Discrimination of the enantiomers of new biphenylic derivatives in chiral micellar aggregates
Francesca
Ceccacci
a,
Luisa
Giansanti
a,
Giovanna
Mancini
*bc,
Paolo
Mencarelli
a and
Alessandro
Sorrenti
a
aDipartimento di Chimica, Università degli Studi di Roma “La Sapienza”, P. le A. Moro 5, 00185, Roma, Italy. E-mail: paolo.mencarelli@uniroma1.it; Fax: +3906490421; Tel: +390649913078
bCNR, Istituto di Metodologie Chimiche, Dipartimento di Chimica “La Sapienza”, P. le A. Moro 5, 00185, Roma, Italy. E-mail: giovanna.mancini@uniroma1.it; Fax: +3906490421; Tel: +390649913769
cCentro di Eccellenza Materiali Innovativi Nanostrutturati per Applicazioni, Chimiche Fisiche e Biomediche, Via Elce di Sotto, 06123 Perugia, Italy
First published on 30th October 2006
Abstract
The synthesis and characterization of two new chiral biphenylic derivatives is reported. The rotational barriers have been calculated on simpler homologues. The racemic mixtures of the two compounds have been used as probes of chirality for exploring the sites of chiral recognition in chiral micellar aggregates. Results suggest that one of the sites of chiral discrimination is the hydrophobic part of the aggregates, far from the stereogenic centres.
Introduction
Assemblies formed by chiral amphiphiles have received attention over the last 30–40 years from different points of view. They have been investigated by various approaches as chiral media for enantioselective reactions,1 used as selectors for enantiomer separations2 and regarded as models for investigating the role of chirality in bio-membranes.3In the study of polymolecular aggregates as bio-membrane models it is of crucial interest to identify the non- covalent interactions responsible for the organization of the aggregates, for the transfer of the chiral information from the monomer to the aggregates and for aggregate recognition properties. We have largely investigated micellar aggregates formed by sodium N-dodecanoyl-L-prolinate, 1, as bio-membrane models and found that they are capable of discriminating the enantiomers of dipeptides3j and biphenylic derivatives3d,k used as probes of chiral recognition. In fact, under aggregating conditions, the diastereomeric interactions between chiral assembly and probe enantiomers yielded diastereomeric 1H NMR signals. The modulation of the molecular structure of the chiral probe allowed us to tune the interactions with the aggregates and to identify some sites of chiral recognition.3j,k In the case of biphenylic derivatives the choice of substituents and their position on the aromatic rings, besides determining the mode of association and interaction with the aggregates, also influences the rotational barrier . In the case of a sufficiently low rotational barrier the transfer of the chiral information from the assembly to the probe may be revealed as an imbalance in the 1 : 1 equilibrium ratio of the interconverting enantiomers of the probe, i.e. as a deracemization process.3i The occurrence of a deracemization , resulting from different association constants of the enantiomers with the chiral aggregate, provides an opportunity to investigate chiral recognition also by circular dichroism (CD). This may be an important point in this type of investigation, because often NMR fails to detect enantiodiscrimination due to broad signals of surfactants under aggregating conditions. The biphenylic derivatives we had previously used bear the hydrophobic chain that binds the moiety to the aggregates in an ortho position. In order to further investigate the interactions involved in the chiral recognition process in aggregates formed by 1, we took into consideration, as chiral probes, two new biphenylic derivatives, namely 2-carboxy-2′-methoxy-4′-N-dodecyl-N-methylamino-6-nitrobiphenyl, 2a, and N,N-dimethyl-N-dodecil-N-[4-(2-methoxycarbonyl-6-nitrophenyl)-3-methoxy]-phenylammonium bromide, 3a, both bearing a hydrophobic chain in a para position. Moving the hydrophobic chain from an ortho to a para position should change the orientation of the aromatic portion of the probe inside the aggregates.
