Factors affecting imine coordination in (iminoterpyridine)MX2 (M = Fe, Co, Ni, Zn): synthesis, structures, DFT calculations and ethylene oligomerisation studies
Yohan D. M.
Champouret
a,
Jean-Didier
Maréchal†
ab,
Rajinder K.
Chaggar
a,
John
Fawcett
a,
Kuldip
Singh
a,
Feliu
Maseras
c and
Gregory A.
Solan
*a
aDepartment of Chemistry, University of Leicester, University Road, Leicester, UK LE1 7RH. E-mail: gas8@leicester.ac.uk
bDepartment of Biochemistry, University of Leicester, University Road, Leicester, UK LE1 7RH
cInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans, 16, 43007, Tarragona, Spain
First published on 9th October 2006
Abstract
The bulky arylimino-terpyridine ligands, 6-{(2,6-i-Pr2C6H3)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR}-2,2′:6′,2″-C15H10N3 [R = H (L1), Me (L2)], have been prepared in high yield from the condensation reaction of the corresponding carbonyl compound with one equivalent of 2,6-diisopropylaniline. Interaction of an equimolar ratio of MX2 with aldimine-containing L1 in n-BuOH at 110 °C affords the mononuclear five-coordinate complexes, [(L1)MX2] (M = Fe, X = Cl 1a; M = Co, X = Cl 1b; M = Ni, X = Br 1c; M = Zn, X = Cl 1d), in which the metal centres occupy the terpyridine cavities in L1 with the imino group pendant and exo to the adjacent pyridine nitrogen atom. Similarly, a five-coordinate complex [(L2)ZnCl2] (2d) is accessible from the reaction of ketimine-containing L2 with ZnCl2 with the non-coordinated imine group in 2d, in this case, adopting a pseudo-exo configuration. In contrast, coordination of the imino-nitrogen atom (endo configuration) results on reaction of (DME)NiBr2 (DME = 1,2-dimethoxyethane) with L2 to furnish the six-coordinate complex trans-[(L2)NiBr2] (2c). With [(L2)MCl2] (M = Fe 2a; M = Co 2b) both solution and solid state IR spectroscopy suggest that the imine group prefers to adopt a bound conformation. Quantum mechanical calculations (DFT) have been performed on [(Lx)MX2] (Lx = L1, L2; M = Fe, Co, Ni, Zn; X = Cl, Br) and support the synthetic results with the exo conformations energetically preferred for 1a–1d and 2d, while a distinct energetic preference is observed for the endo conformation in 2c. For 2a and 2b, however, the closeness in energies for endo and exo configurations suggests dynamic behaviour is likely. The iron species 1a and 2a display low activities (on treatment with excess MAO) for ethylene oligomerisation at one bar ethylene pressure affording highly linear α-olefins (>98%); the cobalt (1b, 2b) and nickel (1c, 2c) species are inactive. Single crystal X-ray diffraction studies have been performed on 1a, 1b, 1d, 2c and 2d.
CR}-2,2′:6′,2″-C15H10N3 [R = H (L1), Me (L2)], have been prepared in high yield from the condensation reaction of the corresponding carbonyl compound with one equivalent of 2,6-diisopropylaniline. Interaction of an equimolar ratio of MX2 with aldimine-containing L1 in n-BuOH at 110 °C affords the mononuclear five-coordinate complexes, [(L1)MX2] (M = Fe, X = Cl 1a; M = Co, X = Cl 1b; M = Ni, X = Br 1c; M = Zn, X = Cl 1d), in which the metal centres occupy the terpyridine cavities in L1 with the imino group pendant and exo to the adjacent pyridine nitrogen atom. Similarly, a five-coordinate complex [(L2)ZnCl2] (2d) is accessible from the reaction of ketimine-containing L2 with ZnCl2 with the non-coordinated imine group in 2d, in this case, adopting a pseudo-exo configuration. In contrast, coordination of the imino-nitrogen atom (endo configuration) results on reaction of (DME)NiBr2 (DME = 1,2-dimethoxyethane) with L2 to furnish the six-coordinate complex trans-[(L2)NiBr2] (2c). With [(L2)MCl2] (M = Fe 2a; M = Co 2b) both solution and solid state IR spectroscopy suggest that the imine group prefers to adopt a bound conformation. Quantum mechanical calculations (DFT) have been performed on [(Lx)MX2] (Lx = L1, L2; M = Fe, Co, Ni, Zn; X = Cl, Br) and support the synthetic results with the exo conformations energetically preferred for 1a–1d and 2d, while a distinct energetic preference is observed for the endo conformation in 2c. For 2a and 2b, however, the closeness in energies for endo and exo configurations suggests dynamic behaviour is likely. The iron species 1a and 2a display low activities (on treatment with excess MAO) for ethylene oligomerisation at one bar ethylene pressure affording highly linear α-olefins (>98%); the cobalt (1b, 2b) and nickel (1c, 2c) species are inactive. Single crystal X-ray diffraction studies have been performed on 1a, 1b, 1d, 2c and 2d.
1. Introduction
In spite of the rich variety of transition metal complexes of 2,2′:6′,2″:6″,2‴-quaterpyridine (A, Fig. 1) that have been reported,1–5 the coordination chemistry of closely related 2,6-linked oligopyridylimine N,N,N,N-ligands such as 6,6′-bis(arylimino)-2,2′-bipyridine (B, Fig. 1) is not nearly as well developed. Nevertheless, the role of B in both supramolecular chemistry6 and metal-mediated alkene polymerisation/oligomerisation catalysis7 has been documented. On the other hand 6-arylimino-2,2′:6,2″-terpyridine (Lx, Fig. 1), a hybrid of A and B, has to the knowledge of the authors not been investigated as a support for metal complexes.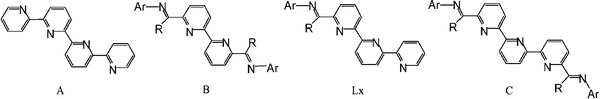 | ||
| Fig. 1 Quaterpyrine (A), bis(imino)bipyridine (B), terpyridylimine (Lx) and bis(imino)terpyridine (C); R = H or methyl, Ar = aryl group. | ||
Recently, we have been interested in the coordination chemistry and catalytic potential of divalent late transition metal complexes containing the more extended oligopyridylimine ligand, 6,6″-bis(arylimino)-2,2′:6′,2″-terpyridine (C, Fig. 1).8 By judicious choice of imino-carbon substituent (R) in C, 1 : 1 coordination compounds incorporating the metal centre in either a tridentate terpyridyl (R = H) or a tetradentate iminoterpyridyl (R = Me) cavity have been structurally identified for Ni(II). By contrast, attempted isolation of a 1 : 1 complex featuring Zn(II) and the ketimine derivative of C (R = Me) furnished only a dinuclear Zn2{bis(arylimino)terpyridine}-containing species. This variation in bonding capacity has prompted us to target related ligand frames in which bimetallic formation is unlikely.
Herein, the use of the shorter chain mono-imine Lx (Fig. 1) is investigated as a support for a series of late transition metal(II) halides. In particular, we employ a combined synthetic and theoretical approach to study the factors that influence the capacity of 6-{(2,6-i-Pr2C6H3)NCR}-2,2′:6′,2″-C15H10N3 [L1 (R = Haldimine), L2 (R = Meketimine)] to act as tri- or tetra-dentate (or both) ligands in complexes based on the 3d metals, iron, cobalt, nickel and zinc. In addition, several of these systems are probed as precatalysts for the oligomerisation of ethylene and their performances related to the bonding modes possible for Lx.
2. Results and discussion
2.1. Ligand synthesis
Ligands 6-{(2,6-i-Pr2C6H3)NCR}-2,2′:6′,2″-C15H10N3 [R = H (L1), Me (L2)] can be prepared by treating the corresponding carbonyl compound, 6-{OCR}-2,2′:6′,2″-C15H10N3 (R = H, Me), with 2,6-diisopropylaniline in absolute ethanol in the presence of a catalytic amount of glacial acetic acid. To achieve satisfactory yields, the synthesis of aldimine-containing L1 required milder conditions and shorter reaction times (50 °C, 12 hours) while for L2 higher temperatures and longer reaction times (80 °C, 72 hours) were found to be more suitable (Scheme 1). Alternatively, L2 can be prepared by reacting 6-{OCMe}-2,2′:6′,2″-C15H10N3 in neat 2,6-diisopropylaniline at 160 °C over a 30 minute reaction time.9 The precursor carbonyl compounds are not commercially available and are synthesised by using either a palladium(0)-mediated cross-coupling of 6-bromo-2,2′-bipyridine9 with 2-(n-Bu3Sn)-6-{C(R)CH2CH2O}-C5H3N (R = H, Me) or by treating the lithiated derivative of 6-bromo-2,2′:6′-2″-terpyridine with Me2NCRO (R = H, Me) using conditions established by Tanaka et al. for the formyl derivative 6-{OCH}-2,2′:6′,2″-C15H10N3 (Scheme 1).10
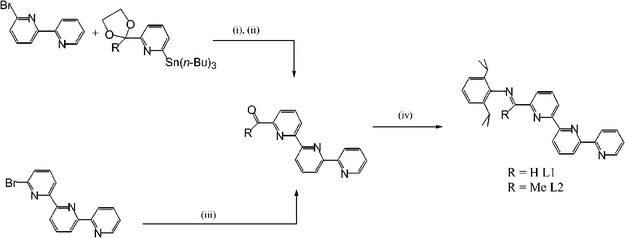 | ||
| Scheme 1 Reagents and conditions: (i) Pd(PPh3)4 (6 mol%), toluene, 90 °C, 72 h; (ii) HCl (4 M), 60 °C, 12 h; (iii) n-BuLi, −78 °C, Me2NC(O)R (R = H, Me), diethyl ether–hexane–tetrahydrofuran; (iv) 2,6-i-Pr2C6H3NH2, cat. H+, ethanol, 50–80 °C, 12–72 h. | ||
2.2 Synthesis of complexes
The reaction of L1 or L2 with one equivalent of MX2 [MX2 = FeCl2, CoCl2, (DME)NiBr2, ZnCl2] in n-butanol at 110 °C gave complexes [(L1)MX2] (M = Fe, X = Cl 1a; M = Co, X = Cl 1b; M = Ni, X = Br 1c; M = Zn, X = Cl 1d) and [(L2)MX2] (M = Fe, X = Cl 2a; M = Co, X = Cl 2b; M = Ni, X = Br 2c; M = Zn, X = Cl 2d) in high yield, respectively (Scheme 2). All products have been characterised by FAB mass spectrometry, IR spectroscopy and, in the cases of 1a–c and 2a–c by magnetic measurements and diamagnetic 1d/2d by 1H NMR spectroscopy (see Table 1 and Experimental section). In addition, crystals of 1a, 1b, 1d, 2c and 2d have been the subject of single crystal X-ray diffraction studies.![Reagents and conditions: (i) MX2 [MX2 = FeCl2, CoCl2, ZnCl2, (DME)NiBr2], n-butanol, 110 °C (Ar = 2,6-i-Pr2C6H3).](/image/article/2007/NJ/b610562a/b610562a-s2.gif) | ||
| Scheme 2 Reagents and conditions: (i) MX2 [MX2 = FeCl2, CoCl2, ZnCl2, (DME)NiBr2], n-butanol, 110 °C (Ar = 2,6-i-Pr2C6H3). | ||
| Compound | Colour |
v(CN)a/cm−1 |
μ eff b/μB | 1H NMR spectrumc | FAB mass spectrum |
|---|---|---|---|---|---|
| a Recorded on a Perkin-Elmer Spectrum One FT-IR spectrometer on solid samples. b Recorded on an Evans balance at room temperature. c Recorded in CDCl3 (1d) and CD3CN (2d) at ambient temperature. d Sample is diamagnetic. e Broad paramagnetically shifted peaks. | |||||
| 1a | Purple-blue | 1637 | 5.2 | e | 546 [M]+, 511 [M − Cl]+ |
| 1b | Blue-green | 1644 | 4.3 | e | 514 [M − Cl]+, 479 [M − 2Cl]+ |
| 1c | Orange | 1639 | 2.5 | e | 559 [M − Br]+, 478 [M − 2Br]+ |
| 1d | Pale yellow | 1644 | d | 1.23 (d, 12H, 3JHH 7.0 Hz, CH(CH3)2), 3.12 (sept, 2H, 3JHH 7.0 Hz, CH(CH3)2), 7.1–7.2 (m, 3H, ArH), 7.6 (m, 1H, PyH), 8.04 (dd, 1H, 3JHH 7.6 Hz, 4JHH 1.4 Hz, PyH), 8.16 (app. t, 2H, 3JHH 7.6 Hz, 3JHH 7.6 Hz, PyH), 8.2–8.4 (m, 4H, PyH), 8.74 (d, 1H, 3JHH 7.3 Hz, PyH), 9.02 (d, 1H, 3JHH 5.0 Hz, PyH), 9.53 (s, 1H, HCN). | 519 [M − Cl]+, 484 [M − 2Cl]+ |
| 2a | Dark purple | 1594 | 5.3 | e | 525 [M − Cl]+, 490 [M − 2Cl]+ |
| 2b | Blue | 1569 | 4.6 | e | 528 [M − Cl]+, 493 [M − 2Cl]+ |
| 2c | Orange | 1575 | 2.6 | e | 573 [M − Br]+, 492 [M − 2Br]+ |
| 2d | Pale yellow | 1634 | d | 1.07 (d, 12H, 3JHH 7.0 Hz, CH(CH3)2), 2.56 (s, 3H, CH3CN), 2.93 (sept, 2H, 3JHH 7.0 Hz, CH(CH3)2), 7.4–7.5 (m, 3H, ArH), 7.6 (m, 1H, PyH), 7.8 (m, 1H, 1.4 Hz, PyH), 8.25 (ddd, 1H, 3JHH 7.9 Hz, 3JHH 7.9 Hz, 4JHH 1.5 Hz, PyH), 8.40 (d, 1H, 3JHH 7.9 Hz, PyH), 8.4–8.6 (m, 5H, PyH), 8.66 (d, 1H, 3JHH 7.9 Hz, PyH). | 533 [M − Cl]+, 498 [M − 2Cl]+ |
Crystals of 1 suitable for the X-ray determinations were grown from acetonitrile (1a, 1b) or from a mixed chloroform–acetonitrile (1d) solution. The structures of 1a, 1b and 1d belong to an isomorphous series and will be discussed together. A view of 1a is depicted in Fig. 2; selected bond distances and angles for 1a, 1b and 1d are listed in Table 2. Each structure consists of a five-coordinate metal centre surrounded by two chloride ligands and three pyridyl groups from L2 with the imino group non-coordinated. With the geometric parameter (τ) in 1a, 1b and 1d being close to 0.5 [τ = 0.39 (1a), 0.51 (1b), 0.46 (1d)],11 geometries based on distorted trigonal bipyramidal or distorted square pyramidal could both be applicable. Nevertheless, in 1a and 1d a slight preference towards distorted square pyramidal (τ < 0.5) is observed, as is the case for other experimental and calculated five-coordinate structures determined in this work (vide infra). The pyridyl nitrogen–metal distances are unsymmetrical with the central N(2)py–M(1) distance being the shortest [2.1081(19) (1a), 2.0302(15) (1b), 2.0729(18) (1d) Å] and the distance involving the imine-substituted pyridyl [N(3)py] group the longest [2.2214(19) (1a), 2.1937(16) (1b), 2.2574(18) (1d) Å]. Between structures the nitrogen–cobalt distances in 1b are shorter than the corresponding values in 1a and 1d, consistent with the ionic radii for a high spin dipositive cobalt ion being smaller than for iron and zinc. The pendant imino group in each structure [C(16)–N(4) 1.263(3)–1.264(3) Å] is exo and almost coplanar to the adjacent coordinated pyridine groups [tors: N(3)–C(15)–C(16)–N(4) 174.6° (1a), 174.0° (1b), 168.2° (1d)] with the N-aryl unit essentially orthogonal to these planes.
 | ||
| Fig. 2 Molecular structure of 1a including the atom numbering scheme. All hydrogen atoms, apart from H16, have been omitted for clarity. | ||
| 1a (M = Fe) | 1b (M = Co) | 1d (M = Zn) | |
|---|---|---|---|
| M(1)–N(1) | 2.175(2) | 2.1514(16) | 2.1935(19) |
| M(1)–N(2) | 2.1081(19) | 2.0302(15) | 2.0729(18) |
| M(1)–N(3) | 2.2214(19) | 2.1937(16) | 2.2574(18) |
| M(1)–Cl(1) | 2.2868(8) | 2.2630(7) | 2.2429(6) |
| M(1)–Cl(2) | 2.3081(7) | 2.2733(7) | 2.2548(7) |
| C(16)–N(4) | 1.264(3) | 1.263(2) | 1.263(3) |
| N(1)–M(1)–N(2) | 74.48(8) | 76.64(6) | 75.28(7) |
| N(1)–M(1)–N(3) | 148.97(8) | 153.29(6) | 150.47(7) |
| N(2)–M(1)–N(3) | 74.53(7) | 76.71(6) | 75.24(7) |
| Cl(1)–M(1)–Cl(2) | 114.93(3) | 117.76(3) | 117.37(3) |
| Cl(1)–M(1)–N(1) | 97.41(6) | 95.34(5) | 97.68(5) |
| Cl(1)–M(1)–N(2) | 125.48(6) | 122.84(5) | 119.87(5) |
| Cl(1)–M(1)–N(3) | 99.22(5) | 97.38(4) | 98.39(5) |
| Cl(2)–M(1)–N(1) | 94.55(6) | 93.95(5) | 95.48(5) |
| Cl(2)–M(1)–N(2) | 119.39(6) | 119.20(5) | 122.72(5) |
| Cl(2)–M(1)–N(3) | 101.84(5) | 100.68(5) | 98.88(5) |
The preference of the MX2 unit in 1 to occupy uniquely the terpyridyl cavity within L1 has also been observed in the related 1 : 1 bis(aldimino)terpyridyl [{(2,6-i-Pr2C6H3NCH)2terpy}MX2] (M = Fe, Ni, Zn, X = halide) complexes.8 However when a sequence of only two pyridine units is present within the oligopyridylimine chain, imine coordination can be achieved. For example, in the bis(aldimino)bipyridyl series, [{(2,6-i-Pr2C6H3NCH)2bipy}MCl2] (M = Fe,7 Ni12), the MX2 unit fills the tridentate dipyridylimine pocket leaving the remaining aldimine unit unbound. Nevertheless, an exo conformation for the pendant CHNAr unit is a feature of all the above structural types contrasting with the pseudo-endo disposition observed by the non-coordinated pyridine nitrogen atoms (one per quaterpyridine) in octahedral [(quaterpyridine)2Fe](ClO4)2.3b
Complexes 1a–d all show molecular ion peaks and/or fragmentation peaks corresponding to the loss of halide ions. In their infrared spectra (in solution or in solid state) ν(CN) bands are seen at ca. 1640 cm−1 corresponding to a non-coordinated imine group and in a similar region to that for the free ligand L1.8 Complexes 1a–c are paramagnetic and exhibit magnetic moments of 5.2 μB, 4.3 μB and 2.5 μB (Evans balance at ambient temperature), values that are typical of high spin configurations corresponding to four, three, and two unpaired electrons, respectively. In the 1H NMR spectrum of diamagnetic 1d, signals characteristic for the aryl-H and pyridyl-H protons are seen along with a singlet at δ 9.53 for the aldimine CHN proton which is shifted by ca. δ 1.2 downfield in comparison with the corresponding signal in free L1. The presence of only one doublet for the CHMe2 protons suggests that free rotation around the aryl–nitrogen bond is operational further supporting the non-coordination of the imine.
Crystals of 2c suitable for X-ray determination were grown by prolonged standing in chloroform. A view of 2c is shown in Fig. 3; selected bond distances and angles are listed in Table 3. The molecular structure comprises a single nickel atom bound by both L2 and two terminal bromide ligands so as to form a distorted octahedral geometry. The bromide ligands are disposed mutually trans [Br(1)–Ni(1)–Br(2) 175.74(10)°] with all the four nitrogen donor atoms of L2 filling the equatorial belt. The pyridyl nitrogen–nickel distances are unequally disposed with the internal pyridyl distances being the shortest [Ni(1)–N(2) 1.953(12) Å, Ni(1)–N(3) 1.972(10) Å] and the external pyridyl lengths the longest [Ni(1)–N(1) 2.064(12) Å]. Of the four nitrogen donor moieties, the imino nitrogen forms the longest distance to nickel [Ni(1)–N(4) 2.191(12) Å] with its N-aryl group located almost orthogonal to the terpyridylimine plane. The tetradentate bonding mode exhibited by L2 in 2c is in contrast to the tridentate mode adopted by L1 in 1 but resembles that found in the 1 : 1 complexes trans-[(quaterpyridine)Ni(L)2](NCA) (L = NCMe, NCA = PF6;4d L = OH2, NCA = BF44i) and the ketimine-containing complex trans-[{(2,6-i-Pr2C6H3NCMe)2terpy}NiBr2].9
 | ||
| Fig. 3 Molecular structure of 2c including the atom numbering scheme. All hydrogen atoms have been omitted for clarity. | ||
| Ni(1)–N(1) | 2.064(12) | Ni(1)–Br(1) | 2.579(2) |
| Ni(1)–N(2) | 1.953(12) | Ni(1)–Br(2) | 2.514(2) |
| Ni(1)–N(3) | 1.972(10) | C(16)–N(4) | 1.313(16) |
| Ni(1)–N(4) | 2.191(12) | C(16)–C(17) | 1.504(19) |
| N(1)–Ni(1)–N(2) | 79.9(4) | N(2)–Ni(1)–Br(1) | 88.5(3) |
| N(1)–Ni(1)–N(3) | 157.0(5) | N(2)–Ni(1)–Br(2) | 88.8(3) |
| N(1)–Ni(1)–N(4) | 154.1(4) | N(3)–Ni(1)–N(4) | 77.3(4) |
| N(2)–Ni(1)–N(3) | 77.4(5) | N(3)–Ni(1)–Br(1) | 86.5(3) |
| N(2)–Ni(1)–N(4) | 154.1(4) | N(3)–Ni(1)–Br(2) | 96.2(3) |
| Br(1)–Ni(1)–Br(2) | 175.74(10) | N(4)–Ni(1)–Br(1) | 95.4(3) |
| N(1)–Ni(1)–Br(1) | 89.3(3) | N(4)–Ni(1)–Br(2) | 88.4(3) |
| N(1)–Ni(1)–Br(2) | 87.0(3) | ||
Crystals of 2d suitable for X-ray determination were grown from a mixed chloroform–acetonitrile solution. A view of 2d is shown in Fig. 4; selected bond distances and angles are listed in Table 4. The structure of 2d resembles 1d with a five-coordinate zinc centre surrounded by two chloride ligands and three pyridyl groups from L2 with the CMeNAr group non-coordinated. The geometry at the metal centre is also distorted square pyramidal [τ = 0.35]11 with the N(2) atom defining the apical site and N(1), N(3), Cl(1) and Cl(2) the basal ones. However unlike 1d, the non-coordinated imine group adopts a pseudo-exo configuration [tors.: N(3)–C(15)–C(16)–N(4) 146.3°] with respect to the adjacent coordinated pyridine group. The deviation from planarity is likely due to steric hindrance that can occur between the imino-methyl and the chloride ligands. The effect of this steric interaction appears also to impact on the N(3)–Zn(1) distance with a noticeably longer bond in 2d when compared with 1d [2.308(2) (2d) vs. 2.2574(18) (1d) Å].
 | ||
| Fig. 4 Molecular structure of 2d including the atom numbering scheme. All hydrogen atoms have been omitted for clarity. | ||
| Zn(1)–N(1) | 2.193(2) | Zn(1)–Cl(2) | 2.2322(9) |
| Zn(1)–N(2) | 2.069(2) | C(16)–N(4) | 1.281(4) |
| Zn(1)–N(3) | 2.308(2) | C(16)–C(17) | 1.465(4) |
| Zn(1)–Cl(1) | 2.2654(9) | ||
| N(1)–Zn(1)–N(2) | 75.18(9) | N(1)–Zn(1)–Cl(2) | 95.59(7) |
| N(1)–Zn(1)–N(3) | 150.70(9) | N(2)–Zn(1)–Cl(1) | 110.85(7) |
| N(2)–Zn(1)–N(3) | 75.69(9) | N(2)–Zn(1)–Cl(2) | 119.32(7) |
| Cl(1)–Zn(1)–Cl(2) | 129.60(3) | N(3)–Zn(1)–Cl(1) | 100.75(6) |
| N(1)–Zn(1)–Cl(1) | 92.49(7) | N(3)–Zn(1)–Cl(2) | 95.82(6) |
In the IR spectra for 2a–d (recorded in the solid state), ν(CN) bands are seen between 1634–1569 cm−1 with the band for 2d (1634 cm−1) falling at the higher end of the range and consistent with a non-coordinated imine group. In contrast, 2c shows no CN stretch above 1575 cm−1 while in 2a and 2b only very weak bands at ca. 1630 cm−1 are apparent suggesting that the bound imine is the preferred configuration. The FAB mass spectra of 2a–2d each show fragmentation peaks corresponding to the loss of one and two halide ions. The high spin nature of 2a–c is confirmed from magnetic measurements which indicate moments of 5.3 μB, 4.6 μB and 2.6 μB (Evans balance at ambient temperature), their magnitude being consistent with the presence of four, three, and two unpaired electrons, respectively.
In the 1H NMR spectrum of diamagnetic 2d, sharp signals are seen for the aryl-H and pyridyl-H protons along with a singlet at δ 2.56 corresponding to the CMe
N group; shifted by ca. 0.3 ppm downfield in comparison with free L2. The isopropyl methyl protons are seen as a single doublet at δ 1.07 suggesting that as with 1d the N-aryl group in 2d can freely rotate, indicating that the imine is not bound. By contrast the 1H NMR spectrum of 2c shows broad paramagnetically shifted peaks between δ
−4.1 and 130.9 (see Experimental section). Some degree of assignment can be made on comparison of the chemical shifts and relaxation times with previously reported nickel(II)–pyridyl systems.13 For example, the single pyridyl-Hα proton can be identified as the most downfield signal (δ 130.9), the six pyridyl-Hβ and pyridyl-Hβ′ protons to the signals at δ 92.9, 82.5, 72.3, 66.8, 59.5, 35.0 while the pyridyl-Hγ protons are assigned to the more upfield signals at δ 18.0, 16.9, 13.3. A related assignment of the signals for 2a and 2b, however, proved problematic due to the poor solubility of the complexes and extreme broadness of the signals. Nevertheless, the pyridyl-Hα protons could be identified as peaks at δ 103.4 and δ 95.2 for 2a and 2b, respectively.
2.3 Density functional theory calculations
Given our recent findings on the flexibility of coordination behaviour exhibited by the more extended oligopyridylimine C (see Fig. 1),8 the capacity of Lx to adopt both tri- (with imino group exo) or tetra-dentate (with imino group endo) bonding modes on coordination to an MX2 unit was not entirely surprising. In this previous study the nature of the imino-carbon substituent (Me vs. H) was found to be highly influential on the bonding mode displayed by the ligand while the role of the 3d metal centre (or possibly the halide employed) was not clear. To complement the synthetic work undertaken herein and to compare with our previous studies, DFT calculations have been performed on [(Lx)MX2] (M = Fe, Co, Ni, Zn; X = Cl, Br) for both L1 and L2 with the intent of understanding more fully the factors that influence the binding mode of Lx.In order to evaluate the validity of our DFT approach, geometry optimisations have initially been performed on selected complexes that have been the subject of X-ray determinations in this work. Specifically, optimised structures of trans-[(L2)NiBr2] (opt-2cendo), [(L1)ZnCl2] (opt-1dexo) and [(L2)ZnCl2] (opt-2dexo) were compared with the X-ray data for 2c, 1d and 2d, respectively. The calculated structures are in good agreement with their respective crystallographic counterparts with their overall structural features well reproduced: octahedral for opt-2cendo, distorted square pyramidal for opt-1dexo and distorted square pyramidal for opt-2dexo (Table 5).
| M = Fe, X = Cl | M = Co, X = Cl | M = Ni, X = Br | M = Zn, X = Cl | M = Ni, X = Cl | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a exo | 2a exo | 1a endo | 2a endo | 1b exo | 2b exo | 1b endo | 2b endo | 1c exo | 2c exo | 1c endo | 2c endo | 1d exo | 2d exo | 1d endo | 2d endo | 1c ′ exo | 2c ′ exo | 1c ′ endo | 2c ′ endo | |
| a τ = structural index parameter = (β − α)/60 where β = the largest L–M–L angles and α = second largest (see ref. 11). | ||||||||||||||||||||
| Bond lengths | ||||||||||||||||||||
| M(1)–N(1) | 2.25 | 2.25 | 2.23 | 2.22 | 2.19 | 2.2 | 2.15 | 2.17 | 2.13 | 2.13 | 2.12 | 2.11 | 2.25 | 2.27 | 2.32 | 2.31 | 2.13 | 2.13 | 2.13 | 2.13 |
| M(1)–N(2) | 2.13 | 2.12 | 2.19 | 2.2 | 2.1 | 2.07 | 2.14 | 2.12 | 2.02 | 1.99 | 2.04 | 2.03 | 2.19 | 2.16 | 2.23 | 2.22 | 2.02 | 1.99 | 2.04 | 2.04 |
| M(1)–N(3) | 2.28 | 2.35 | 2.18 | 2.18 | 2.2 | 2.26 | 2.12 | 2.1 | 2.16 | 2.17 | 2.03 | 2.02 | 2.29 | 2.41 | 2.23 | 2.21 | 2.15 | 2.16 | 2.04 | 2.03 |
| M(1)–N(4) | 4.66 | 4.95 | 2.29 | 2.32 | 4.63 | 4.89 | 2.24 | 2.26 | 4.65 | 4.87 | 2.29 | 2.25 | 4.65 | 4.99 | 2.55 | 2.47 | 4.63 | 4.85 | 2.29 | 2.24 |
| M(1)–X(1) | 2.32 | 2.34 | 2.46 | 2.47 | 2.33 | 2.35 | 2.48 | 2.46 | 2.48 | 2.51 | 2.6 | 2.63 | 2.32 | 2.31 | 2.38 | 2.42 | 2.34 | 2.38 | 2.45 | 2.48 |
| M(1)–X(2) | 2.34 | 2.36 | 2.54 | 2.51 | 2.33 | 2.36 | 2.53 | 2.51 | 2.47 | 2.5 | 2.64 | 2.65 | 2.32 | 2.33 | 2.4 | 2.4 | 2.35 | 2.38 | 2.48 | 2.48 |
| C(16)–N(4) | 1.29 | 1.29 | 1.29 | 1.29 | 1.28 | 1.29 | 1.29 | 1.29 | 1.29 | 1.29 | 1.29 | 1.29 | 1.28 | 1.29 | 1.29 | 1.29 | 1.28 | 1.29 | 1.29 | 1.29 |
| Bond angles | ||||||||||||||||||||
| N(1)–M(1)–N(2) | 75.3 | 75.5 | 74.4 | 73.8 | 75.5 | 76.0 | 75.8 | 76.3 | 78.6 | 79.2 | 78.5 | 78.8 | 73.4 | 73.5 | 72.0 | 72.5 | 78.6 | 79.2 | 78.5 | 78.6 |
| N(1)–M(1)–N(3) | 150.6 | 151.6 | 146.8 | 146.7 | 152.7 | 153.4 | 146.8 | 152.3 | 157.3 | 159.7 | 156.4 | 157.4 | 146.9 | 146.8 | 144.6 | 146.0 | 157.3 | 159.8 | 156.4 | 157.0 |
| N(2)–M(1)–N(3) | 75.4 | 76.6 | 72.4 | 73.3 | 77.2 | 78.4 | 73.9 | 76.1 | 78.7 | 80.5 | 78.0 | 78.6 | 73.7 | 74.5 | 72.6 | 73.5 | 78.7 | 80.6 | 77.9 | 78.4 |
| X(1)–M(1)–X(2) | 135.9 | 146.2 | 173.5 | 171.1 | 129.0 | 143.5 | 174.8 | 171.4 | 147.0 | 161.1 | 174.8 | 168.8 | 127.6 | 133.9 | 162.6 | 168.6 | 145.3 | 161.0 | 175.6 | 171.3 |
| X(1)–M(1)–N(1) | 94.6 | 91.9 | 85.4 | 85.2 | 91.5 | 89.2 | 86.1 | 89.7 | 91.7 | 89.4 | 88.9 | 87.8 | 96.0 | 94.1 | 85.2 | 85.2 | 92.1 | 89.4 | 87.9 | 89.0 |
| X(1)–M(1)–N(2) | 116.2 | 114.6 | 95.0 | 89.8 | 119.7 | 115.2 | 92.3 | 86.8 | 110.7 | 100.4 | 84.9 | 89.7 | 120.0 | 123.8 | 101.5 | 101.5 | 113.3 | 99.7 | 90.3 | 85.6 |
| X(1)–M(1)–N(3) | 97.5 | 95.1 | 98.1 | 99.4 | 100.4 | 97.5 | 95.6 | 90.9 | 96.0 | 94.1 | 89.4 | 92.6 | 97.9 | 96.3 | 102.2 | 102.2 | 96.2 | 93.9 | 92.3 | 89.2 |
| X(2)–M(1)–N(1) | 94.1 | 90.5 | 88.7 | 86.4 | 91.1 | 87.7 | 89.1 | 86.8 | 92.8 | 89.3 | 88.8 | 89.9 | 95.7 | 91.5 | 88.7 | 88.7 | 93.0 | 89.5 | 89.9 | 88.9 |
| X(2)–M(1)–N(2) | 107.8 | 98.6 | 86.0 | 85.0 | 110.2 | 99.3 | 84.7 | 84.8 | 102.2 | 97.8 | 83.8 | 85.2 | 112.3 | 101.5 | 92.0 | 92.0 | 101.3 | 98.7 | 85.5 | 85.5 |
| X(2)–M(1)–N(3) | 95.6 | 98.56 | 88.2 | 86.0 | 100.0 | 101.1 | 87.5 | 88.5 | 92.2 | 93.7 | 88.5 | 87.6 | 99.2 | 103.2 | 92.3 | 92.3 | 92.2 | 93.7 | 88.1 | 89.2 |
| τ α | 0.25 | 0.09 | — | — | 0.40 | 0.17 | — | — | 0.17 | 0.03 | — | — | 0.32 | 0.22 | — | — | 0.20 | 0.02 | — | — |
With regard to opt-2dexo, the structural arrangement of the aryl and imine moieties is maintained when compared to 2d, though more displaced towards idealised square pyramidal [τ = 0.35 (2d) vs. 0.22 (opt-2dexo)]. This discrepancy is likely due to the gas phase conditions employed for the calculations which are exempt of crystal packing interactions that are evident in 2d. While agreement between experimental and theoretical angles for opt-1dexo, opt-2cendo and opt-2dexo are good with discrepancies less than 6°, bond lengths are slightly more divergent with average differences around 0.1 Å (Table 5). Nevertheless, the expected variations in the metal ligand distances (e.g., M–N distances: Fehigh spin ≈ Zn > Cohigh spin > Ni) between the different 3d metal complexes employed for all experimentally characterised structures are particularly well reproduced in these theoretical calculations. Due to the size of the system, it was viewed that our approach leads to good agreement with the experimental data and demonstrates that using the B3LYP functional with this basis set constitutes a viable method for studying systems described in this work. To extend the investigation, we have applied this theoretical approach to the two other X-ray characterised complexes 1a and 1b along with 1c, 2a, 2b and the chloride analogues of 1c and 2c, [(Lx)NiCl2] [Lx = L1 (1c′), L2 (2c′)].
Optimised structures for 1a, 1b, 1c, 1d, 1c′, 2a, 2b, 2c, 2d and 2c′ have been determined based on the imine nitrogen atom being bound (opt-1aendo, opt-1bendo, opt-1cendo, opt-1dendo, opt-1c′endo, opt-2aendo, opt-2bendo, opt-2cendo, opt-2dendo, opt-2c′endo) and unbound (opt-1aexo, opt-1bexo, opt-1cexo, opt-1dexo, opt-1c′exo, opt-2aexo, opt-2bexo, opt-2cexo, opt-2dexo, opt-2c′exo); selected bond lengths and angles are listed in Table 5. By inspection of the computed structures it is evident that the endo conformations adopt a common octahedral structure, trans-[(Lx)MX2], regardless of Lx, metal or halide (Fig. 5a). In contrast, the exo conformations fall into three distinct structural types: (i) with the imine co-planar to the adjacent pyridine and the metal centre adopting a distorted square pyramidal geometry (range for τ: 0.17–0.40) (Fig. 5b), (ii) with the imine unit out-of-the plane formed by the adjacent pyridine and the metal centre exhibiting a distorted square based pyramidal geometry (range for τ: 0.09–0.22) (Fig. 5c), (iii) with the imine co-planar to the adjacent pyridine, the halides disposed in a linear fashion and the metal centre displaying a more idealised square pyramidal geometry (τ = 0.02–0.03) (Fig. 5d). The particular type of exo conformation exhibited by a complex (Fig. 5b–d) is found to depend not only on Lx but also on the metal centre. The nature of the halide (Cl vs. Br) ligand appears not to be influential in these systems. For all the L1-containing complexes (opt-1exo), the structures adopted are based on the conformation shown in Fig. 5b with the two nickel versions displaying the more idealised square pyramidal geometries [τ = 0.17 (opt-1cexo), 0.20 (opt-1c′exo)]. On the other hand, complexes bound by the ketimine ligand L2 can adopt two of the exo structures, the precise type being determined by the metal centre with the iron, cobalt and zinc complexes (opt-2aexo, opt-2bexo, opt-2dexo) preferring the structure in Fig. 5c while the nickel systems (opt-2cexo, opt-2c′exo) prefer that in Fig. 5d.
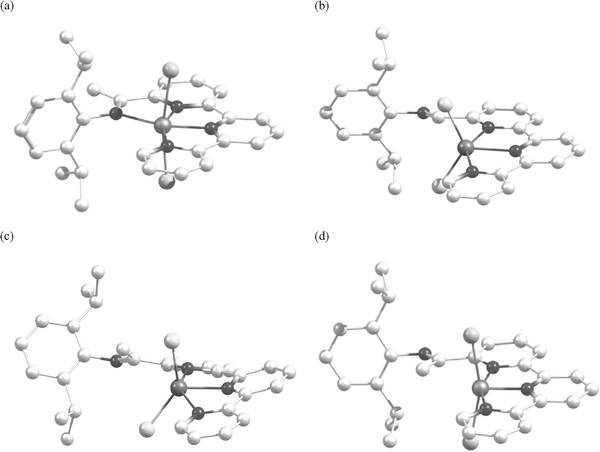 | ||
| Fig. 5 Common optimised structural types for 1 and 2: (a) opt-2endo (also applicable to opt-1endo) (FeCl2, CoCl2, NiBr2, NiCl2, ZnCl2); (b) opt-1exo (FeCl2, CoCl2, NiBr2, NiCl2, ZnCl2); (c) opt-2exo (FeCl2, CoCl2, ZnCl2); (d) opt-2exo (NiBr2, NiCl2). | ||
To investigate the relative stability of endoversus exo structural types for a particular Lx–metal ion combination and to probe the potential for conformation conversions, the difference in energy between endo and exo structures for [(L1)MX2] (1) and [(L2)MX2] (2) was determined (Table 6).
| ΔE | ΔE | ||
|---|---|---|---|
| opt-1a | 6.7 | opt-2a | 0.2 |
| opt-1b | 9.7 | opt-2b | −0.5 |
| opt-1c | 2.1 | opt-2c | −2.5 |
| opt-1d | 7.5 | opt-2d | 3.3 |
| opt-1c′ | 1.6 | opt-2c′ | −3.1 |
The agreement between computed energy differences and the available experimental data is satisfactory. For species opt-1a, opt-1b, opt-1c, opt-1d and opt-2d, the experimentally observed exo-form is favoured by a value between 2.1 and 9.7 kcal mol−1. For opt-2c, the experimentally observed endo form is computed to be more stable by 2.5 kcal mol−1. Interestingly, for opt-2a and opt-2b, where experimental data supports the presence of the endo form, a small energy difference of 0.5 kcal mol−1 at most between the computed structures would suggest the coexistence between two species in equilibrium. Furthermore the fact that the calculations allow the least stable isomers to be computed, additional useful information can be gleaned. As highlighted above the calculations show that the behaviour depends little on the nature of the halide (Cl or Br). With regard to energy differences two major trends are apparent. The first trend is that the L1-containing systems (opt-1) always favour the exo form with respect to the corresponding L2-containing systems (opt-2), with the difference in preference oscillating between 4 and 10 kcal mol−1. The second trend is that the nickel complexes (opt-1c, opt-1c′, opt-2c, opt-2c′) have a larger preference towards the endo form than the rest of the species, which can be quantified at ≥5 kcal mol−1. It is also worth mentioning that both trends are additive. For example, opt-2c and opt-2c′ are the only cases where the endo form is clearly favoured because of the joint presence of the ketimine L2 ligand frame and the nickel metal centre.
These trends identified in the calculations can be qualitatively rationalised. With regard to the role played by the ligands themselves, both steric and electronic effects are likely operational. The reduced donor capability of an aldimine (L1) nitrogen over a ketimine (L2) nitrogen supports the preference of L1 to adopt the exo form while the improved donor capacity of L2 allows endo configurations to be accessed.14 In terms of steric variations, the smaller steric hindrance in L1 imparted by the H imino-carbon substituent when occupying the position closer to the metal means exo configurations are preferential, while for L2 the bulkier methyl substituent is less likely to adopt the exo configuration with the result that the endo arrangement will prevail. With respect to the metal centre and the resultant coordination geometries displayed, a consideration of the dn configuration is useful. For the complexes displaying five-coordinate geometries only in the case of the nickel(II) species are they adequately described as square pyramidal, while the zinc(II) species along with the high spin iron(II) and cobalt(II) systems as described as distorted square pyramidal.15 This deviation of nickel (d8) can be rationalised by consideration of the energy gain that would occur when the three non-bonding d-orbitals are fully occupied in the square pyramidal case (opt-1cexo, opt-1c′exo, opt-2cexo, opt-2c′exo). For Fe (d6), Co (d7) and Zn (d10) the nature of the dn configurations has no significant impact on the resting distorted square pyramidal geometry. Moreover, it appears that the stability of a high spin d8-square pyramidal geometry relative to the high spin d8-octahedral geometry is delicately balanced with the presence of the ketimine moiety in 2c being sufficient to drive the endo formation. A similar but less dramatic effect is also apparent with the iron and cobalt derivatives. In the case of the zinc complexes, endo configurations do not form which is likely due to electron–electron repulsions between the fully occupied d-shell and the incoming imine-nitrogen donor.
2.4 Screening of complexes as catalysts for ethylene oligomerisation/polymerisation
Late transition metal complexes of the first row (e.g., Fe, Co, Ni) have, in recent years, been widely employed as catalysts for alkene polymerisation and/or oligomerisation.16 In particular, complexes containing multidentate nitrogen donor ligands incorporating bulky aryliminepyridine moieties have been central to many of the key developments, one such example being the [{(2,6-isopropylphenylimino)bipyridine)}FeCl2]/MAO system reported by Gibson et al. that has been shown to be highly active for alkene oligomerisation.7 With the intent of probing the effect of an additional pyridine group within the ligand frame in the latter system, we have screened the two iron complexes (1a and 2a), along with the cobalt (1b, 2b) and nickel (1d, 2d) species, as precatalysts for the oligomerisation/polymerisation of ethylene.On activation with 400 equivalents of methylaluminoxane (MAO) only the iron complexes 1a and 2a showed any activity for ethylene oligomerisation; the cobalt and nickel systems were inactive. The results for 1a and 2a employing one atmosphere of ethylene are summarised in Table 7.
| Entry | Pre-catalyst | Massb/g | Activity/g mmol−1 h−1 bar−1 | Internal olefinc (%) | External olefinc (%) | Av. chain length,c Cn | α d |
|---|---|---|---|---|---|---|---|
| a General conditions: 1 bar ethylene Schlenk test carried out in toluene (40 cm3) at ambient temperature using 4.0 mmol MAO (Al : M = 400 : 1), 0.01 mmol precatalyst, over one hour. Reactions were terminated by addition of dilute HCl. b Mass of oligomer isolated. c Oligomerisation product percentages and average chain length, Cn, calculated via integration of 1H NMR spectra. d Determined by GC; α = (rate of propagation)/((rate of propagation) + (rate of chain transfer)) = (moles of Cn+2)/(moles of Cn). | |||||||
| 1 | 1a | 0.040 | 4 | 1 | 99 | 16.4 | 0.84 |
| 2 | 2a | 0.100 | 10 | 2 | 98 | 16.2 | 0.72 |
Several points emerge from inspection of the data. Both complexes show only low activity for ethylene oligomerisation with the ketimine species (2a) showing slightly higher activity when compared with the aldimine species (1a). The oligomeric products are typical of iron-based catalysts giving greater than 98% linear α-olefins (C6–C26). Schulz–Flory distribution of oligomers are observed in both cases with values ca. 0.76.17
The explanation for the low activity of these iron systems (1a/MAO or 2a/MAO) when compared to the highly active [{(2,6-diisopropylphenylimino)bipyridine)}FeCl2]/MAO7 is uncertain but could be attributable to two reasons or a combination of both. Firstly, the imino arm 1a/2a could be coordinated in the putative cationic active catalyst and thus block the approach of an incoming ethylene monomer. Indeed, this explanation has been suggested as one possible reason for the inactivity of [{bis(2,6-diisopropylphenylimino)bipyridine}FeCl2]/MAO towards ethylene.7 Alternatively if the imino group does not coordinate, the low activity could be due to the ligand support acting more like a substituted terpyridine ligand in a manner similar to that seen in the sterically encumbered [(6,6″-diarylterpyridine)FeCl2]/MAO systems in which only very low activities are reported; the steric bulk of the 6-position in this case inhibiting monomer approach.18 Nevertheless, it is likely that the nature of the imino carbon substituent plays a key role in dictating which pathway results in a manner akin to that seen for the precatalytic species 1 and 2.
3 Experimental
3.1 General
All reactions, unless otherwise stated, were carried out under an atmosphere of dry, oxygen-free nitrogen, using standard Schlenk techniques or in a nitrogen purged glove box. Solvents were distilled under nitrogen from appropriate drying agents and degassed prior to use.19 The infrared spectra were recorded on a Perkin-Elmer Spectrum One FT-IR spectrometer on solid samples. The ES (electrospray) and the FAB mass spectra were recorded using a micromass Quattra LC mass spectrometer and a Kratos Concept spectrometer with methanol or NBA as the matrix, respectively. 1H and 13C NMR spectra were recorded on a Bruker ARX spectrometer (250 or 300 MHz) at ambient temperature; chemical shifts (ppm) are referred to the residual protic solvent peaks and coupling constants measured in Hertz (Hz). Oligomer products were analysed by GC, using a Perkin-Elmer Autosystem XL chromatograph equipped with a flame ionisation detector and 30 m PE-5 column (0.25 mm thickness), injector temperature 45 °C and the following temperature programme: 45 °C/7 min, 45–195 °C/10 °C min−1, 195 °C/5 min, 195–225 °C/10 °C min−1, 225 °C/5 min, 225–250 °C/10 °C min−1, 250 °C/22 min. Magnetic susceptibility studies were performed using an Evans balance (Johnson Matthey) at room temperature. The magnetic moment was calculated following standard methods20 and corrections for underlying diamagnetism were applied to the data.21 Elemental analyses were performed at the Science Technical Support Unit, London Metropolitan University.The reagents, 2,6-diisopropylaniline, MAO (10 wt% in toluene), metal dichlorides and (DME)NiBr2 (DME = 1,2-dimethoxyethane) were purchased from Aldrich Chemical Co. and used without further purification. The compounds, 6-{(2,6-i-Pr2C6H3)NCH}2−2,2′:6′,2″-C15H10N3 (L1)9 and 6-bromo-2,2′:6′,2″-terpyridine22 were prepared according to previously reported procedures. All other chemicals were obtained commercially and used without further purification.
3.2 Synthesis of 6-{(2,6-i-Pr2C6H3)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CMe}-2,2′:6′,2″-C15H10N3 (L2)
CMe}-2,2′:6′,2″-C15H10N3 (L2)
Prepared via a two-step procedure:
O). 13C {1H} NMR (62.5 MHz, CDCl3): δ 24.8 (CH3CO), 120.0 (Py), 120.1 (Py), 120.4 (Py), 120.5 (Py), 122.8 (Py), 122.9 (Py), 135.9 (Py), 136.7 (Py), 137.0 (Py), 148.2 (Py), 151.9 (Py), 153.5 (Py), 154.4 (Py), 154.5 (Py), 155.9 (Py), 199.4 (CO). IR (cm−1) 1700 (C = O), 1564, 1426, 1078, 790, 748, 668. ESIMS: m/z 276 [M + H]+. HRMS (FAB): Calcd for C17H14N3O [M + H]+ 276.113 69, found 276.113 64.
N), 2.81 (sept, 2H, CH(Me)2), 7.0–7.2 (m, 3H, Ar–H), 7.2–7.3 (m, 1H, Py-H), 7.7–7.9 (m, 1H, Py-H), 7.91 (dd, 3JH−H 7.9, 3JH−H 7.9, 2H, Py-H), 8.35 (d, 3JH−H 7.9, 1H, Py-H), 8.42 (d, 3JH−H 7.9, 1H, Py-H), 8.53 (d, 3JH−H 7.9, 1H, Py-H), 8.59 (d, 3JH−H 7.9, 1H, Py-H), 8.67 (m, 2H, Py-H). 13C {1H} NMR (75 MHz, CDCl3): δ 16.3 (CH3CN), 21.9 (CH3), 22.2 (CH3), 27.3 (CH), 120.1 (Py), 120.2 (Py), 121.0 (Py), 122.5 (Ar), 122.8 (Ar), 134.8 (Py), 136.3 (Py), 136.8 (Py), 142.0 (Ar), 148.1 (Py), 154.4 (Py), 154.5 (Py), 155.1 (Py), 155.9 (Py), 166.1 (CN). IR (cm−1) 2952, 1630 (CN), 1562, 1422, 1360, 1263, 1075, 990, 777, 744. ESIMS: m/z 435 [M + H]+. HRMS (FAB): Calcd for C29H31N4 [M + H]+ 435.254 90, found 435.254 87.
3.3 Synthesis of [{6-iminoformyl-2,2′:6′,2″-terpyridine-(2,6-diisopropylanil)}MX2] (1)
CH}-2,2′:6′,2″-C15H9N3}FeCl2] (1a) as a purple-blue powder. Yield: 68% (0.088 g, 0.162 mmol). Recrystallisation from a hot acetonitrile solution gave 1a as purple plates. Anal. Calcd for C28H28N4FeCl2: C, 61.43; H, 5.12; N, 10.24. Found C, 60.69; H, 5.06; N, 10.30%.
CH}-2,2′:6′,2″-C15H9N3}CoCl2] (1b) as a blue-green powder. Yield: 70% (0.091 g, 0.166 mmol). Recrystallisation from a hot acetonitrile solution gave 1b as green blocks. Anal. Calcd for C28H28N4CoCl2: C, 61.09; H, 5.09; N, 10.18. Found C, 60.76; H, 5.15; N, 10.12%.
CH}-2,2′:6′,2″-C15H9N3}NiBr2] (1c) as an orange solid. Yield: 80% (0.122 g, 0.190 mmol). Anal. Calcd for C28H28N4NiBr2: C, 52.62; H, 4.39; N, 8.77. Found C, 52.89; H, 4.71; N, 8.64%.
CH}-2,2′:6′,2″-C15H9N3}ZnCl2] (1d) as a pale yellow solid. Yield: 65% (0.086 g, 0.155 mmol). Recrystallisation from an acetonitrile–chloroform solution gave 1d as pale yellow blocks. Anal. Calcd for C28H28N4ZnCl2: C, 60.39; H, 5.03; N, 10.06. Found C, 60.51; H, 4.99; N, 10.21%.
3.4 Synthesis of [{6-iminoacetyl-2,2′:6′,2″-terpyridine-(2,6- diisopropylanil)}MX2] (2)
CMe}-2,2′:6′,2″-C15H9N3}FeCl2] (2a) as a dark-purple solid. Yield: 72% (0.096 g, 0.171 mmol). 1H NMR (CDCl3, 293 K): δ 103.4 (<1.6 ms, Hα).
CMe}-2,2′:6′,2″-C15H9N3}CoCl2] (2b) as a blue solid. Yield: 70% (0.094 g, 0.166 mmol). 1H NMR (CDCl3, 293 K): δ 95.2 (br, Hα).
CMe}-2,2′:6′,2″-C15H9N3}NiBr2] (2c) as an orange solid. Yield: 75% (0.117 g, 0.179 mmol). Recrystallisation from a chloroform solution at 0 °C gave 2c as orange plates. 1H NMR (CDCl3, 293 K): δ 130.9 (<0.1 ms, Hα), 92.9 (2.07 ms, HβHβ′), 82.5 (2.25 ms, HβHβ), 72.3 (2.24 ms, HβHβ′), 66.8 (2.54 ms, HβHβ′), 59.5 (2.59 ms, HβHβ′), 35.0 (3.66 ms, Hβ′β′), 18.0 (7.37 ms, Hγ), 16.9 (6.30 ms, Hγ), 13.3 (8.20 ms, Hγ), 12.0–4.1 (Ar–H, CMeN, Ar–CHMe2). Anal. Calcd for C29H30N4NiBr2: C, 46.69; H, 4.02; N, 7.26. Found C, 47.09; H, 3.97; N, 7.11%.
CMe}-2,2′:6′,2″-C15H9N3}ZnCl2] (2d) as a yellow solid. Yield: 65% (0.088 g, 0.155 mmol). Prolonged standing at room temperature of an acetonitrile–chloroform mixture containing the complex gave 2d as pale yellow blocks. Anal. Calcd for C29H30N4ZnCl2: C, 61.01; H, 5.26; N, 9.82. Found C, 61.21; H, 5.12; N, 9.97%.
3.5 Ethylene oligomerisation
An oven dried 200 ml Schlenk vessel equipped with magnetic stir bar was evacuated and backfilled with nitrogen. The vessel was charged with the precatalyst (0.01 mmol) and dissolved or suspended in toluene (40 ml). MAO (4.0 mmol, 400 eq.) was introduced and the reaction mixture left to stir for 5 minutes. The vessel was purged with ethylene and the contents magnetically stirred under 1 bar ethylene pressure at room temperature for the duration of the test. After 1 h, the test was terminated by the addition of dilute aqueous hydrogen chloride (5 ml). The organic phase was separated and dried over magnesium sulfate and filtered. GC analysis was performed by taking an aliquot of the solution. For analysis of the oligomers by 1H NMR spectroscopy, the solvent was removed on a rotary evaporator and the residue dissolved in CDCl3.3.6 Density functional calculations
Quantum mechanical calculations have been carried out using the Gaussian 03 package of programs.23 Density functional theory (DFT) was applied, in particular the functional Becke’s three-parameter hybrid exchange method combined with the LYP correlation functional (B3LYP).24 The quasi-relativistic effective core potential (ECP) LANL2DZ was used for all metal atoms.25 The basis set for both atoms in the valence double-ζ contraction was associated with this ECP. The valence double-ζ with polarisation 6-31 G(d) basis was used for N and Cl and the minimal basis STO-3G for C and H.26,273.7 Crystallography
Data for 1a, 1b, 1d, 2c and 2d were collected on a Bruker APEX 2000 CCD diffractometer. Details of data collection, refinement and crystal data are listed in Table 8. The data were corrected for Lorentz and polarisation effects and empirical absorption corrections applied. Structure solution by direct methods or Patterson (2d) and structure refinement based on full-matrix least-squares on F2 employed SHELXTL version 6.10.28 Hydrogen atoms were included in calculated positions (C–H = 0.96 Å) riding on the bonded atom with isotropic displacement parameters set to 1.5 Ueq (C) for methyl H atoms and 1.2 Ueq (C) for all other H atoms. All non-H atoms were refined with anisotropic displacement parameters. Atoms C(25) and C(26) in 1b were disordered and modelled.| Complex | 1a | 1b | 1d | 2c | 2d |
|---|---|---|---|---|---|
| a Data in common: graphite-monochromated Mo-Kα radiation, λ = 0.710 73 Å; R1 = ∑∥Fo| − |Fc∥/∑|Fo|, wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2, w−1 = [σ2(Fo)2 + (aP)2], P = [max(Fo2,0) + 2(Fc2)]/3, where a is a constant adjusted by the program; goodness of fit = [∑(Fo2 − Fc2)2/(n − p)]1/2 where n is the number of reflections and p the number of parameters. | |||||
| Formula | C28H28Cl2N4Fe·CH3CN | C28H28Cl2N4Co·CH3CN | C28H28Cl2N4Zn·CHCl3 | C29H30Br2N4Ni·3.5CHCl3 | C29H30Cl2N4Zn |
| M | 588.35 | 591.43 | 676.18 | 1070.89 | 570.84 |
| Crystal size/mm3 | 0.22 × 0.17 × 0.05 | 0.17 × 0.25 × 0.35 | 0.20 × 0.16 × 0.11 | 0.35 × 0.23 × 0.05 | 0.18 × 0.16 × 0.13 |
| T/K | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | P2(1)/c | P2(1)/c | P2(1)/c | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 10.0666(12) | 10.089(3) | 10.3842(7) | 33.824(8) | 10.2739(19) |
| b/Å | 17.260(2) | 17.124(5) | 16.8912(11) | 10.759(3) | 10.5553(19) |
| c/Å | 16.987(2) | 16.991(5) | 17.4972(12) | 23.446(6) | 12.623(2) |
| α/° | 90 | 90 | 90 | 90 | 94.301(3) |
| β/° | 96.542(2) | 96.459(5) | 96.1660(10) | 100.613(4) | 101.922(3) |
| γ/° | 90 | 90 | 90 | 90 | 96.325(3) |
| U/Å3 | 2932.3(6) | 2916.8(15) | 3051.3(4) | 8386(3) | 1324.4(4) |
| Z | 4 | 4 | 4 | 8 | 2 |
| D c/Mg m−3 | 1.333 | 1.347 | 1.472 | 1.696 | 1.431 |
| F(000) | 1224 | 1228 | 1384 | 4264 | 592 |
| μ(Mo-Kα)/mm−1 | 0.724 | 0.799 | 1.269 | 3.068 | 1.155 |
| Reflections collected | 20994 | 22391 | 23670 | 31356 | 10435 |
| Independent reflections | 5155 | 5730 | 5986 | 8219 | 5134 |
| R int | 0.0383 | 0.0285 | 0.0366 | 0.1464 | 0.0362 |
| Restraints/parameters | 0/367 | 0/407 | 0/374 | 0/454 | 0/330 |
| Final R indices (I > 2σ(I)) | R 1 = 0.0408, wR2 = 0.1039 | R 1 = 0.0336, wR2 = 0.0920 | R 1 = 0.067, wR2 = 0.0884 | R 1 = 0.1088, wR2 = 0.2653 | R 1 = 0.0440, wR2 = 0.1004 |
| All data | R 1 = 0.0497, wR2 = 0.1089 | R 1 = 0.0409, wR2 = 0.0947 | R 1 = 0.0471, wR2 = 0.0919 | R 1 = 0.2232, wR2 = 0.3250 | R 1 = 0.0567, wR2 = 0.1055 |
| Goodness of fit on F2 (all data) | 1.045 | 1.036 | 0.974 | 0.893 | 1.003 |
CCDC reference numbers 615609–615613.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b610562a
4 Conclusions
The capacity of Lx to behave as either a tri- or tetra-dentate ligand has been probed through the use of a combined theoretical and synthetic study of a series of 1 : 1 3d metal halide complexes, [(Lx)MX2] (M = Fe, Co, Ni, Zn). It is apparent that not only the imino-carbon substituent but also the nature of the metal centre influences the bonding mode adopted by Lx. For nickel, both tetra- and tri-dentate bonding modes are possible while for zinc only tridentate bonding is evident. The steric/electronic attributes of the particular imine donor (ketimine vs. aldimine) coupled with stabilisation effects imparted by specific dn configurations has been offered as an explanation for these trends. Moreover it has been shown that lability of the ketimine donor is most likely to occur for the high spin iron and cobalt complexes although no clear evidence for this has been identified in the complexes prepared. Nevertheless, the propensity for imine coordination has been suggested as an explanation for the very low catalytic activities for ethylene oligomerisation displayed by the iron systems.Acknowledgements
We thank the University of Leicester for financial assistance.References
- (a) C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005 CrossRef CAS; (b) E. C. Constable, Adv. Inorg. Chem. Radiochem., 1986, 30, 69 CAS.
- (a) E. C. Constable, Polynuclear Transition Metal Helicates, in Comprehensive Supramolecular Chemistry, ed., J. P. Sauvage and M. W. Hosseini, Elsevier, Oxford, 1996, pp. 213–252 Search PubMed; (b) J. M. Lehn, J. P. Sauvage, J. Simon, R. Ziessel, C. Piccinni-Leopardi, G. Germain, J. P. Declercq and M. Van Meerssche, Nouv. J. Chim., 1983, 7, 413 Search PubMed; (c) E. C. Constable, M. J. Hannon, P. A. Martin, P. R. Raithby and D. A. Tocher, Polyhedron, 1992, 11, 2967 CrossRef CAS; (d) K. T. Potts, M. Keshavarz-K, F. S. Tham, H. D. Abruna and C. R. Arana, Inorg. Chem., 1993, 32, 4422 CrossRef CAS.
- (a) D. B. Dell’Amico, F. Calderazzo, M. Curiardi, L. Labella and F. Marchetti, Inorg. Chem., 2004, 43, 5459 CrossRef CAS and ref. therein; (b) D. B. Dell Amico, F. Calderazzo, U. Englert, L. Labella and F. Marchetti, J. Chem. Soc., Dalton Trans., 2001, 357 RSC.
- (a) D. Xiao, Y. Hou, E. Wang, J. Lu, Y. Li, L. Xu and C. Hu, Inorg. Chem. Commun., 2004, 7, 437 CrossRef CAS; (b) W. Henke, S. Kremer and D. Reinen, Z. Anorg. Allg. Chem., 1982, 491, 124 CAS; (c) C.-W. Chan, C.-M. Che and S.-M. Peng, Polyhedron, 1993, 12, 2169 CrossRef CAS; (d) E. C. Constable, S. M. Elder, J. Healy and D. A. Tocher, J. Chem. Soc., Dalton Trans., 1990, 1669 RSC; (e) E. C. Constable, S. M. Elder and D. A. Tocher, Polyhedron, 1992, 11, 1337 CrossRef CAS; (f) C.-M. Che, C.-W. Chan, S.-M. Yang, C.-X. Guo, C.-Y. Lee and S.-M. Peng, J. Chem. Soc., Dalton Trans., 1995, 2961 RSC; (g) E. N. Maslen, C. L. Raston and A. H. White, J. Chem. Soc., Dalton Trans., 1975, 323 RSC; (h) E. C. Constable, M. J. Hannon, P. Harverson, M. Neuburger, D. R. Smith, V. F. Wanner, L. A. Whall and M. Zehnder, Polyhedron, 2000, 19, 23 CrossRef CAS; (i) E. C. Constable, S. M. Elder, M. J. Hannon, A. Martin, P. R. Raithby and D. A. Tocher, J. Chem. Soc., Dalton Trans., 1996, 2423 RSC.
- J.-P. Gisselbrecht, M. Gross, J.-M. Lehn, J. P. Sauvage, R. Ziessel, C. Piccinni-Leopardi, J. M. Arrieta, G. Germain and M. van Meerssche, Nouv. J. Chim., 1984, 8, 661 Search PubMed.
- R. Ziessel, A. Harriman, J. Suffert, M.-T. Youinou, A. De Cian and J. Fischer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2509 CrossRef CAS.
- G. J. P. Britovsek, S. P. D. Baugh, O. Hoarau, V. C. Gibson, D. F. Wass, A. J. P. White and D. J. Williams, Inorg. Chim. Acta, 2003, 345, 279 CrossRef CAS.
- Y. D. M. Champouret, J.-D. Maréchal, I. Dadhiwala, J. Fawcett, D. Palmer and G. A. Solan, Dalton Trans., 2006, 2350 RSC.
- Y. D. M. Champouret, R. K. Chaggar, I. Dadhiwala, J. Fawcett and G. A. Solan, Tetrahedron, 2006, 62, 79 CrossRef CAS.
- J. Uenishi, T. Tanaka, N. Takakazu, K. Nishiwaki, S. Wakabayashi, S. Oae and H. Tsukube, J. Org. Chem., 1993, 58, 4382 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. von Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- M. J. Al-Khatib, J. Fawcett and G. A. Solan, unpublished results.
- E. Szajna, P. Dobrowolski, A. L. Fuller, A. M. Arif and L. M. Berreau, Inorg. Chem., 2004, 43, 3988 CrossRef CAS.
- For use of the ν(CO) stretch as a guide to relative donor capability of imines/pyridines see: (a) M. Brockmann, H. tom Dieck and J. Klaus, J. Organomet. Chem., 1986, 301, 209 CrossRef CAS; (b) S. Morton and J. F. Nixon, J. Organomet. Chem., 1985, 282, 123 CrossRef CAS; (c) L. Gonsalvi, J. A. Gaunt, H. Adams, A. Castro, G. J. Sunley and A. Haynes, Organometallics, 2003, 22, 1047 CrossRef CAS.
- (a) A. R. Rossi and R. Hoffmann, Inorg. Chem., 1975, 2, 365 CrossRef; (b) S. Alvarez and J. Ciera, Angew. Chem., Int. Ed., 2006, 45, 3012 CrossRef CAS.
- For reviews see: (a) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS; (b) G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428 CrossRef CAS; (c) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS; (d) S. Mecking, Angew. Chem., Int. Ed., 2001, 40, 534 CrossRef CAS.
- (a) P. J. Flory, J. Am. Chem. Soc., 1940, 62, 1561 CrossRef CAS; (b) G. V. Schulz, J. Phys. Chem. B, 1939, 43, 25 Search PubMed.
- Y. Nakayama, Y. Baba, H. Yasuda, K. Kawakita and N. Ueyama, Macromolecules, 2003, 36, 7953 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth Heinemann, London, 4th edn, 1996 Search PubMed.
- F. E. Mabbs and D. J. Machin, Magnetism and Transition Metal Complexes, Chapman and Hall, London, 1973 Search PubMed.
- (a) C. J. O’Connor, Prog. Inorg. Chem., 1982, 29, 203 CAS; (b) Handbook of Chemistry and Physics, ed. R. C. Weast, CRC Press, Boca Raton, FL, 70th edn, 1990, p. E134 Search PubMed.
- G. R. Newkome, D. C. Hager and G. E. Kiefer, J. Org. Chem., 1986, 51, 850 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, Gaussian Inc., Wallingford, CT, 2004 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- G. M. Sheldrick, SHELXTL Version 6.10, Bruker AXS Inc., Madison, Wisconsin, USA, 2000 Search PubMed.
Footnote |
| † Present address: Institute of Biochemistry and Biophysics Molecular and Cellular Bât 430, Université Paris Sud, XI 91405 Orsay Cedex, France. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |