical shift of the (S)-CHDT isotopomer was 49 ppb downfield of the (R)-CHDT resonance. The results suggest that (−)-α-isosparteine might be a useful derivatising agent for determining enantiomeric excesses of chiral methyl groups as long as the reagent used for transferring the methyl group to the alkaloid is a good electrophile. The proviso arises because incomplete methylation and some racemisation were apparent with the rather unreactive bis(p-toluenesulfonamide); but experiments in which α-isosparteine was alkylated with more electrophilic reagents such as unlabelled methyl p-toluenesulfonate or methyl d3-triflate showed reasonably rapid, complete methylation of the alkaloid under quite mild conditions.
Würthwein and Hoppe have reported quantum chemical DFT calculations on the enantioselective lithiation of O-alkyl and O-alk-2-enyl carbamates by isopropyllithium in the presence of (−)-sparteine 307 and (−)-α-isosparteine 303.104 After optimising several geometries of interaction between the carbamate, the chiral ligand and the organolithium base, they found that the sparteine-mediated kinetically-controlled abstraction of the pro-S proton from carbamate 333 had the lowest activation barrier, and led to the (S)-lithiated derivative 334, in line with experimental observations. (−)-α-Isosparteine favoured abstraction of the pro-R proton, but the selectivity was predicted to be significantly less. The preference for sparteine-mediated abstraction of the pro-S proton from the allylic carbamate 335 was also less marked, although the energies of the transition states were calculated to be lower than those from 333, as one would expect for the formation of an allyllithium product. Hoppe has also continued to develop experimental aspects of the (−)-sparteine-assisted lithiation of allylic carbamates; recent publications include a study of the configurational stability of lithiated geranyl and neryl N,N-diisopropylcarbamates,105 the preparation and reactions of enantioenriched α-carbamoyloxy crotylboronates,106 and applications in the synthesis of bicyclic γ-lactones,107 stereohomogeneous cyclopropanecarbaldehydes and cyclopropyl ketones108 and (−)-α-kainic acid.109
The remarkable success of the ligand (+)-330 as a surrogate for the rare alkaloid (+)-sparteine ent-307 in mediating various enantioselective transformations has prompted O'Brien to synthesise and test the related ligands 336,110 all of which were prepared from (−)-cytisine 322 by minor modifications of his original synthesis of 330111 (cf.ref. 7m). In general, however, increasing the steric size of the N-alkyl substituent adversely affected the enantioselectivity in a range of reactions (mostly deprotonations) normally assisted by (−)-sparteine; the original surrogate (+)-330 gave the highest selectivity, although obviously in the opposite sense to the parent alkaloid. These results were then tested by computational studies in which the sense of asymmetric induction, but not the adverse steric effect of the N-alkyl substituent, was correctly predicted for the enantioselective deprotonation of N-Boc-pyrrolidine 337 with isopropyllithium in the presence of the enantiomers of 336, chosen for ease of comparison with (−)-sparteine itself.112 The steric crowding thus probably inhibits the formation of the prelithiation complex rather than increasing the activation energy of proton transfer in the transition state. In addition, modest enantioselectivity was predicted for three other ligands 338–340, which were not tested experimentally. Other workers have since prepared several analogues of the O'Brien ligand bearing a range of different N-alkyl substituents, and found that they could assist the deprotonation of prochiral phosphane–borane complexes with varying degrees of enantioselectivity to give largely the (R)-products 341 after the lithiated intermediates were trapped with benzophenone.113 Finally, while O'Brien's ligand is effectively missing the D ring of sparteine, Kozlowski and co-workers wondered whether the A ring could also be dispensed with.114 However, experimental and computational results with the bispidine ligand 342, the simplest chiral component of sparteine, showed that an intact A ring was necessary for the asymmetric lithiation-substitution of N-Boc-pyrrolidine.
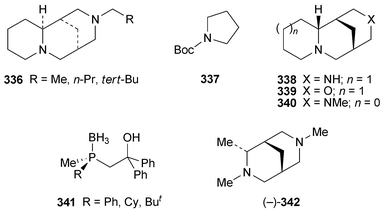
13.3 Synthesis and other chemical transformations
A previously reported synthesis of racemic tashiromine (±)-302 and its 5-epimer by Dieter and Watson115 (cf.ref. 7n) has been modified to produce optically active products116 (Scheme 27). Asymmetric deprotonation of N-Boc-pyrrolidine 337 with sec-butyllithium and (−)-sparteine 307 as chiral ligand followed by treatment with copper(I) cyanide generated an intermediate 2-pyrrolidinylcuprate, which was coupled with the vinyl iodide 343 to give almost exclusively (95 : 5) the (R)-2-alkenylpyrrolidine 344 in 83% yield. Removal of the protecting group and cyclisation afforded the exo-methylene product 345, hydroboration of which produced a mixture of the desired products as the borane complexes. After decomplexation with hydrochloric acid, the mixture of diastereomers (4 : 1; 85% yield) was separated by column chromatography to give ent-302, the unnatural (+)-enantiomer of tashiromine, and (+)-5-epi-tashiromine 346. The optical purities of the products, estimated from the measured specific rotations, matched that of the intermediate alkenylpyrrolidine 344. The method illustrated in the Scheme was also suitable for the preparation of related pyrrolizidine alkaloids.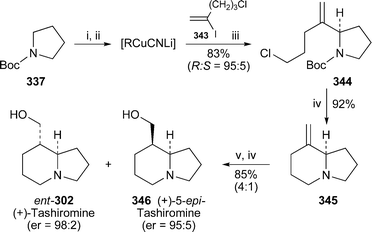 | ||
| Scheme 27 Reagents and conditions: i, (−)-sparteine 307, Et2O, −78 °C, then sec-BuLi, 1 h; ii, add CuCN·2LiCl (1 equiv.) in THF, −78 °C, 30 min; iii, 343, −78 °C to rt, overnight; iv, Me3SiCl in MeOH, 25 °C, overnight, then aq. NaHCO3; v, 9-BBN (0.5 M in THF), 60 °C, 1 h, then BH3·THF, rt, 30 min, then aq. NaOH (10 M), aq. H2O (35%), 0 °C, 1 h. | ||
The thermal epimerisation of (−)-lupinine 347 to (+)-epilupinine 348 in the presence of a strong base has been known for decades. The putative mechanism, which proceeds via the corresponding aldehydes 349 and 350, is shown in Scheme 28. Sparatore et al. have now obtained evidence for this unusual transformation.117 From the residues of several epimerisation attempts they managed to isolate a solid, the elemental analysis of which corresponded to the hemihydrate of a compound with two hydrogen atoms fewer than either of the two alkaloids. An enolic structure was apparent from the IR and NMR spectra of the new compound, which was soluble in ethanol but not in dry diethyl ether. Furthermore, evaporation of the ethanol solution yielded an oil, the spectra of which showed enhanced aldehyde signals, but which reverted to the ether-insoluble solid on standing. Reduction of the solid with sodium borohydride gave a mixture of lupinine and epilupinine, while mild oxidation of the alkaloids gave identical mixtures of the oily products lupinal 349 and epilupinal 350, which were also transformed into the ether-insoluble solid over time. Finally, they found that the conversion of lupinine into epilupinine, formerly considered to be unidirectional, could also be driven in the reverse direction; the extent of the conversion of 347 into 348 was about 30% in xylene at 165 °C (sealed tube, 6 h) in the presence of sodium metal, while that in the opposite direction was about 11–14%. From the observations, the authors concluded that the solid was the inner salt 351, which is derived from the common enol form 352 of both lupinal and epilupinal. Since the conversion of lupinine into epilupinine could be improved to as much as 85% by the addition of a quantity of the mixed aldehydes 349–350 to the equilibrating mixture of alkaloids, it appears that successful transformation depends on bimolecular transfer of hydride between the alkoxides 353 and 354 and the aldehyde intermediates, as shown in the final line of the Scheme. Loss of aldehyde by precipitation as the insoluble inner salt 351 is thus detrimental to the epimerisation process.
 | ||
| Scheme 28 Proposed mechanism for the base-induced equilibration of lupinine 347 and epilupinine 348. | ||
In a concise synthesis of (±)-lupinine rac-347 by Chang et al., a formal [3 + 3] cycloaddition of methyl acrylate to the anion of the α-sulfonylacetamide 355, prepared in two steps from 5-aminopentanol, gave the glutarimide 356 in 62% yield118 (Scheme 29). A further four steps via intermediate 357 yielded the vinylogous sulfonamide 358, treatment of which with aqueous acetic acid effected cyclisation to a mixture of quinolizidine isomers 359 in 78% yield. Reduction of the aldehyde and desulfonylation then completed the synthesis of (±)-lupinine.
 | ||
| Scheme 29 Reagents and conditions: i, H2CCHCO2Me, NaH, THF, rt, 10 h; ii, PCC, Celite, CH2Cl2, rt, 18 h; iii, HC(OMe)3, p-TsOH, MeOH, rt, 2 h; iv, NBS, Na2CO3, MeCN, rt, 90 min; v, LiAlH4, THF, 0 °C, 30 min, then reflux, 2 h; vi, AcOH–H2O (3 : 1), reflux, 4 h; vii, NaBH4, MeOH, rt, 2 h; viii, Na/Hg (6%), Na3PO4, MeOH, rt, 2 h. | ||
Martin's concise approach for making simple quinolizidine alkaloids by an iminium ion cascade (see Section 8, Scheme 22) has been used in a spectacularly short synthesis of (±)-epilupinine rac-34868 (Scheme 30). Condensation of the monoprotected dialdehyde 253 with the amine-bearing allylsilane 360 afforded the putative bicyclic iminium ion 361, which was intercepted with triethylsilane to furnish the 1-vinylquinolizidine 362 in 75% yield as a single diastereomer. Although this intermediate has been used in a previous synthesis of the alkaloid,119 Martin's team found improved conditions for achieving the ozonolysis and subsequent reduction to give (±)-348 in 88% yield.
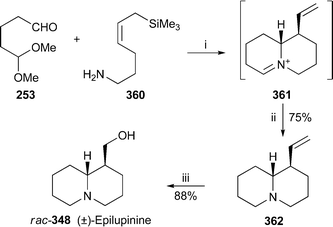 | ||
| Scheme 30 Reagents and conditions: i, 4 Å molecular sieves, MeCN, 0 °C, then rt, 12 h, then TFA, 0 °C, 2 h and rt, 12 h; ii, Et3SiH, MeCN, reflux, 24 h; iii, TFA, then O3, Et2O, −78 °C, 5 min, then LiAlH4, THF, rt, 12 h. | ||
Ma and Ni employed Sharpless asymmetric epoxidation and double ring-closing metathesis as key steps in a new synthetic approach to various azabicyclic alkaloid skeletons.120 In their lengthy but imaginative synthesis of both enantiomers of epilupinine and lupinine, for example, asymmetric epoxidation of (E)-hexa-2,5-dienol (E)-363 in the presence of L-(+)-diethyl tartrate produced the epoxide (2S,3S)-364 in 86% yield and an ee of 94.6% (Scheme 31). Regiospecific SN2 opening of the epoxide ring with vinylcuprate gave the diol 365, which was converted in a further eight steps into the tetraene (3S,4R)-366. The double metathesis was then induced by treatment with the Grubbs second-generation catalyst to produce a mixture of the tetrahydroquinolizin-4(H)-ones 367 and 368 in 89% yield and a ratio of 1.96 : 1, together with a trace of the alternative metathesis product 369 (4%). The structure of the major isomer was confirmed by X-ray crystallographic analysis of the diol derivative 370, which was obtained from 367 in three steps. Hydrogenation of the mixture of fused bicyclic compounds followed by reduction of the lactam with lithium aluminium hydride afforded (+)-epilupinine (+)-348 in 95% yield and 92.7% ee. The synthesis of (−)-epilupinine ent-348 simply required D-(−)-diethyl tartrate at the start of the reaction sequence; the epoxide ent-364 was obtained in 95.3% ee, and the ee of the final alkaloid was 91.8% ee. For the syntheses of lupinine, the initial reactant was (Z)-hexa-2,5-dienol (Z)-363, epoxidation of which with L-(+)- or D-(−)-diethyl tartrate produced 371 and ent-371, respectively, both in 90% ee. The former was converted via the tetraene (3R,4R)-372 into (−)-lupinine (−)-347 in 82% yield and 86.5% ee, while the latter was transformed in analogous fashion into (+)-lupinine ent-347 (ee 85.9%). The versatility of the double ring-closing metathesis that lies at the heart of this reaction sequence was further demonstrated by the conversion of a range of other tetraenes 373 into 1-azabicyclo[m.n.0]alkadienones (m, n = 6, 7).
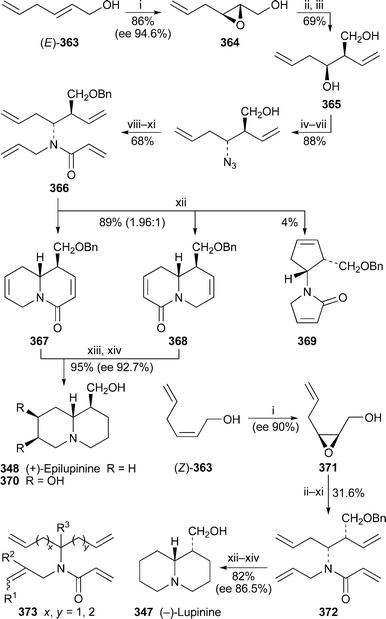 | ||
| Scheme 31 Reagents and conditions: i, L-(+)-DET, Ti(OPri)4, ButOOH, 4 Å molecular sieves, CH2Cl2, −20 °C, 4 h; ii, (H2CCH)2CuMgBr, Et2O, −78 °C to −25 °C, 16 h; iii, NaIO4, THF–H2O (1 : 1), 3 h; iv, TBDMSCl, NEt3, DMAP, CH2Cl2, −78 °C, then rt, 3 h; v, MeSO2Cl, NEt3, CH2Cl2, −78 °C, then rt, 15 h; vi, NaN3, HMPA, 40 °C, 2 h; vii, Bu4NF, THF; viii, NaH, THF, rt, 30 min, then BnBr, rt, 3.5 h; ix, LiAlH4, THF, −78 °C, then rt, 2 h; x, H2CCHCH2Br, K2CO3, DMF, 0 °C, 30 min; xi, H2CCHCOCl, NEt3, CH2Cl2, −78 °C to rt; xii, Grubbs II catalyst (5 mol%), CH2Cl2, reflux, 2 h; xiii, H2 (1 atm), Pd/C, MeOH + AcOH (2 drops), 30 °C, 24 h; xiv, LiAlH4, THF, reflux, 4 h. | ||
The selective N-alkylation of 6-bromo-2-pyridone 374 with the bromomethylpiperidin-2-one 375 proved to be a non-trivial step in a new synthesis of the topical alkaloid cytisine 322 by Gallagher and co-workers121 (Scheme 32). Basing their procedure on a known protocol for the N-alkylation of pyridones, they used factorial experimental design methods to optimise conditions (solvent, temperature, substrate concentration, quantities of additives; see caption to Scheme) for carrying out the reaction, which eventually produced the desired product 376 in 61% yield together with the O-alkyl isomer 377 (25%) and the elimination product 378 (14%). The crucial ring closure of 376, entailing palladium-catalysed intramolecular arylation of the lactam enolate, gave the expected tricyclic product 379 in only 44% yield—a disappointing but understandable result in view of the fact that 376 needs to adopt a conformation 376′ with an axial substituent in order for cyclisation to occur. Selective reduction of the saturated lactam and a final debenzylation completed the synthesis of racemic cytisine (±)-322.
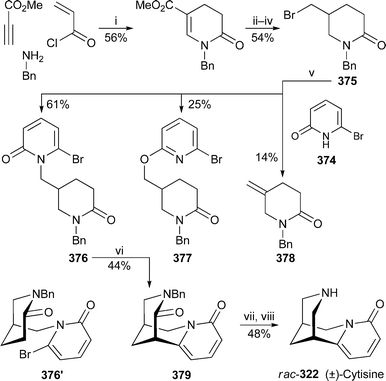 | ||
| Scheme 32 Reagents and conditions: i, HCCCO2Me + BnNH2, Et2O, rt, then H2CCHCOCl, THF, 70 °C; ii, H2 (1 atm), Pd/C, Na2CO3, EtOH, 20 °C; iii, LiAlH4, THF, −10 °C; iv, PBr3, PhMe, 110 °C; v, 374 (0.2 M; 1 equiv.), 375 (0.2 M, 1 equiv.), NaH (2 equiv.), LiBr (4 equiv.), DME–DMF (24 : 1), 10 d; vi, Pd(OAc)2 (5 mol%), (±)-BINAP (7.5 mol%), KHMDS (2 equiv.), THF, 70 °C; vii, BH3·THF, THF, 0 °C to rt; viii, H2 (1 atm), Pd(OH)2/C, HCl (1 equiv.), MeOH. | ||
The first reported synthesis of the unnatural dextrorotatory enantiomer of cytisine, by Honda et al.,122 began with the (4R)-4-hydroxy-L-proline ester 380, which was converted in three steps into the enol triflate 381 (Scheme 33). Palladium-catalysed coupling with 2-tributylstannyl-6-methoxypyridine 382 gave the dihydropyrrole 383, from which the pyrrolidine 384 was obtained by stereoselective hydrogenation and removal of the N-Boc group. The central transformation, however, was the reductive deamination of 384 by samarium diiodide, the resulting intermediate then recyclising to produce the lactam 385 in 78% yield. Protection of the lactam by N-benzylation was followed by acylation of the corresponding enolate with ethyl chloroformate, the product 386 being obtained as a 1 : 1 mixture of diastereomers that resisted attempts at equilibration to the more stable 3,5-cis-isomer. The authors were forced to reduce the mixture with lithium aluminium hydride to give the primary alcohols 387 and 388 in isolated yields of 43% and 48%, respectively. Mesylation of the latter product preceded thermal cyclisation to give 389 in 89% yield. Removal of the N-benzyl group by hydrogenolysis concluded the synthesis of (+)-cytisine, ent-322. Incidentally, the N-methyl analogue of 388 is the unusual lupin alkaloid jussiaeiine A, the synthesis of which was also reported in this publication.
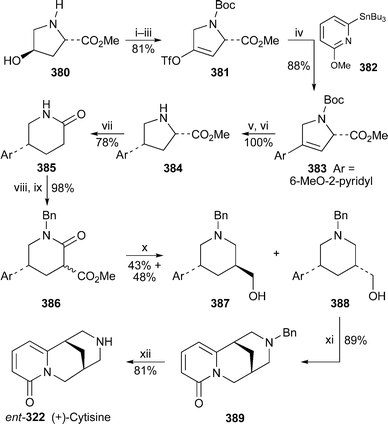 | ||
| Scheme 33 Reagents and conditions: i, (Boc)2O, NEt3, CH2Cl2, rt; ii, (COCl)2, DMSO, NEt3, CH2Cl2, −40 °C to rt; iii, LiHMDS, 5-Cl-2-NTf2-pyridine, THF, −78 °C to −20 °C; iv, 382, Pd(PPh3)4, LiCl, CuI, THF, 65 °C, 4 h; v, H2 (1 atm), 10% Pd/C, MeOH, rt, 2 h; vi, TFA, CH2Cl2, 0 °C, then rt, 2 h; vii, SmI2, THF–HMPA, MeOH, 0 °C, then rt, 40 min; viii, NaH, BnBr, THF–HMPA, 0 °C to rt, 90 min; ix, LDA, THF, −78 °C, then ClCO2Et, 1 h; x, LiAlH4, THF, rt, 12 h; xi, MeSO2Cl, NEt3, CH2Cl2, 0 °C, 30 min, then PhMe, reflux, 3 h; xii, H2 (1 atm), 20% Pd(OH)2/C, NH4+ HCO2−, MeOH, reflux, 1 h. | ||
A clever synthesis of (±)-sparteine rac-307 by Fleming and co-workers123 capitalises on the almost-but-not-quite symmetrical constitution of the alkaloid by adopting a bidirectional approach in which both ‘halves’ of the target are simultaneously elaborated from a meso precursor (Scheme 34). Initial Diels–Alder reaction between dimethyl bromomesaconate 390 and the diene 391 gave a 75 : 25 mixture of the adducts 392 and 393, base-catalysed cyclisation of which produced the two meso cyclopropanes 394 and 395 in the same ratio. Cleavage of the strained bond between the two ester groups with lithium in liquid ammonia afforded the bis-enolate 396, essentially still a meso intermediate, protonation of which turned out to be the step on which the success of the synthesis hinged. Upon treatment with ammonium chloride, the bis-enolate gave a mixture of the desymmetrised product 397 and its meso isomer 398 in a ratio of 30 : 70; the alternative meso product was not observed. The relative stereochemistry of 398 was confirmed by X-ray crystallography. On the other hand, quenching with methanol produced a ratio of 76 : 24 in favour of the desired isomer 397, which could be isolated in 68% yield by recrystallisation and chromatography of the mother liquors. The two tethered moieties of the target were then unmasked by ozonolysis of 397, which afforded the diketone 399 in 98% yield as long as acetaldehyde was added to the mixture to trap the intermediate carbonyl oxide (formed by breakdown of the molozonide) and prevent epimerisation adjacent to the ketone. The bis-oxime derivative 400, separated by chromatography and crystallisation from a mixture of geometric isomers, then underwent Beckmann rearrangement to give the bis-lactam 401. Reduction with lithium aluminium hydride led to the diol 402, the corresponding chloride of which cyclised to produce (±)-sparteine rac-307. The authors make the point that, if an enantiomerically enriched acid could be found for protonating the bis-enolate 396 not only stereoselectively but also enantioselectively, their route would be eminently suitable for accessing either enantiomer of the alkaloid. The wistful envoi to the article reads “if only there were such a reagent…”.
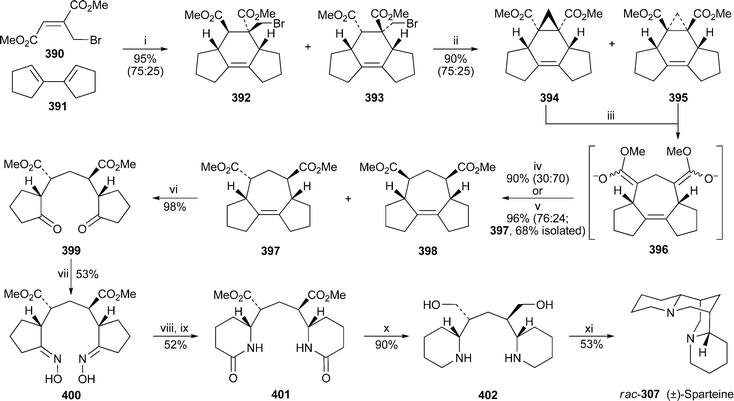 | ||
| Scheme 34 Reagents and conditions: i, Me2AlCl (1 M in hexanes), CH2Cl2, −78 °C, then rt, 12 h; ii, NaOMe, PhMe, reflux, 12 h; iii, Li, NH3, −78 °C, 0.5 h, then isoprene; iv, quench with NH4Cl; v, quench with MeOH; vi, O3, Me2CO, MeCHO, −78 °C, then PPh3, 1 h, then rt, 12 h; vii, NH2OH·HCl, py, EtOH, 0 °C, 2 d; viii, MeSO2Cl, NEt3, CH2Cl2, −20 °C to rt; ix, THF–H2O (2 : 1), 60 °C, 24 h; x, LiAlH4, THF, reflux, 12 h; xi, CCl4, PPh3, NEt3, MeCN, rt, 18 h. | ||
Recognition of the near-symmetry of the target alkaloid is also apparent in the concise route to (−)-sparteine by O'Brien's team124 (Scheme 35). Reaction between (R)-α-methylbenzylamine 403 and ethyl 7-iodohept-2-enoate 404, presumably by tandem substitution–intramolecular conjugate addition, produced a separable mixture of the piperidines 405 and 406 in yields of 24% and 45%, respectively. The latter compound was converted into the enoate 407 by alkylation of its enolate with chloromethyl ethyl ether followed by basic elimination of ethanol. Similarly, reaction between (S)-α-methylbenzylamine ent-403 and 404 produced ent-406, the enolate of which participated in conjugate addition to 407 to give the adduct 408. While this compound could not be separated from unreacted ent-406, debenzylation of the mixture by transfer hydrogenation with ammonium formate and Pearlman's catalyst allowed the isolation of the bislactam 409 as a single diastereomer in 36% yield over the two steps. Compound 409 is actually a known natural product, 10,17-dioxosparteine, although the authors do not mention this fact. Finally, reduction of 409 with lithium aluminium hydride afforded (−)-sparteine 307 in 88% yield. This very effective synthetic strategy has obvious potential for the preparation of novel sparteine analogues in either enantiomeric form as well as other alkaloids of the sparteine class, and further developments are sure to be forthcoming.
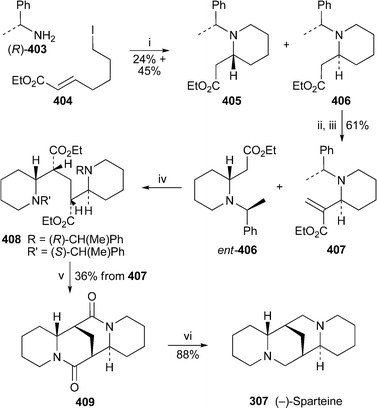 | ||
| Scheme 35 Reagents and conditions: i, NEt3, EtOH, reflux, 16 h, then chromatography; ii, LiHDMS, THF, −78 °C, 1 h, then EtOCH2Cl, −78 °C to rt over 4 h; iii, KOBut, THF, −78 °C, 8.5 h; iv, ent-406 + LDA, THF, −78 °C, 20 min, then 0 °C, 5 min, then −78 °C, 30 min, add 407, −78 °C to −30 °C over 5.5 h, then −30 °C, 3 h, quench with aq. HCl (1 M); v, NH4+ HCO2−, Pd(OH)2/C, EtOH, reflux, 14 h, then crystallise from Et2O; vi, LiAlH4, THF, reflux, 16 h. | ||
In 2002, Passarella et al. reported a synthesis of racemic aloperine from commercially available piperidine-2-ethanol125 (cf.ref. 7n). A simple and environmentally benign modification of this route has now provided access to (+)-aloperine 410.126 The revised route exploits the authors' recently published enzymatic resolution of the N-Boc derivative of racemic piperidine-2-ethanol (±)-411127 which, in a nutshell, entails selective acetylation of (±)-411 with vinyl acetate and lipase PS to produce the (R)-enantiomer selectively, after which treatment of the crude (S)-alcohol–(R)-acetate mixture with vinyl isobutyrate and porcine pancreatic lipase selectively esterifies the (S)-alcohol. In the present case, chromatographic separation of the esters gave the (R)-acetate 412 (45%, 63% ee), the (S)-isobutyrate (30%, 83% ee) and recovered racemic precursor (24%). Hydrolysis of the (R)-acetate to the alcohol with sodium carbonate in methanol followed by a second cycle of esterification with vinyl acetate and lipase PS, chromatographic purification and basic hydrolysis afforded (R)-alcohol 411 in an improved ee of 90%. This compound could be oxidised to the corresponding aldehyde 413 under Swern conditions, but problems in scaling up the reaction forced the authors to examine alternative methods. They found that TEMPO-mediated enzymatic oxidation with either of two different laccases and molecular oxygen in an aqueous buffer (pH 4–5)–ethyl acetate mixture afforded 413 in 90% yield. This enzymatic oxidation was also successfully applied to the subsequently formed secondary propargyl alcohol 414, the ee of the resulting ketone 415 matching that of precursor 411 (90%). The synthesis of (+)-aloperine 410, also obtained in 90% ee, thereafter followed the same course as described previously for (±)-aloperine.
The potency of (−)-cytisine 322 and some of its derivatives as nicotinic receptor agonists has been touched on in a review of naturally occurring nicotinic agonists, antagonists and modulators.128 This important alkaloid continues to inspire the design and synthesis of analogues for biological testing. For example, catalytic hydrogenation of 322 (5 atm, PtO2, H2O, 50 °C) gave a 98% yield of the known but uncommon natural product tetrahydrocytisine 416, alkaline hydrolysis of which followed by esterification (and sometimes preceded by N-alkylation) then afforded a range of bispidine esters 417.129 These products were used as scaffolds with three points of diversity for the combinatorial synthesis of compound libraries expected to possess useful activity as antiarrhythmics or nAChR ligands, among others. Alternatively, 417 (R1 = H) could be condensed with paraformaldehyde to give the diazaadamantane 418 in 87% yield, or with 1,1′-carbonyldiimidazole followed by microwave irradiation to produce the urea 419.130 This provides a useful entry to products possessing the 1,3-diazatricyclo[3.3.1.13,7]decane nucleus, which occurs in rare lupine alkaloids such as acosmine 420. The simpler 3-halogenated cytisines 421 showed improved binding affinity and functional activity towards a range of nAChR subtypes, while halogenation at C-5 led to small decreases in both properties. Substitution at both sites reduced activity almost totally.131 By contrast, cytisine methiodide 422 retained significant activity, but thionocytisine 423, although only weakly potent and effective, showed selectivity for the α4β2 subtype. In another study, intended as part of a search for new α4β2 nAChR partial agonists that might serve as therapeutic aids for smoking cessation, 27 synthetic benzenoid cytisine analogues of general structure 424 were tested for their binding affinity and partial agonist profiles;132 only the methoxy derivative 424 (R3 = OMe) showed a partial agonist profile similar to that of cytisine. In comparison, the synthetic analogue varenicline 425 (Ki 0.06 nM) is sufficiently potent to have been advanced to clinical trials as an anti-smoking aid.133 Other new N(12)-substituted cytisine derivatives prepared for various biological studies include several dithiocarbamates,134 propargyl phosphonamidoates135 and the 1,2,4-thiadiazoles 426.136
14 Alkaloids from marine sources
Synthetic approaches to the spirocyclic core of the VCAM-1 inhibitor halichlorine 427 continue to proliferate. Standing out from a clutch of new methodological investigations and formal syntheses is a new route to the racemic alkaloid itself by Christie and Heathcock,137 the first full total synthesis since Danishefsky's precedent-setting route to (+)-427 was published in 1999.138 Key steps and intermediates in the novel route are shown in Scheme 36. When the acyliminium ion precursor 428, the structure of which was confirmed by X-ray crystallography, was treated with allyltrimethylsilane and titanium tetrachloride, allylation took place mainly on the less hindered face. The acetate derivative 429 of the adduct subsequently underwent cross-metathesis with the Nazarov reagent 430 to give enone 431 in 80% yield. Catalytic hydrogenation of 431 afforded the spiropiperidine 432, the secondary amine site of which was internally protected as a β-lactam. Deacetylation then yielded product 433, also characterised by single-crystal X-ray analysis. Oxidation to the aldehyde and Wittig reaction with the phosphorane 434 afforded the dienone 435, reduction of which with sodium borohydride gave a 5 : 2 mixture of the alcohol 436 and its epimer. With all four stereogenic centres of the target alcohol in place, protection as the triethylsilyl ether followed by reductive cleavage of the β-lactam then afforded aldehyde 437. Elaboration of the quinolizidine core entailed reaction of 437 with trimethyl phosphonoacrylate and lithium thiophenoxide, thereby providing a mixture of (E)- and (Z)-thioethers 438, which cyclised directly to the desired spiroquinolizidine 439 when heated with thiophenoxide ion. After removal of the protecting groups from both alcohol groups and basic hydrolysis of the ester, a disappointing macrolactonisation (32% yield) completed the synthesis of (±)-halichlorine 427. Single-crystal X-ray analysis of the product not only confirmed the structure but also demonstrated cis ring fusion of the dehydroquinolizidine system, as predicted by Trauner et al.139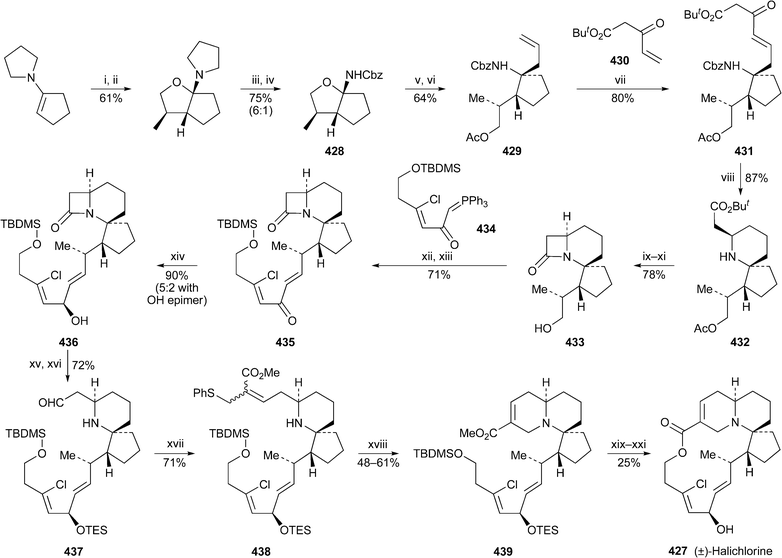 | ||
| Scheme 36 Reagents and conditions: i, MeCH(Br)CO2Et, NEt3, dioxane, reflux, 10 h; ii, LiAlH4, Et2O, 0 °C, 3 h; iii, PhOCOCl, PhMe, −78 °C to 0 °C; iv, BnOCONH2, PhMe, 0 °C to 110 °C; v, H2CCHCH2SiMe3, TiCl4, CH2Cl2, −50 °C to −20 °C, 7 h; vi, Ac2O, DMAP, NEt3, CH2Cl2, rt, 1 h; vii, Grubbs II catalyst, CH2Cl2, 40 °C, 3.5 h; viii, H2 (55 psi), Pd/C, EtOAc, rt, 50 h; ix, TFA, rt, 35 min; x, 1-Me-2-Cl-pyridinium triflate, EtNPri2, MeCN, 70 °C; xi, K2CO3, MeOH, rt, 2 h; xii, TPAP, NMO, CH2Cl2, rt, 10 min; xiii, phosphorane 434, MeOH, 65 °C, 3 d; xiv, NaBH4, CeCl3·7H2O, MeOH, rt; xv, Et3SiCl (TESCl), DMAP, EtNPri2, CH2Cl2, −78 °C to 0 °C; xvi, Red-Alp–KOBut (4 equiv.), MeOBut, rt, 3 h, then SiO2; xvii, trimethyl phosphonoacrylate, PhSLi, THF, 0 °C to rt, 12 h; xviii, PhSH, K2CO3, DMF, 55 °C, 35 h; xix, Bu4NF, THF, 0 °C, 3 h; xx, NaOH, MeOH, H2O, 55 °C, 2 h, then rt, overnight; xxi, EtNCN(CH2)3NHMe·HCl, DMAP + DMAP·HCl, CHCl3–THF, reflux, 10 h. | ||
In an intriguing formal synthesis of (±)-halichlorine by Feldman et al.,140 pyridine was converted in six steps into the 2,6-trans-disubstituted piperidine 440, from which the unstable alkynyliodonium species 441 was obtained by treatment with Stang's reagent, PhI(CN)OTf (Scheme 37). The carbene generated from this intermediate by heating with sodium p-toluenesulfinate cyclised to the indolizidinone 442, which in turn gave the tricyclic product 443 when treated with magnesium bromide or other Lewis acids. Although reductive methylation with lithium naphthalenide and iodomethane favoured the desired methylated diastereomer 444 (>10 : 1), the subsequent destannylation to 445 required a rather circuitous procedure in order to avoid epimerisation. After reductive cleavage of the lactam, N-alkylation of the silyl-protected alcohol 446 with ethyl 2-(bromomethyl)acrylate 447, ring-closing metathesis and removal of the protecting group afforded the spirotricyclic product 448. This compound, previously prepared by Kibayashi and co-workers,141 effectively completes a formal synthesis of racemic halichlorine, since the corresponding enantiomerically pure tert-butyl ester featured in Danishefsky's synthesis of the (+)-alkaloid.138
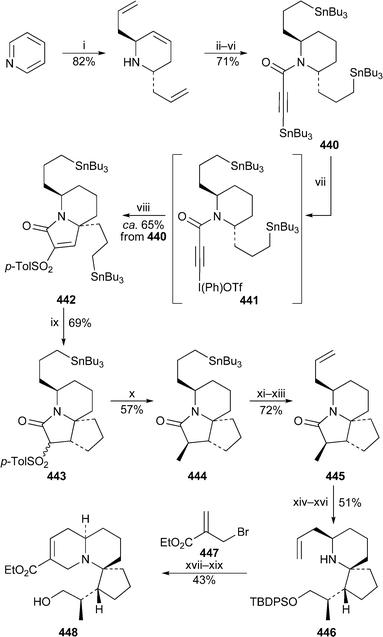 | ||
| Scheme 37 Reagents and conditions: i, (H2CCHCH2)3B, PriOH; ii, Bu3SnH, AIBN, 100 °C (sealed tube), 96 h; iii, H2 (1 atm), PtO2, EtOH, rt, 14 h; iv, Me3SiCCCOCl, EtNPri2, CH2Cl2, −45 °C, 12 h; v, Bu4NF, THF, −45 °C, 30 min, then rt; vi, (Bu3Sn)2O, MgSO4, Et2O, rt, 48 h; vii, PhI(CN)OTf, CH2Cl2, −45 °C, 2.5 h; viii, p-TolSO2Na, DME, reflux, 20 min; ix, MgBr2, PhMe, reflux,14 h; x, Li-naphthalene, THF, −78 °C, 1.5 h, then MeI, −60 °C, 1 h; xi, (PhIO)n, BF3·Et2O, CH2Cl2, 0 °C, 45 min; xii, aq. H2O2 (30%), KHCO3, THF–MeOH (1 : 1), rt, 18 h; xiii, 2-O2NC6H4SeCN, PBu3, THF, rt, 10 h, then aq. H2O2 (30%), 16 h; xiv, CF3SO3Me, ClCH2CH2Cl, 0 °C, then 60 °C, 1 h, then THF–H2O, rt, 16 h; xv, LiAlH4, THF, 0 °C, then rt, 5 h; xvi, TBDPSCl, DMAP, NEt3, CH2Cl2, rt, 2 h; xvii, 447, K2CO3, MeCN, 60 °C, 14 h; xviii, Grubbs II catalyst, CH2Cl2, reflux, 1 h; xix, py·HF, MeCN, 2 h. | ||
In the formal enantioselective synthesis of halichlorine by Zhang et al.,142 lipase-induced acetylation was used to resolve the racemic alcohol 449, the unreacted (1R,2S)-(+)-enantiomer of which was converted into the lactone 450, and thence into the cyclopentanone oxime 451 over a further five steps (Scheme 38). Oxidation produced the nitro compound 452, conjugate addition of which with methyl acrylate gave the adduct 453 as a single diastereomer. Elaboration of the side chain by standard methods yielded the nitro ketone 454 which, upon reduction with nickel boride and hydrazine, afforded the spirocyclic nitrone 455. Further reduction produced the piperidine 456. Finally, manipulation of the protecting groups then gave 457, the stereochemistry of which was established by single-crystal X-ray analysis of the p-iodobenzoate ester. The preparation of 457 also converges with the routes of both Danishefsky138 and Kibayashi,141 and thus completes a formal synthesis of the alkaloid.
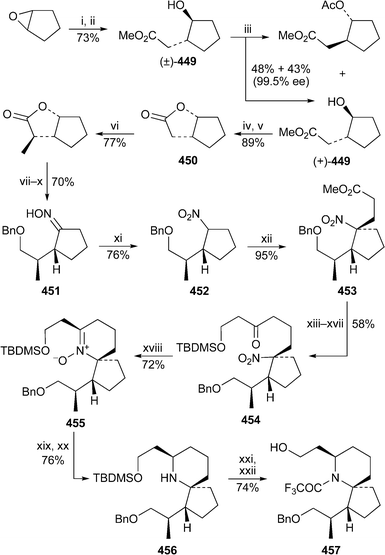 | ||
| Scheme 38 Reagents and conditions: i, Na, CH2(CO2Me)2, MeOH, reflux; ii, LiCl (2 equiv.), H2O (1 equiv.), DMSO, 140 °C, 3 h; iii, lipase PS (5%), H2CCHOAc (2 equiv.), rt, 48 h; iv, MeSO2Cl, NEt3, DMAP, CH2Cl2, 0 °C to rt; v, aq. NaOH (5%), THF, 0 °C, 24 h; vi, LDA, THF, −78 °C, then MeI, 6 h; vii, LiAlH4, THF, reflux, 4 h; viii, NaH (1.3 equiv.), BnBr (1.2 equiv.), THF, rt, 10 h; ix, PCC, CH2Cl2, rt, 4 h; x, NH2OH·HCl, K2CO3, MeOH, rt, 4 h; xi, MCPBA, Na2HPO4, crushed urea, MeCN, 80 °C; xii, H2CCHCO2Me, Triton B, ButOH, THF, rt, 48 h; xiii, NaBH4 dioxane–H2O (1 : 1), rt, 24 h; xiv, MeSO2Cl, NEt3, DMAP, CH2Cl2, 0 °C to rt, 2 h; xv, NaI, NaHCO3, Me2CO, rt, 24 h; xvi, (1,3-dithian-2-yl)CH2CH2OTBDMS + ButLi (1.5 equiv.), HMPA (3 equiv.), THF, −78 °C, 40 min; xvii, MeI, CaCO3, MeCN–H2O (4 : 1), rt, 24 h; xviii, Ni2B, H2NNH2·H2O, EtOH, reflux, 2 h; xix, NaBH4, MeOH, 0 °C to rt; xx, TiCl3, NaOAc, MeOH, H2O, rt, 1.5 h; xxi, (CF3CO)2O, EtNPri2, ClCH2CH2Cl, 0 °C, 40 min; xxii, py·HF, THF, rt, 24 h. | ||
Among the new strategies for preparing the spirocyclic core of halichlorine is a bidirectional approach (cf. Section 15 below) by Stockman and co-workers via the ketone 458.143 Treatment with hydroxylamine gave the tricyclic adduct 459 in 62% yield, and this product was converted in several steps into the model compound 460. The route by de Sousa and Pilli employed enolate chemistry and a Dieckmann cyclisation to produce the spirobicyclic ketone 461, the oxime of which underwent a reasonably efficient Beckmann rearrangement (60%) to give the halichlorine model 462.144 Huxford and Simpkins deprotonated the symmetrical piperidine diester 463 with a chiral base, and showed that subsequent allylation gave the allylated product 464 in 90% ee.145 Further transformations, including a final ring-closing metathesis, produced the spirobicyclic product 465, which is also a halichlorine model, albeit one with incorrect stereochemistry for the alkaloid itself. Clive et al. also used the Simpkins allylation procedure to obtain an enantiomerically enriched sample of 464, after which a series of functional group transformations and a ring-closing metathesis furnished the spirobicyclic product 466.146 Subsequent steps included another ring closure by condensation, and eventually led to the tricyclic product 467, which is a late intermediate in Kibayashi's route to the halichlorine precursor 468.141 Clive's group also showed that both epimers of 469, formed in situ by methanolysis of the Morita–Baylis–Hillman adducts 470, cyclised spontaneously by an SN2′ mechanism to give 471, another plausible intermediate en route to halichlorine.147
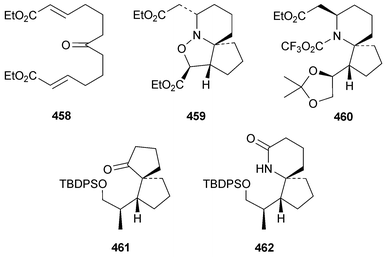
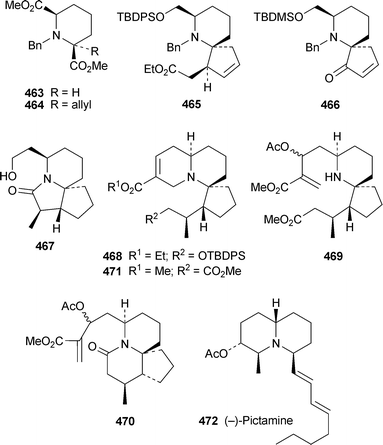
The ascidian alkaloid (−)-pictamine 472 has been found to act as a blocker of α4β2 and α7 neuronal nicotinic acetylcholine receptors (IC50 1.5 and 1.3 µM, respectively).148 The action on the former was irreversible, while acetylcholine-elicited currents in α7 receptors recovered quickly after removal of the alkaloid. Since irreversible antagonists of α4β2 receptors are apparently rare, pictamine may prove to be a valuable tool for selective modification of neuronal activities.
15 Alkaloids from coccinellid beetles
In 2002, Rejzek and Stockman communicated an efficient bidirectional synthesis of the quinolizidine 473 from the symmetrical alcohol 474.149 Experimental details of this work have now been presented in a full paper that also includes an application to the synthesis of the ladybird alkaloid (±)-hippodamine 475150 (Scheme 39). The pivotal conversion of the phthalimide intermediate 476 into the quinolizidine 473 entailed an experimentally tricky tandem deprotection and double conjugate addition, the first phase being achieved with sodium borohydride under carefully optimised conditions (see caption to Scheme). In situ destruction of excess borohydride by addition of acetone prior to acetic acid-induced cyclisation was necessary in order to avoid partial reduction of the conjugated ester groups. The cyclisation proceeds through the isolable 2,6-cis-disubstituted piperidine 477, the stereochemistry of this ring then controlling the outcome of the second cyclisation leading to the 4,6-trans-disubsituted quinolizidine 473. The third ring was formed by Dieckmann condensation followed by ester hydrolysis and decarboxylation, again under carefully optimised conditions, to give the tricyclic ketone 478 in 55% overall yield. Wittig olefination produced the exocyclic methylene compound 479, hydrogenation of which needs to occur on the more hindered face in order to ensure the required equatorial disposition of the target alkaloid's methyl substituent. Salt formation with mesitylenesulfonic acid was reasonably effective in blocking the more exposed face of the alkene; after catalytic hydrogenation, a 3.5 : 1 mixture of (±)-hippodamine 475 and its unnatural epimer 480 was isolated in 93% combined yield. The isomerically pure alkaloid 475 could be obtained in 37% yield after repeated chromatography. An alternative approach involving the addition of methyllithium to ketone 478 gave a good yield of the tertiary alcohol 481, the structure of which was confirmed by X-ray crystallography; but Barton–McCombie deoxygenation via the methylxanthate, expected to be stereoselective, produced a 2.3 : 1 mixture of hippodamine and epi-hippodamine in a poor overall yield. A more successful stereoselective synthesis of epi-hippodamine was achieved by initial hydroboration–oxidation of alkene 479 to give exclusively the primary alcohol 482, the structure of which was also confirmed by X-ray crystallography. Deoxygenation of the corresponding iodide was effected with zinc and acetic acid, completing the synthesis of isomerically pure epi-hippodamine 480 in 11 steps and an overall yield of 17% based on 474. | ||
| Scheme 39 Reagents and conditions: i, phthalimide, PPh3, DIAD, THF, −20 °C, then rt, overnight; ii, OsO4 (2 mol%) in ButOH, NaIO4, THF–H2O, rt, 20 h; iii, (EtO)2POCH2CO2Et, NaH, THF, 0 °C, 2 h, then rt, 48 h; iv, NaBH4 (1.4 equiv.), PriOH–H2O (6 : 1), rt, 24 h, then Me2CO (3.6 equiv.), rt, 12 h, then AcOH (30 equiv.), 80 °C, then work-up, then K2CO3, C6H6, rt, overnight; v, ButOK, C6H6, reflux (Dean–Stark apparatus), 19 h, then HCl (2 M in Et2O), CHCl3, rt, 1 h; vi, LiCl (5 equiv.), H2O (5 equiv.), DMF, reflux, 4 h; vii, Ph3PCH2, THF, rt, 1 h, then 60 °C, 30 min; viii, 2,4,6-Me3C6H2SO3H·2H2O, THF, rt, 1 h, then H2 (1 atm), 10% Pd/C, 2 d; ix, MeLi (1.6 M in Et2O), PhMe, 0 °C, 8 h; x, n-BuLi, THF, −78 °C, 1 h, then CS2, rt, 3 h, then MeI, rt, overnight; xi, Bu3SnH, AIBN, PhMe, reflux, 4 h; xii, 9-BBN (0.5 M in THF), THF, −78 °C, 1 h, then rt, overnight, then H2O2, aq. NaOH, 0 °C, 2 h; xiii, n-BuLi, Ph3CH, THF, 0 °C, 10 min, then p-TsCl, 0 °C, 2.5 h; xiv, NaI, MeCOEt, reflux, 24 h; xv, Zn, AcOH, reflux, 3 h. | ||
16 Acknowledgements
This material is based upon work supported by the National Research Foundation under grant number 2053652. Financial support from the University of the Witwatersrand is also gratefully acknowledged.17 References
- K. Afarinkia and A. Bahar, Tetrahedron: Asymmetry, 2005, 16, 1239 CrossRef CAS.
- T. Ayad, Y. Genisson and M. Baltas, Curr. Org. Chem., 2004, 8, 1211 CAS.
- H. Iida, N. Yamazaki and C. Kibayashi, J. Org. Chem., 1987, 52, 3337 CrossRef CAS.
- Y. Ichikawa, T. Ito and M. Isobe, Chem.–Eur. J., 2005, 11, 1949 CrossRef CAS.
- T. Schimming, K. Jenett-Siems, P. Mann, B. Tofern-Reblin, J. Milson, R. W. Johnson, T. Deroin, D. F. Austin and E. Eich, Phytochemistry, 2005, 66, 469 CrossRef CAS.
- K. Braun, J. Romero, C. Liddell and R. Creamer, Mycol. Res., 2003, 107, 980 Search PubMed.
- (a) J. P. Michael, Nat. Prod. Rep., 2005, 22, 604 Search PubMed; (b) J. P. Michael, Nat. Prod. Rep., 2004, 21, 628 Search PubMed; (c) J. P. Michael, Nat. Prod. Rep., 1999, 16, 681 Search PubMed; (d) J. P. Michael, Nat. Prod. Rep., 1997, 16, 683 Search PubMed; (e) J. P. Michael, Nat. Prod. Rep., 2003, 20, 458 RSC; (f) J. P. Michael, Nat. Prod. Rep., 2005, 22, 610 Search PubMed; (g) J. P. Michael, Nat. Prod. Rep., 2004, 21, 635 Search PubMed; (h) J. P. Michael, Nat. Prod. Rep., 2005, 22, 612 Search PubMed; (i) J. P. Michael, Nat. Prod. Rep., 2003, 20, 466 Search PubMed; (j) J. P. Michael, Nat. Prod. Rep., 2003, 20, 468 Search PubMed; (k) J. P. Michael, Nat. Prod. Rep., 2005, 22, 616 Search PubMed; (l) J. P. Michael, Nat. Prod. Rep., 2005, 22, 620 Search PubMed; (m) J. P. Michael, Nat. Prod. Rep., 2004, 21, 645 Search PubMed; (n) J. P. Michael, Nat. Prod. Rep., 2004, 21, 643 Search PubMed.
- J. McLain-Romero, R. Creamer, H. Zepeda, J. Strickland and G. Bell, J. Anim. Sci., 2004, 82, 2169 Search PubMed.
- S. G. Pyne, Curr. Org. Synth., 2005, 2, 39 Search PubMed.
- S. G. Pyne, A. S. Davis, N. J. Gates, J. P. Hartley, K. B. Lindsay, T. Machan and M. Tang, Synlett, 2004, 2670 CrossRef CAS.
- K. B. Lindsay and S. G. Pyne, J. Org. Chem., 2002, 67, 7774 CrossRef CAS.
- K. B. Lindsay and S. G. Pyne, Aust. J. Chem., 2004, 57, 669 CrossRef CAS.
- R. Ling and P. S. Mariano, J. Org. Chem., 1998, 63, 6072 CrossRef CAS.
- L. Song, E. N. Duesler and P. S. Mariano, J. Org. Chem., 2004, 69, 7284 CrossRef CAS.
- R. Martín, C. Murruzzu, M. A. Pericàs and A. Riera, J. Org. Chem., 2005, 70, 2325 CrossRef CAS.
- See, a. o., (a) H. Zhao, S. Hans, X. Cheng and D. R. Mootoo, J. Org. Chem., 2001, 66, 1761 CrossRef CAS; (b) W.-S. Zhou, W.-G. Xie, Z.-H. Lu and X.-F. Pan, J. Chem. Soc., Perkin Trans. 1, 1995, 2599 RSC; (c) M. Naruse, S. Aoyagi and C. Kibayashi, J. Org. Chem., 1994, 59, 1358 CrossRef CAS.
- G. Heimgärtner, D. Raatz and O. Reiser, Tetrahedron, 2005, 61, 643 CrossRef.
- M. J. Martín-López, R. Rodriguez and F. Bermejo, Tetrahedron, 1998, 54, 11623 CrossRef CAS.
- D. Díez, M. T. Benéitez, M. J. Gil, R. F. Moro, I. S. Marcos, N. M. Garrido, P. Basabe and J. G. Urones, Synthesis, 2005, 565 CAS.
- N. Ikota and A. Hanaki, Chem. Pharm. Bull., 1990, 38, 2712 CAS.
- K. Kadlečiková, V. Dalla, S. Marchalín, B. Decroix and P. Baran, Tetrahedron, 2005, 61, 4743 CrossRef CAS.
- M. Bonanni, M. Marradi, S. Cicchi, C. Faggi and A. Goti, Org. Lett., 2005, 7, 319 CrossRef CAS.
- K. Whitby, T. C. Pierson, B. Geiss, K. Lane, M. Engle, Y. Zhou, R. W. Doms and M. S. Diamond, J. Virol., 2005, 79, 8698 CrossRef CAS.
- L. Cronin and P. V. Murphy, Org. Lett., 2005, 7, 2691 CrossRef CAS.
- R. H. Furneaux, G. J. Gainsford, J. M. Mason and P. C. Tyler, Carbohydr. Res., 2004, 339, 1747 CrossRef CAS.
- F. Chevallier, E. Le Grognec, I. Beaudet, F. Fliegel, M. Evain and J.-P. Quintard, Org. Biomol. Chem., 2004, 2, 3128 RSC.
- N. S. Karanjule, S. D. Markad, T. Sharma, S. G. Sabharwal, V. G. Puranik and D. D. Dhavale, J. Org. Chem., 2005, 70, 1356 CrossRef CAS.
- D. D. Dhavale, S. M. Jachak, N. P. Karche and C. Trombini, Synlett, 2004, 1549 CrossRef CAS.
- L. Gao and R. I. Hollingsworth, Tetrahedron, 2005, 61, 3805 CrossRef CAS.
- J. W. Daly, N. Noimai, B. Kongkathip, N. Kongkathip, J. M. Wilham, H. M. Garraffo, T. Kaneko, T. F. Spande, Y. Nimit, J. Nabhitabhata and T. Chan-Ard, Toxicon, 2004, 44, 805 CrossRef CAS.
- H. Tsuneki, Y. You, N. Toyooka, S. Kagawa, S. Kobayashi, T. Sasaoka, H. Nemoto, I. Kimura and J. A. Dani, Mol. Pharmacol., 2004, 66, 1061 CrossRef CAS.
- E. Klegraf, M. Follmann, D. Schollmeyer and H. Kunz, Eur. J. Org. Chem., 2004, 3346 CrossRef CAS.
- M. Weymann, W. Pfrengle, D. Schollmeyer and H. Kunz, Synthesis, 1997, 1151 CrossRef CAS.
- S. Knauer, B. Kranke, L. Krause and H. Kunz, Curr. Org. Chem., 2004, 8, 1739 CrossRef CAS.
- L. F. Roa, D. Gnecco, A. Galindo, J. L. Terán and S. Bernès, Tetrahedron: Asymmetry, 2004, 15, 847 CrossRef CAS.
- L. F. Roa, D. Gnecco, A. Galindo and J. L. Terán, Tetrahedron: Asymmetry, 2004, 15, 3393 CrossRef CAS.
- R. Settambolo, G. Guazzelli and R. Lazzaroni, Lett. Org. Chem., 2005, 2, 176 Search PubMed.
- N. T. Patil, N. K. Pahadi and Y. Yamamoto, Tetrahedron Lett., 2005, 46, 2101 CrossRef CAS.
- J. Gebauer, P. Dewi and S. Blechert, Tetrahedron Lett., 2005, 46, 43 CrossRef CAS.
- N. Toyooka, K. Tanaka, T. Momose, J. W. Daly and H. M. Garraffo, Tetrahedron, 1997, 53, 9553 CrossRef CAS.
- N. Toyooka, H. Nemoto, M. Kawasaki, H. M. Garraffo, T. F. Spande and J. W. Daly, Tetrahedron, 2005, 61, 1187 CrossRef CAS ; correction in N. Toyooka, H. Nemoto, M. Kawasaki, H. M. Garraffo, T. F. Spande and J. W. Daly, Tetrahedron, 2005, 61, 5139 Search PubMed.
- N. Toyooka, M. Kawasaki, H. Nemoto, J. W. Daly, T. F. Spande and H. M. Garraffo, Heterocycles, 2005, 65, 5 CrossRef CAS ; correction in N. Toyooka, M. Kawasaki, H. Nemoto, J. W. Daly, T. F. Spande and H. M. Garraffo, Heterocycles, 2006, 66, 1317 Search PubMed.
- N. Toyooka, M. Kawasaki and H. Nemoto, Chem. Pharm. Bull., 2005, 53, 555 CrossRef CAS.
- N. Toyooka, A. Fukutome, H. Shinoda and H. Nemoto, Angew. Chem., Int. Ed., 2003, 42, 3808 CrossRef CAS.
- N. Toyooka, A. Fukutome, H. Shinoda and H. Nemoto, Tetrahedron, 2004, 60, 6197 CrossRef CAS.
- N. Toyooka, Toyama Medical and Pharmaceutical University, personal communication. Valuable correspondence with Professor Toyooka is gratefully acknowledged. The corrigendum for reference 42 is given above; that for reference 43 will be published in due course.
- G. Kim and N. Kim, Tetrahedron Lett., 2005, 46, 423 CrossRef CAS.
- N. Toyooka, A. Fukutome, H. Nemoto, J. W. Daly, T. F. Spande, H. M. Garraffo and T. Kaneko, Org. Lett., 2002, 4, 1715 CrossRef CAS.
- F. A. Davis and B. Yang, J. Am. Chem. Soc., 2005, 127, 8398 CrossRef CAS.
- X. Pu and D. Ma, J. Org. Chem., 2003, 68, 4400 CrossRef CAS.
- W. Zhu, D. Dong, X. Pu and D. Ma, Org. Lett., 2005, 7, 705 CrossRef CAS.
- R. Kumareswaran, J. Gallucci and T. V. RajanBabu, J. Org. Chem., 2004, 69, 9151 CrossRef CAS.
- J. P. Michael, C. B. de Koning and C. W. van der Westhuyzen, Org. Biomol. Chem., 2005, 3, 836 RSC.
- B.-F. Chen, M.-R. Tasi, C.-Y. Yang, J.-K. Chang and N.-C. Chang, Tetrahedron, 2004, 60, 10223 CrossRef CAS.
- L. S. Santos and R. A. Pilli, Tetrahedron Lett., 2001, 41, 6999 CrossRef.
- L.-R. Sun, S.-X. Wang and X. Li, Zhongguo Yaowu Huaxue Zazhi, 1997, 7, 129 (Chem. Abstr., 1999, 130, 2168) Search PubMed.
- L.-R. Sun, X. Li and S.-X. Wang, J. Asian Nat. Prod. Res., 2005, 7, 127–130 (Chem. Abstr., 2005, 143, 302433) Search PubMed.
- S. M. Allin, W. R. S. Barton, W. R. Bowman and T. McInally, Tetrahedron Lett., 2001, 42, 7887–7890 CrossRef CAS.
- H. Nakano, E. Nakajima, S. Hiradate, Y. Fujii, K. Yamada, H. Shigemori and K. Hasegawa, Phytochemistry, 2004, 65, 587 CrossRef CAS.
- H. Nakano, E. Nakajima, Y. Fujii, H. Shigemori and K. Hasegawa, Plant Growth Regul., 2004, 44, 207 CrossRef CAS.
- B. B. Snider and B. J. Neubert, Org. Lett., 2005, 7, 2715 CrossRef CAS.
- A. R. Carroll, G. Arumugan, R. J. Quinn, J. Redburn, G. Guymer and P. Grimshaw, J. Org. Chem., 2005, 70, 1889 CrossRef CAS.
- P. Klausmeyer, G. N. Chmurny, T. G. McCloud, K. D. Tucker and R. H. Shoemaker, J. Nat. Prod., 2004, 67, 1732 CrossRef CAS.
- Z. Jin, S. P. Li, Q. M. Wang and R. Q. Huang, Chin. Chem. Lett., 2004, 15, 1164 CAS.
- K. N. Chaudhuri, B. Ghosh, D. Tepfer and S. Jha, Plant Cell Rep., 2005, 24, 25 Search PubMed.
- Z. Xi, R. Zhang, Z. Yu, D. Ouyang and R. Huang, Bioorg. Med. Chem. Lett., 2005, 15, 2673 CrossRef CAS.
- T. G. Back, M. D. Hamilton, V. J. J. Lim and M. Parvez, J. Org. Chem., 2005, 70, 967 CrossRef CAS.
- S. M. Amorde, A. S. Judd and S. F. Martin, Org. Lett., 2005, 7, 2031 CrossRef CAS.
- S. N. Kazmi, Z. Ahmed, W. Ahmed and A. Malik, Heterocycles, 1989, 29, 1901 CrossRef CAS.
- D. L. Comins, X. Zheng and R. R. Goehring, Org. Lett., 2002, 4, 1611 CrossRef CAS.
- A. O. Maldaner and R. A. Pilli, Synlett, 2004, 1343 CAS.
- H. Morita, Y. Hirasawa, T. Shinzato and J. Kobayashi, Tetrahedron, 2004, 60, 7015 CrossRef CAS.
- Y. Hirasawa, H. Morita and J. Kobayashi, Heterocycles, 2004, 64, 515 CrossRef CAS.
- T. G. Back and M. D. Hamilton, Org. Lett., 2002, 4, 1779 CrossRef CAS.
- M. Zaja and S. Blechert, Tetrahedron, 2004, 60, 9629 CrossRef CAS.
- M. Atobe, N. Yamazaki and C. Kibayashi, Tetrahedron Lett., 2005, 46, 2669 CrossRef CAS.
- W. J. Moran, K. M. Goodenough, P. Raubo and J. P. A. Harrity, Org. Lett., 2003, 5, 3427 CrossRef CAS.
- K. M. Goodenough, W. J. Moran, P. Raubo and J. P. A. Harrity, J. Org. Chem., 2005, 70, 207 CrossRef CAS.
- P. Mai Le, M.-T. Martin, N. Van Hung, D. Guénard, T. Sévenet and N. Platzer, Magn. Reson. Chem., 2005, 43, 283 CrossRef.
- K. Asres, F. Sporer and M. Wink, Biochem. Syst. Ecol., 2004, 32, 915 CrossRef CAS.
- G. C. Kite, Biochem. Syst. Ecol., 2005, 33, 39 CrossRef CAS.
- M. Qi, X. Ge, M. Liang and R. Fu, Anal. Chim. Acta, 2004, 527, 69 CrossRef CAS.
- M. de Cortes Sánchez, P. Altares, M. M. Pedrosa, C. Burbano, C. Cuadrado, C. Goyoaga, M. Muzquiz, C. Jiménez-Martínez and G. Dávila-Ortiz, Food Chem., 2005, 90, 347 CrossRef CAS.
- J. K. Przybylak, D. Ciesiolka, W. Wysocka, P. M. García-López, M. A. Ruiz-López, W. Wysocki and K. Gulewicz, Ind. Crops Prod., 2005, 21, 1 CrossRef CAS.
- X. Chen, C. Yi, X. Yang and X. Wang, J. Chromatogr., B, 2004, 812, 149 CrossRef CAS.
- P. M. García López, P. G. de la Mora, W. Wysocka, B. Maiztegui, M. E. Alzugaray, H. Del Zotto and M. I. Borelli, Eur. J. Pharmacol., 2004, 504, 139 CrossRef CAS.
- Y. Yu, P. Ding and D. Chen, Anal. Chim. Acta, 2004, 523, 15 CrossRef CAS.
- C. Yi, P. Li, Y. Tao and X. Chen, Microchim. Acta, 2004, 147, 237 CrossRef CAS.
- L.-L. Yin and X.-Z. Zhu, Pharmacol., Biochem. Behav., 2005, 80, 419 CrossRef CAS.
- E. Górnicka, J. E. Rode, E. D. Raczyńska, B. Dasiewicz and J. C. Dobrowolski, Vib. Spectrosc., 2004, 36, 105 CrossRef CAS.
- E. D. Raczyńska, M. Makowski, E. Górnicka and M. Darowska, Int. J. Mol. Sci., 2005, 6, 143 Search PubMed.
- A. Zellagui, S. Rhouati, J. Creche, G. Toth, A. A. Ahmed and P. W. Paré, Rev. Latinoam. Quím., 2004, 32, 109 (Chem. Abstr., 2005, 143, 302279) Search PubMed.
- B. Jasiewicz, W. Boczoń and J. Kurek, Collect. Czech. Chem. Commun., 2004, 69, 2068 CrossRef CAS.
- R. Kolanoś, W. Wysocka, T. Borowiak, G. Dutkiewicz and T. Brukwicki, J. Mol. Struct., 2005, 737, 75 CrossRef CAS.
- B. Jasiewicz, E. Sikorska, I. V. Khmelinskii, B. Warżajtis, U. Rychlewska, W. Boczoń and M. Sikorski, J. Mol. Struct., 2004, 707, 89 CrossRef CAS.
- B. Jasiewicz, W. Boczoń, A. Mumot, B. Warżajtis and U. Rychlewska, J. Mol. Struct., 2005, 737, 239 CrossRef CAS.
- S. K. Kang, Y.-M. Lee, Y.-I. Kim, Y. Kim, K. Seff and S.-N. Choi, Inorg. Chim. Acta, 2004, 357, 2602 CrossRef CAS.
- Y.-M. Lee, S.-M. Park, S. K. Kang, Y.-I. Kim and S.-N. Choi, Bull. Korean Chem. Soc., 2004, 25, 823 CAS.
- A. Johansson, M. Vestergren, M. Håkansson, B. Gustafsson and S. Jagner, New J. Chem., 2004, 28, 1000 RSC.
- M. Vestergren, J. Eriksson, G. Hilmersson and M. Håkansson, J. Organomet. Chem., 2003, 682, 172 CrossRef CAS.
- C. Strohmann, K. Strohfeldt, D. Schildbach, M. J. McGrath and P. O'Brien, Organometallics, 2004, 23, 5389 CrossRef CAS.
- B. D. Allen and D. J. O'Leary, J. Am. Chem. Soc., 2003, 125, 9018 CrossRef CAS.
- B. D. Allen, J.-C. Cintrat, N. Faucher, P. Berthault, B. Rousseau and D. J. O'Leary, J. Am. Chem. Soc., 2005, 127, 412 CrossRef CAS.
- E.-U. Würthwein and D. Hoppe, J. Org. Chem., 2005, 70, 4443 CrossRef.
- W. Zeng, R. Fröhlich and D. Hoppe, Tetrahedron, 2005, 61, 3281 CrossRef CAS.
- E. Beckmann and D. Hoppe, Synthesis, 2005, 217 CAS.
- M. Özlügedik, J. Kristensen, J. Reuber, R. Fröhlich and D. Hoppe, Synthesis, 2004, 2303.
- R. Kalkofen, S. Brandau, B. Wibbeling and D. Hoppe, Angew. Chem., Int. Ed., 2004, 43, 6667 CrossRef CAS.
- (a) M. M. Martinez and D. Hoppe, Org. Lett., 2004, 6, 3743 CrossRef CAS; (b) M. M. Martinez and D. Hoppe, Eur. J. Org. Chem., 2005, 1427 CrossRef CAS.
- M. J. Dearden, M. J. McGrath and P. O'Brien, J. Org. Chem., 2004, 69, 5789 CrossRef CAS.
- M. J. Dearden, C. R. Firkin, J.-P. R. Hermet and P. O'Brien, J. Am. Chem. Soc., 2002, 124, 11870 CrossRef CAS.
- P. O'Brien, K. B. Wiberg, W. F. Bailey, J.-P. R. Hermet and M. J. McGrath, J. Am. Chem. Soc., 2004, 126, 15480 CrossRef CAS.
- M. J. Johansson, L. Schwartz, M. Amedjkouh and N. Kann, Tetrahedron: Asymmetry, 2004, 15, 3531 CrossRef CAS.
- P.-W. Phuan, J. C. Ianni and M. C. Kozlowski, J. Am. Chem. Soc., 2004, 126, 15473 CrossRef CAS.
- R. K. Dieter and R. T. Watson, Tetrahedron Lett., 2002, 43, 7725 CrossRef CAS.
- R. K. Dieter, N. Chen and R. T. Watson, Tetrahedron, 2005, 61, 3221 CrossRef CAS.
- A. Sparatore, B. Tasso, V. Boido and F. Sparatore, Helv. Chim. Acta, 2005, 88, 245 CrossRef CAS.
- M.-Y. Chang, H.-M. Tai, C.-H. Lin and N.-C. Chang, Heterocycles, 2005, 65, 395 CrossRef CAS.
- G. Pandey, G. D. Reddy and D. Chakrabarti, J. Chem. Soc., Perkin Trans. 1, 1996, 219 RSC.
- S. Ma and B. Ni, Chem.–Eur. J., 2004, 10, 3286 CrossRef CAS.
- C. Botuha, C. M. S. Galley and T. Gallagher, Org. Biomol. Chem., 2004, 2, 1825 RSC.
- T. Honda, R. Takahashi and H. Namiki, J. Org. Chem., 2005, 70, 499 CrossRef CAS.
- (a) T. Buttler and I. Fleming, Chem. Commun., 2004, 2404 RSC; (b) T. Buttler, I. Fleming, S. Gonsior, B.-H. Kim, A.-Y. Sung and H.-G. Woo, Org. Biomol. Chem., 2005, 3, 1557 RSC.
- J.-P. R. Hermet, M. J. McGrath, P. O'Brien, D. W. Porter and J. Gilday, Chem. Commun., 2004, 1830 RSC.
- D. Passarella, M. Angoli, A. Giardini, G. Lesma, A. Silvani and B. Danieli, Org. Lett., 2002, 4, 2925 CrossRef CAS.
- A. Barilli, F. Belinghieri, D. Passarella, G. Lesma, S. Riva, A. Silvani and B. Danieli, Tetrahedron: Asymmetry, 2004, 15, 2921 CrossRef CAS.
- M. Angoli, A. Barilli, G. Lesma, D. Passarella, S. Riva, A. Silvani and B. Danieli, J. Org. Chem., 2003, 68, 9525 CrossRef CAS.
- J. W. Daly, Cell. Mol. Neurobiol., 2005, 25, 513 CrossRef CAS.
- A. I. Ivachtchenko, S. E. Tkachenko, Y. B. Sandulenko, V. Y. Vvedensky and A. V. Khvat, J. Comb. Chem., 2004, 6, 828 CrossRef CAS.
- A. I. Ivachtchenko, A. Khvat, S. E. Tkachenko, Y. B. Sandulenko and V. Y. Vvedensky, Tetrahedron Lett., 2004, 45, 6733 CrossRef CAS.
- R. W. Fitch, Y. Kaneko, P. Klaperski, J. W. Daly, G. Seitz and D. Gündisch, Bioorg. Med. Chem. Lett., 2005, 15, 1221 CrossRef CAS.
- J. W. Coe, M. G. Vetelino, C. G. Bashore, M. C. Wirtz, P. R. Brooks, E. P. Arnold, L. A. Lebel, C. B. Fox, S. B. Sands, T. I. Davis, D. W. Schulz, H. Rollema, F. D. Tingley, III and B. T. O'Neill, Bioorg. Med. Chem. Lett., 2005, 15, 2974 CrossRef CAS.
- J. W. Coe, P. R. Brooks, M. G. Vetelino, M. C. Wirtz, E. P. Arnold, J. Huang, S. B. Sands, T. I. Davis, L. A. Lebel, C. B. Fox, A. Shrikhande, J. H. Heym, E. Schaeffer, H. Rollema, Y. Lu, R. S. Mansbach, L. K. Chambers, C. C. Rovetti, D. W. Schulz, F. D. Tingley, III and B. T. O'Neill, J. Med. Chem., 2005, 48, 3474 CrossRef CAS.
- T. S. Zhivotova, A. M. Gazaliev, M. K. Ibraev, S. D. Fazylov and R. Z. Kasenov, Russ. J. Appl. Chem., 2004, 77, 1321 CrossRef CAS.
- S. D. Fazylov, A. M. Gazaliev, A. B. Karimova and S. Z. Kudaibergenova, Russ. J. Gen. Chem., 2004, 74, 1133 CrossRef CAS.
- V. A. Saprykina, V. I. Vinogradova, R. F. Ambartsumova, T. F. Ibragimov, A. Sultankulov and K. M. Shakhidoyatov, Chem. Nat. Compd., 2004, 40, 582 CrossRef CAS.
- H. S. Christie and C. H. Heathcock, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12079 CrossRef CAS.
- D. Trauner, J. B. Schwarz and S. J. Danishefsky, Angew. Chem., Int. Ed., 1999, 38, 3542 CrossRef CAS.
- D. Trauner, D. G. Churchill and S. J. Danishefsky, Helv. Chim. Acta, 2000, 83, 2344 CrossRef CAS.
- K. S. Feldman, A. L. Perkins and K. M. Masters, J. Org. Chem., 2004, 69, 7928 CrossRef CAS.
- Y. Matsumura, S. Aoyagi and C. Kibayashi, Org. Lett., 2004, 6, 965 CrossRef CAS.
- H.-L. Zhang, G. Zhao, Y. Ding and B. Wu, J. Org. Chem., 2005, 70, 4954 CrossRef CAS.
- L. G. Arini, P. Szeto, D. L. Hughes and R. A. Stockman, Tetrahedron Lett., 2004, 45, 8371 CrossRef CAS.
- A. L. de Sousa and R. A. Pilli, Org. Lett., 2005, 7, 1617 CrossRef CAS.
- T. Huxford and N. S. Simpkins, Synlett, 2004, 2295 CAS.
- D. L. J. Clive, J. Wang and M. Yu, Tetrahedron Lett., 2005, 46, 2853 CrossRef CAS.
- D. L. J. Clive, M. Yu and Z. Li, Chem. Commun., 2005, 906 RSC.
- H. Tsuneki, Y. You, N. Toyooka, T. Sasaoka, H. Nemoto, J. A. Dani and I. Kimura, Biol. Pharm. Bull., 2005, 28, 611 CrossRef CAS.
- M. Rejzek and R. A. Stockman, Tetrahedron Lett., 2002, 43, 6505 CrossRef CAS.
- M. Rejzek, R. A. Stockman and D. L. Hughes, Org. Biomol. Chem., 2005, 3, 73 RSC.
| This journal is © The Royal Society of Chemistry 2007 |