Biphenyl derivative 2a features functional groups in the ortho positions that are known to determine a rotational barrier that easily allows enantiomer interconversion at ambient temperature as we found for 2-carboxy-2′-methoxy-6-nitrobiphenyl, 4, for which a rotational barrier of 22 kcal mol–1 was measured.4 The presence of a dialkylamino group in the para position of 2a, should lead to an even lower barrier since it is known that the presence of an electron-donating substituent in the para position on one phenyl ring, combined with electron-withdrawing groups on the other, decreases the rotational barrier of biphenyls.5
Biphenyl derivative 3a features a similar substitution pattern, with two modifications: the presence of the carboxymethyl group instead of the carboxylic group and the presence of a trialkyl ammonium group instead of the dialkylamino group in the para position. Both modifications should substantially increase the rotational barrier with respect to 2a. The former due to an increase of the steric hindrance at the ortho positions, the latter because it is known that the presence of an electron-withdrawing substituent in the para position enhances the rotational barrier .5
Here we report on the preparation of 2a, and 3a, on the theoretical calculation of the rotational barriers for the simpler homologues 2-carboxy-2′-methoxy-4′-N,N-dimethylamino-6-nitrobiphenyl, 2b and N,N,N-trimethyl-N-[4-(2-methoxycarbonyl-6-nitrophenyl)-3-methoxy]-phenylammonium3b, and on the discrimination of biphenylic enantiomers of 2a, and 3a, in micellar aggregates formed by 1, observed by 1H NMR.
Results and discussion
Synthesis of biphenylic derivatives
Biphenylic derivatives 2a and 3a were prepared by mild condition Ulmann coupling of 2-bromo-3-nitromethylbenzoate with 5-N-dodecyl-N-methylamino-2-iodoanisole (Scheme 1). The following deprotection of the carboxylic group by alkaline hydrolysis yielded derivative 2a, whereas quaternization of the amino function with bromomethane yielded derivative 3a. The halogenation of N-dodecyl-N-methyl-m-anisidine according to a described procedure6 to yield 5-N-dodecyl-N-methylamino-2-iodoanisole was regiospecific; halogenation of position 5 was verified by NOESY NMR experiments. | ||
| Chart 1 | ||
 | ||
| Scheme 1 Synthetic pattern. (i) Cu/DMF at 343 K; (ii) 10% NaOH under reflux; (iii) CH3Br in acetone. | ||
Theoretical calculation of the rotational barrier of biphenilic derivatives
In the past we have measured the rotational barrier of biphenylic derivatives by various techniques:3k,4 (a) by measuring the racemization rate of one of the enantiomer after separation by HPLC on a chiral phase at low temperature, (b) by dynamic HPLC, exploiting the on-column racemization ; (c) by dynamic NMR, by investigating the response of diastereomeric signals to temperature. In this work we did not succeed in separating biphenylic enantiomers by HPLC, and dynamic NMR experiments could not be performed in aggregating conditions (i.e. in aqueous samples of (1) because of the poor resolution of spectra below 292 K; moreover, the use of other chiral auxiliaries did not yield well resolved diastereomeric signals. In view of the fact that the rotational barriers of 2a and 3a were experimentally not available to us, but considering that their knowledge is fundamental for the correct interpretation of chiral recognition experiments, we decided to evaluate the rotational barrier of their simpler homologues, 2b and 3b, by theoretical calculation.In previous papers we showed that the rotational barrier of a substituted biphenyl can be reliably evaluated by DFT calculations at the B3LYP/6-31G(d) level of theory. The results obtained for the rotational barriers, at such level of theory, were in good agreement with the experimental values.3k,4 Our observation of the good behaviour of the DFT approach was in accordance with results obtained by other authors in the calculations of rotational barriers in biphenyls.7 Therefore the same DFT approach, at the same level of theory, was used in this work for the calculations of the rotational barriers of 2b and 3b (all the calculations were carried out by using the Gaussian 98 or Gaussian 03 packages8).
The ground state structures and the transition states for the enantiomerization of 2b and 3b were fully optimized, and each stationary point found was characterized by a frequency calculation. For the structures featuring one imaginary frequency and therefore found to be a saddle point , the normal mode corresponding to the imaginary frequency, was animated by using the visualization program Molden.9 In this way it was verified that the displacements that compose the mode lead to the two enantiomeric structures.
For compound 2b six different minima were found for the non-planar ground state (Fig. 1), and two different structures, (E) and (Z), were found for the transition state of the enantiomerization pathway (Fig. 2). The minima differ for the biphenyl dihedral angle and for the rotation of the substituents. In Table 1 total energies, relative energies and biphenyl dihedral angles for the structures obtained from the DFT calculations on 2b are reported.
 | ||
| Fig. 1 Structures of the six minima found for 2b. | ||
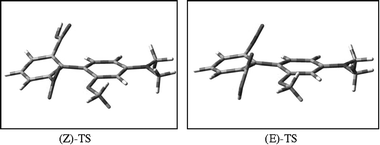 | ||
| Fig. 2 Structures of the two transition states found for the enantiomerization of 2b. | ||
| E a (a.u.) | E rel /kcal mol–1 | Dihedral angle, degree | |
|---|---|---|---|
| a Zero-point corrections included. | |||
| Minimum 1 | –1104.5510 94 | 0.00 | 126.1 |
| Minimum 2 | –1104.5502 16 | 0.55 | 125.5 |
| Minimum 3 | –1104.5493 93 | 1.07 | 73.2 |
| Minimum 4 | –1104.5489 34 | 1.36 | –82.6 |
| Minimum 5 | –1104.5484 40 | 1.66 | –116.5 |
| Minimum 6 | –1104.5470 84 | 2.52 | –82.7 |
| (Z)-TS | –1104.5202 34 | 19.36 | 176.7 |
| (E)-TS | –1104.5153 58 | 22.42 | –1.9 |
For compound 3b five different minima were found for the non-planar ground state (Fig. 3), and four structures, two of (E) configuration and two of (Z) configuration, were found for the transition state of the enantiomerization pathway (Fig. 4). The minima differ for the biphenyl dihedral angle and for the rotation of the substituents. In particular, the more stable minima 1 and 2 present the methoxycarbonyl moiety in the more stable Z conformation, whereas minima 3, 4, and 5 present the less stable E conformation. This feature is also found for the transition states in that in the least stable (E)-TS2 transition state , the methoxycarbonyl moiety is in the less stable E conformation. In Table 2 the total energies, relative energies and biphenyl dihedral angles for the structures obtained from the DFT calculations on 3b are reported.
 | ||
| Fig. 3 Structures of the five minima found for 3b. | ||
 | ||
| Fig. 4 Structures of the four transition states found for the enantiomerization of 3b. | ||
| E a (a.u.) | E rel /kcal mol–1 | Dihedral angle, degree | |
|---|---|---|---|
| a Zero-point corrections included. | |||
| Minimum 1 | –1183.475 232 | 0.00 | –87.4 |
| Minimum 2 | –1183.474 762 | 0.29 | 86.4 |
| Minimum 3 | –1183.457 565 | 11.09 | –92.4 |
| Minimum 4 | –1183.457 448 | 11.16 | 86.0 |
| Minimum 5 | –1183.457 031 | 11.42 | 92.4 |
| (Z)-TS1 | –1183.433 711 | 26.06 | 176.6 |
| (Z)-TS2 | –1183.433 187 | 26.38 | 177.8 |
| (E)-TS1 | –1183.433 047 | 26.47 | –7.3 |
| (E)-TS2 | –1183.421 584 | 33.66 | 2.2 |
Since six minima and two transition states were found for 2b, and five minima and four transition states were found for 3b, the rotational barriers for the enantiomerization of 2b and 3b, were obtained as the difference between the Boltzmann averaged total energy of the transition states and the Boltzmann averaged total energy of the minima. The results are reported in Table 3.
| 2b/kcal mol–1 | 3b/kcal mol–1 |
|---|---|
| 19.00 | 26.13 |
The values obtained for the rotational barriers confirm our expectations (see above): the rotational barrier of 2b is 3 kcal mol–1 lower than that of 4, whereas 3b features a barrier which is 4 kcal mol–1 higher than that of 4. Our computational finding thus agrees with what was experimentally known about the effect of para substituents on the rotational barriers of biphenyls.5 In order to illustrate the consequence of such barriers on the enantiomerization rate, it is useful to translate the barrier values into the corresponding reaction half-time (τ1/2). At 298 K, a rotational barrier of 19 kcal mol–1 corresponds to a τ1/2 of about 1 s. In other words, the racemization reaction of 2b is complete in about 10 s. At variance, a rotational barrier of 26 kcal mol–1 corresponds, at 298 K, to a τ1/2 of about 19 days; therefore, at room temperature, 3b requires more than 6 months to completely racemize. These data should be taken into consideration when interpreting the results of chiral recognition experiments in micellar aggregates.
Chiral recognition in micellar aggregates
Recognition experiments were carried out by 1H NMR and Circular Dichroism on aqueous solutions of 4.0 mM biphenylic derivative in the presence of 40.0 mM anionic surfactant 1. The concentration ratio was such to allow a complete solubilisation of the solute . Because the association of the probes with the chiral aggregate did not induce excessive growth of the aggregates we obtain reasonable resolved NMR spectra of the aqueous solutions, so that, in both cases, we could observe splitting of some signals in the aromatic region, due to the discrimination of biphenylic enantiomers by the chiral micellar aggregates (Figs. 5 and 6). In fact, in the spectrum relative to the aqueous solution of 2a the only triplet we expect to observe is the signal due to the proton in position 4, whereas the spectrum reported in Fig. 5 shows two triplets (at 7.05 and 7.52 ppm). 2D 1H NMR experiments allowed us to assign signals as reported in Fig. 5, and therefore to assess that the triplet at 7.05 ppm is due to the superimposition of two diastereomeric doublets (in a ∼1 : 1 ratio) relative to the protons in the 5′ position (or 6′) of enantiomeric biphenyls.† Analogously, the 1H NMR spectrum relative to the aqueous solution of 3a reported in Fig. 6 shows splitting of signals due to protons in the 6′ (or 5′) position and to protons in the 3′ position, still in a ∼1 : 1 ratio. Therefore experimental evidence indicates that enantiomers of both biphenyls are discriminated by the chiral micellar aggregates, however, NMR evidence did not allow us to ascertain deracemization . In fact, integration of NMR signals does not allow, generally, the appreciation of small differences, moreover, due to the small chemical shift differences in diastereomeric NMR signals, it was not possible to integrate them precisely. | ||
| Fig. 5 Aromatic region of the 1H NMR spectrum of an aqueous solution 4.0 mM in 2a and 40.0 mM in 1. | ||
 | ||
| Fig. 6 Aromatic region of the 1H NMR spectrum of an aqueous solution 4.0 mM in 3a and 40.0 mM in 1. | ||
Because enantiopure biphenylic derivatives feature high molar ellipticities, even a small imbalance in the enantiomer equilibrium may be easily revealed by circular dichroism measurements. The CD spectra of the aqueous samples of 2a and 3a in chiral micellar aggregates did not show any band demonstrating the absence of any imbalance in the enantiomer equilibrium.
Considering the relatively high rotational barrier that was calculated for 3b, experimental evidence of an imbalance in the enantiomer equilibrium, even in the presence of a differential binding of the two enantiomers to the chiral micellar environment, was hardly expected. Therefore the absence of a band in the CD spectrum of the aggregate solutions containing 3a does not rule out the possibility that there is a differential binding. On the other hand, considering that 2b features a rotational barrier low enough to allow a very fast racemization , we could reasonably expect that, should the interactions with the aggregates favour the association of one enantiomer with the aggregate, the transfer of the chiral information from the aggregate to 2a could manifest itself as an imbalance of the enantiomer equilibrium. Thus, the absence of a deracemization phenomenon in the aqueous solution of 2a in chiral micellar aggregates, clearly indicates that there is no appreciable difference in the extent of binding of 2a enantiomers to the chiral aggregates.
The effect of the aromatic systems of 2a and 3a on the chemical shift of resonances due to 1 gives information on the binding site of the chirality probe. In Fig. 7 we report the comparison of the NMR spectrum of aqueous 0.10 M 1 in the absence of solute (Fig. 7a) and in the presence of 2a (Fig. 7b) and 3a (Fig. 7c), respectively. The chemical shift differences (expressed in Hz) observed between the spectra performed in the absence and in the presence of biphenylic derivative are reported in Table 4. All resonances relative to 1 protons are upfield shifted due to the association of the biphenylic derivatives with the aggregates; however inspection of Table 4 suggests that there are differences in the site of association of the two chirality probes, in fact in the case of 2a the signal relative to a head group proton (δZanti) features the highest chemical shift variation, demonstrating that this proton is in the shielding cone of the aromatic system. In the case of 3a the most shifted signals are those relative to the hydrophobic chain (Chain and 11-CH3), demonstrating a preferential site of binding of the aromatic system in this region.
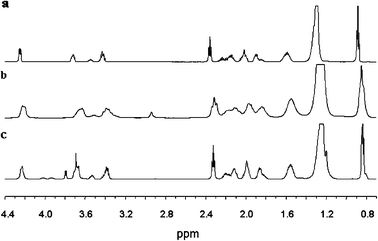 | ||
| Fig. 7 1H NMR spectrum of (a) an aqueous solution 0.1 M in 1; (b) an aqueous solution 0.1 M in 1 in the presence of 2a; (c) an aqueous solution 0.1 M in 1 in the presence of 3a. | ||
| α | δ Z anti | δ E syn | δ Z syn | 1-CH2 | 2-CH2 | Chain | 11-CH3 | |
|---|---|---|---|---|---|---|---|---|
| 1 + 2a | 14 | 36 | 10 | 22 | 22 | 20 | 18 | 17 |
| 1 + 3a | 11 | 25 | 8 | 25 | 20 | 19 | 30 | 29 |
The finding of enantiodiscrimination in a hydrophobic site of binding supports our hypothesis3j that chiral recognition may occur in a chiral environment induced in an internal region of the aggregate by remote stereogenic centers. In fact, this result is in agreement with our previous observation of enantiodiscrimination of dipeptides in the hydrophobic region of aggregates formed by sodium N-dodecanoyl-L-prolinate.
Conclusions
We have explored the chiral recognition capabilities of a bio-membrane model by using two new biphenylic derivatives as probes of chirality. 1H NMR experiments allowed us to observe enantiodiscrimination in both cases and to have information on the sites of binding. The finding of enantiodiscrimination in a hydrophobic site of binding supports our hypothesis that chiral recognition in polymolecular aggregates might occur in a region of the aggregate remote from the stereogenic centers. Therefore the translation of the chiral information from the monomer to the aggregates is due to a sum of recognition processes responsible of the organization of the whole aggregate.Experimental
2-Bromo-3-nitromethylbenzoate
The compound was prepared and characterized as previously described.3d,10N-dodecyl-m-anisidine
A mixture of 9.0 g (60 mmol) of m-anisidine and 6.3 g (25 mmol) of n-bromododecane in 20 mL of isopropanol was kept under reflux until complete disappearance of the alkyl bromide (∼7 h), the reaction being monitored by TLC (SiO2, hexane). After neutralisation with a saturated K2CO3 aqueous solution, the mixture was extracted with Et2O and the organic solution was washed with brine and then dried over Na2SO4. Removal of solvent under reduced pressure gave a brown oil that, after purification on silica gel (hexane/Et2O 95/5), yielded 6.3 g (87%) of a white solid (m.p. found 305–307 K).1H NMR (CDCl3) ppm: 0.885 (3H, t, CH3, 3J = 6.8 Hz); 1.268 (18H, m); 1.616 (2H, m, CH2); 3.083 (2H, t, N–CH2, 3J = 7.0 Hz); 3.769 (3H, s, O–CH3); 6.209 (3H, m, 2, 4, 6); 7.065 (1H, t, 5, Jo = 8.1 Hz).
N-dodecyl-N-methyl-m-anisidine11
A slurry of 1.1 g (3.8 mmol) of N-dodecyl-m-anisidine and 0.55 g (14 mmol) of NaBH4 in 10 mL of THF was added slowly to an efficiently stirred solution of 0.9 mL of 37% aqueous formaldehyde, 1.8 mL of 3M H2SO4 and 5 mL of THF at 263 K, keeping temperature below 293 K. After the evolution of gas ceased, the solution was made strongly basic with NaOH pellets and the supernatant was decanted and saved. The white solid residue was treated with 20 mL of water and the resultant solution was extracted with Et2O. The combined organic solutions were washed with brine and dried over Na2SO4. Purification on silica gel (hexane/Et2O 95/5) of the brown oil, obtained by removal of the solvent under reduced pressure, gave 0.98 g (85%) of a pale yellow oil.1H NMR (CDCl3) ppm: 0.915 (3H, t, CH3, 3J = 6.4 Hz); 1.220–1.420 (18H, m); 1.586 (2H, m, CH2); 2.932 (3H, s, N–CH3,); 3.300 (2H, t, N–CH2, 3J = 7.6 Hz); 3.811 (3H, s, O–CH3); 6.309 (3H, m, 2, 4, 6); 7.149 (1H, t, 5, Jo = 7.9 Hz).
5-N-dodecyl-N-methylamino-2-iodoanisole 6
A solution of 0.9 g (3.5 mmol) of I2 in 70 mL of CCl4 was added over one hour, under inert atmosphere and in the dark, to an heterogeneous mixture of 1.1 g (3.5 mmol) of N-dodecyl-N-methyl-m-anisidine, 5 mL of CCl4, 0.35 g of CaCO3 and 16 mL of water. The reaction was monitored by TLC (SiO2, hexane/Et2O 95/5). After 18 hours the organic phase was washed with 7 mL of a saturated Na2S2O3 aqueous solution, with brine and dried over Na2SO4. Removal of the solvent under reduced pressure gave a dark yellow oil (in a 95% yield) that was used immediately in order to avoid degradation.1H NMR (CDCl3) ppm: 0.877 (3H, t, CH3, 3J = 6.3 Hz); 1.150–1.450 (18H, m); 1.556 (2H, m, CH2); 2.914 (3H, s, N–CH3,); 3.276 (2H, t, N–CH2, 3J = 7.3 Hz); 3.844 (3H, s, O–CH3); 6.094 (1H, dd, 4, Jo = 8.8 Hz, Jm = 2.5 Hz); 6.163 (1H, d, 6, Jm = 2.5 Hz); 7.466 (1H, d, 3, Jo = 8.8 Hz).
2-Carboxymethyl-2′-methoxy-4′-N-dodecyl-N-methylamino-6-nitrobiphenyl
A solution of 1.4 g (3.2 mmol) of 5-N-dodecyl-N-methylamino-2-iodoanisole and 0.09 g (0.34 mmol) of 2-bromo-3-nitromethylbenzoate in 12 mL of anhydrous DMF was heated gently in an inert atmosphere and 1.0 g of activated Cu0 powder was added when the temperature reached 343 K. The reaction was monitored by TLC (SiO2, hexane/Et2O 6/4) and was complete after 3 hours. After filtration of the reaction mixture, the solvent was removed under reduced pressure and the residue dissolved in 100 mL of Et2O, treated with 10% NH3 aqueous solution (2 × 5 mL), with brine and dried over Na2SO4. After removal of the solvent under reduced pressure, purification of the obtained brown oil on silica gel (hexane/ Et2O 7/3) yielded 0.062 g (37%) of a red oil.1H NMR (CDCl3) ppm: 0.870 (3H, t, CH3, 3J = 6.6 Hz); 1.198–1.444 (18H, m); 1.500–1.700 (2H, m, CH2); 2.955 (3H, s, N–CH3,); 3.305 (2H, t, N–CH2, 3J = 6.9 Hz); 3.641 (3H, s, O–CH3); 3.683 (3H, s, COOCH3); 6.176 (1H, d, 3′, Jm = 2.2 Hz); 6.274 (1H, dd, 5′, Jo = 8.4 Hz, Jm = 2.2 Hz); 7.426 (1H, t, 4, Jo = 8.1 Hz); 7.800–7.920 (2H, m, 3, 5).
2-Carboxy-2′-methoxy-4′-N-dodecyl-N-methylamino-6-nitrobiphenyl, 2a
A solution of 0.14 g (0.29 mmol) of 2-carboxymethyl-2′-methoxy-4′-N-dodecyl-N-methylamino-6-nitrobiphenyl in 3 mL of ethanol and 3 mL of 10% NaOH aqueous solution was kept under reflux for 6 hours. The mixture was then acidified to pH 4 with 3 M HCl and extracted with Et2O; the organic phase was washed with brine and dried over Na2SO4. After removal of the solvent under reduced pressure, purification of the reddish oil on silica gel (hexane/AcOEt 1/1) yielded 0.13 g (96%) of a red solid that melts at a low temperature (around ambient temperature). 1H NMR (CDCl3) ppm: 0.875 (3H, t, CH3,); 1.200–1.380 (18H, m); 1.500–1.660 (2H, m, CH2); 2.913 (3H, s, N–CH3,); 3.265 (2H, t, N–CH2, 3J = 7.3 Hz); 3.652 (3H, s, O–CH3); 6.259 (1H, d, 3′, Jm = 1.8 Hz); 6.307 (1H, dd, 5′, Jo = 8.5 Hz); 6.936 (1H, d, 6′, Jo = 8.5 Hz) 7.428 (1H, t, 4, Jo = 7.9 Hz); 7.839 (1H, dd, 5, Jo = 8.1 Hz, Jm = 1.1 Hz); 7.958 (1H, dd, 3, Jo = 8.1 Hz, Jm = 1.1 Hz).13C NMR (CDCl3) ppm: 14.11; 22.67; 26.64; 27.14; 29.34; 29.47; 29.62; 29.65; 31.90; 38.82; 53.50; 55.08; 76.62; 77.04; 77.46; 96.21; 105.47; 112.28; 126.13; 127.08; 129.95; 132.39; 132.83; 134.94; 150.55; 151.13; 157.12; 171.57. Elemental analysis calculated for C27H38N2O5: C 68.91%, H 8.14%, N 5.95%; found: C 69.70%, H 8.71%, N 5.61%.
N,N-dimethyl-N-dodecil-N-[4-(2-carboxymethyl-6-nitrophenyl)-3-methoxy]-phenylammonium bromide, 3a
A solution of 0.12 g (0.025 mmol) of 2-carboxymethyl-2′-methoxy-4′-N-dodecyl-N-methylamino-6-nitrobiphenyl in 3 mL of acetone was kept under stirring in a CH3Br saturated atmosphere for seven days. The solution was then concentrated to one third of its volume and Et2O was added dropwise until complete precipitation of a white solid (90% yield); m.p. 393–395 K.1H NMR (CD3OD) ppm: 0.861 (3H, t, CH3); 1.060–1.600 (20H, m); 3.636 (3H, s, N–CH3); 3.742 (3H, s, OCH3); 3.850 (3H, s, COOCH3); 4.036 (2H, m, CH2); 7.287 (1H, d, Jo = 8.5 Hz); 7.450–7.540 (2H, m); 7.743 (1H, t, Jo = 7.9 Hz); 8.100–8.190 (2H, m).
13C NMR (CD3OD) ppm: 14.40; 23.64; 24.31; 26.87; 29.53; 29.96; 30.25; 30.38; 30.53; 30.62; 53.01; 55.21; 55.29; 57.45; 70.64; 105.27; 114.01; 127.94; 128.78; 130.64; 131.36; 131.79; 134.68; 135.01; 146.93; 151.89; 159.45; 167.45; 211.69.
Elemental analysis calculated for C29H43BrN2O5: C 60.10%, H 7.48%, N 4.83%; found: C 60.38%, H 8.11%, N 4.47.
Samples preparation
Samples of 4.0 mM biphenylic derivative in 40.0 mM aqueous surfactant 1 were prepared by adding to the proper amounts of surfactant and biphenylic derivative 0.700 mL of D2O. The solutions were sonicated and gently heated to obtain clear solutions.Acknowledgements
This work has been carried out as part of the project “The use of surfaces and vesicles for the amplification of homochirality in polypeptide chains” of COST action D27. We acknowledge contributions from CNR, Dipartimento di Progettazione Molecolare.References
- (a) C. A. Bunton, L. Robinson and M. F. Stam, Tetrahedron Lett., 1971, 121–124 CrossRef CAS; (b) J. M. Brown and C. A Bunton, J. Chem. Soc., Chem. Commun., 1974, 969–971 RSC; (c) H. Ihara, S. Ono, H. Shosenji and K. Yamada, J. Org. Chem., 1980, 45, 1623–1625 CrossRef CAS; (d) R. A. Moss, Y.-S. Lee and K. W. Alwis, J. Am. Chem. Soc., 1980, 102, 6646–6648 CrossRef CAS; (e) H. Ihara, N. Kunikiyo, T. Kunimasa, M. Nango and N. Kuroki, Chem. Lett., 1981, 667–670 CAS; (f) J. M. Brown, R. L. Elliott, C. G. Griggs, G. Helmchen and G. Nill, Angew. Chem., Int. Ed. Engl., 1981, 20, 890–892 CrossRef; (g) R. Ueoka, Y. Matsumoto, T. Yoshino, N. Watanabe, K. Omura and Y. Murakami, Chem. Lett., 1986, 1743–1746 CAS; (h) R. Ueoka, Y. Matsumoto, R. A. Moss, S. Swarup, A. Sugii, K. Harada, J.-I. Kikuchi and Y. Murakami, J. Am. Chem. Soc., 1988, 110, 1588–1595 CrossRef CAS; (i) H. Ihara, K. Igata, Y. Okubo and M. Nango, J. Chem. Soc., Chem. Commun., 1989, 1900–1902 RSC; (j) M. C. Cleji, P. Scrimin, P. Tecilla and U. Tonellato, Langmuir, 1996, 12, 2956–2960 CrossRef CAS; (k) Y. Zhan and W. Wu, Tetrahedron: Asymmetry, 1997, 8, 3575–3578 CrossRef CAS; (l) M. J. Diego-Castro and H. C. Hailes, Chem. Commun., 1998, 1549–1550 RSC.
- (a) D. Tickle, A. George, K. Jennings, P. Camilleri and A. J. Kirby, J. Chem. Soc., Perkin Trans. 2, 1998, 467–474 RSC; (b) J. K. Rugutt, E. Billiot and I. M. Warner, Langmuir, 2000, 16, 3022–3029 CrossRef CAS; (c) T. J. M. De Bruin, A. T. M. Marcelis, H. Zuilhof, L. M. Rodenburg, H. A. G. Niederlander, A. Koudijs, P. E. M. Overdevest, A. Van Der Padt and E. J. R. Sudholter, Chirality, 2000, 12, 627–636 CrossRef CAS.
- (a) E. M. Arnett and J. M. Gold, J. Am. Chem. Soc., 1982, 104, 636–639 CrossRef CAS; (b) J.-H. Fuhrhop, P. Schnieder, J. Rosenberg and E. Boekema, J. Am. Chem. Soc., 1987, 109, 3387–3390 CrossRef CAS; (c) S. Pathirana, W. C. Neely, L. J. Myers and V. Vodyanoy, J. Am. Chem. Soc., 1992, 114, 1404–1405 CrossRef CAS; (d) J. Bella, S. Borocci and G. Mancini, Langmuir, 1999, 15, 8025–8031 CrossRef CAS; (e) D. Izhaky and L. Addadi, Chem.–Eur. J., 2000, 6, 869–874 CrossRef CAS; (f) T. Kawasaki, M. Tokuhiro, N. Kimizuka and T. Kunitake, J. Am. Chem. Soc., 2001, 123, 6792–6800 CrossRef CAS; (g) S. Lalitha, A. Sampath Kumar, K. J. Stine and D. F. Covey, J. Supramol. Chem., 2001, 1, 53–61 CrossRef CAS; (h) H. Nakagawa, M. Yoshida, Y. Kobori and K.-I. Yamada, Chirality, 2003, 15, 703–708 CrossRef CAS; (i) S. Borocci, F. Ceccacci, L. Galantini, G. Mancini, D. Monti, A. Scipioni and M. Venanzi, Chirality, 2003, 15, 441–447 CrossRef CAS; (j) C. Bombelli; S. Borocci, F. Lupi, G. Mancini, L. Mannina, A. L. Segre and S. Viel, J. Am. Chem. Soc., 2004, 126, 13354–13362 CrossRef CAS; (k) R. Andreani, C. Bombelli, S. Borocci, J. Lah, G. Mancini, P. Mencarelli, G. Vesnaver and C. Villani, Tetrahedron: Asymmetry, 2004, 15, 987–994 CrossRef CAS; (l) F. Ceccacci, G. Mancini, A. Sferrazza and C. Villani, J. Am. Chem. Soc., 2005, 127, 13762–13763 CrossRef CAS.
- F. Ceccacci, G. Mancini, P. Mencarelli and C. Villani, Tetrahedron: Asymmetry, 2003, 14, 3117–3122 CrossRef CAS.
- C. Wolf, W. A. König and C. Roussel, Liebigs Ann., 1995, 781–786.
- F. B. Dains, A. W. Magers and W. L. Steiner, J. Am. Chem. Soc., 1930, 52, 1570–1572 CrossRef CAS.
- (a) P. U. Biedermann, V. Schurig and I. Agranat, Chirality, 1997, 9, 350–353 CrossRef CAS; (b) F. Grein, J. Phys. Chem. A, 2002, 106, 3823–3827 CrossRef CAS.
- (a) M. J. Frisch, G. W Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, Jr, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98 (Revision A.7), Gaussian, Inc., Pittsburgh PA, 1998 Search PubMed; (b) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, Jr, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision B.05), Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed.
- G. Schaftenaar and J. H. Noordik, J. Comput. Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS.
- R. W. Stoughton and R. Adams, J. Am. Chem. Soc., 1932, 54, 4426–4434 CrossRef CAS.
- A. G. Giumanini, G. Chiavari, M. M. Musini and P. Rossi, Synthesis, 1980, 743–746 CrossRef CAS.
Footnote |
| † 1 Because of line width enlargement, splitting of long range coupling that allows a definite assignment in organic solvents is lost in aggregating conditions. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |