Carbazole-based 1D and 2D hemicyanines: synthesis, two-photon absorption properties and application for two-photon photopolymerization 3D lithography
Jie
Gu
a,
Wang
Yulan
ab,
Wei-Qiang
Chen
a,
Xian-Zi
Dong
a,
Xuan-Ming
Duan
*ac and
Satoshi
Kawata
c
aLaboratory of Organic Nanophotonics, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing, 100080, P. R. China. E-mail: xmduan@mail.ipc.ac.cn; Fax: +86 01 82543597; Tel: +86 10 8254596
bDepartment of Chemistry, Beijing University of Chemical Technology, Beijing, 100029, P. R. China
cDepartment of Applied Physics, Osaka University, Yamadaoka 2-1, Suita, Osaka, 565-0871, Japan
First published on 5th October 2006
Abstract
One and two dimensional (1D and 2D) carbazole based hemicyanines, where methyl pyridinium, methyl indolium and methyl benzothiazolium were used as acceptor group, were synthesized by Knoevenagel condensation. One-photon absorption, fluorescence and two-photon fluorescence spectra were investigated. The experimental results indicated that the different ionic acceptors affect their one-photon and two-photon properties. Among them, 2D methyl pyridinium carbazole derivatives exhibited low quantum yields and large two-photon absorption cross sections more than 1600 GM. The synthesized compounds were used as photoinitiator of two-photon photopolymerization (TPP), and three-dimensional (3D) microstructure was successfully fabricated by TPP 3D lithography. They could be utilized as effective two-photon polymerization photoinitiators.
Introduction
In the past decade, two-photon absorption (TPA) has attracted increasing attention due to its various applications such as optical data storage, optical power limiting, laser up-conversion, two-photon laser scanning fluorescence microscopy and photodynamic therapy.1–5 As one of the most important applications of TPA, 3D micro-fabrication based on two-photon photopolymerization (TPP) has been attracting a great deal of interest due to its possible application in the fields of micro-electromechanical system (MEMS) and 3D photonic devices.6–8 Depending on the desired application, chromophores combining a large TPA cross section (δ) and high TPP initiating efficiency, which can favor 3D micro-fabrication processes based on TPP, are needed.On one hand, some principles for designing TPA materials had been established, while many structural designs of molecules have been estimated for this purpose. Prasad et al.9–11 have pointed out that 1D D–π–A type molecules containing fluorene or dithienothiophene as the rigid π-conjugate backbone have large δ. Marder et al.12,13 have demonstrated that symmetric stilbenes containing donor (D) or acceptor (A) groups linked by a π conjugate bridge (D–π–D or A–π–A) exhibit large δ. The structures D–π–A–π–D and A–π–D–π–A are also characterized by large δ. Ray and Leszczynski have found the δ of ionic chromophores to be larger than those of the corresponding neutral molecules.14 Some ionic chromophores were involved in two-photon absorption studies.14–19 Meanwhile, some efforts have proved that 2D and multidimensional charge transfer system tend to realize larger δ than the corresponding 1D charge transfer compounds.20–22
On the other hand, hemicyanine system was widely investigated as second-order nonlinear optical materials in related to their large dipole moment and good stability for off-diagonal tensor distribution to first hyperpolarizibitity (β) and without undesirable losses of transparency in the visible region.23 Although a number of reports recently showed that 2D intramolecular charge transfer (2D-ICT) organic salts have larger second-order nonlinearities,24,25 few efforts have been made to investigate TPA of 2D-ICT organic salts.17,18
Since carbazole is very easy to be modified at the 3- and 6-position to form a C2v symmetric molecule, here, we report C2v symmetric carbazole-based 2D-ICT salts by introducing ionic groups at the 3- and 6-position of carbazole ring, to elongate π-conjugation system compared to its 1D analogue. Within this context, we investigate one-photon and two-photon photophysical properties of 1D-ICT organic salts (1a–e) and 2D-ICT organic salts (2a–e), where the pyridinium, indolium and benzothiazolium cations were used as acceptor group, the N-alkyl-carbazolyl as donor group and a carbon–carbon double bond as π-conjugated bridge. Furthermore the V-shaped 2D-ICT salt 2c was used in TPP 3D lithography as a photoinitiator.
Experimental
Materials
Carbazole, sodium hydride, trichlorophosphine oxide, methyl iodine, ethyl bromide, pentyl bromide, 2-methyl benzothiazole, 4-methyl pyridine, methyl tosylate, phenyl hydrazine, methyl isopropyl ketone, methyl acrylate and all solvents were obtained from Beijing Chemical Reagent Company and used without further purification. Dipentaerythritol hexaacrylate (DEP-6A, trade name: Light Acrylate DEP-6A) was obtained from Kyoeisha Chemical Co., Ltd, Japan.Instruments and measurements
1H NMR spectra were recorded on a Varian Gemini-300 spectrometer using CD3OD as solvent and all shifts are referenced to TMS. The fine splitting of pyridinyl or phenyl ring patterns is ignored and the signals are reported as simple doublets, with J values referring to the two most intense peaks. Mass spectra were measured on Shimadzu LCMS2010. Elemental analyses were performed on Flash EA1112. UV-Visible spectra were obtained on a Shimadzu UV-2550 UV-Vis spectrophotometer. All steady-state fluorescence spectra were measured on a Hitachi F2500 spectrofluorometer. Two-photon induced excited fluorescence (TPEF) spectra were recorded on SD2000 spectrometer (Ocean Optical), excited by a mode-locked Ti-sapphire femtosecond laser (Tsunami, Spectra-Physics) with an oscillating wavelength, pulse width and repetition rate of 780 nm, 80 fs and 82 MHz, respectively.Two-photon initiated photopolymerization
The same mode-locked Ti-sapphire laser was used in two-photon polymerization 3D lithography. The photocurable resin (PR1) was prepared by mixing 49.9 wt% of methyl acrylate as monomer, 50.0 wt% of DEP-6A as cross-linker and 0.1 wt% of 2c as photoinitiator. The lasing source was tightly focused by a 100× oil-immersion objective lens with a high numerical aperture (N. A. = 1.4, Olympus). The focal point was focused on the liquid photopolymerisable resin which was placed on the xyz-step motorized stage controlled by a computer. After laser fabrication, the unpolymerized resins were washed out using ethanol. The obtained microstructures were characterized with scanning electron microscopy (SEM; Hitachi S-4300FEGd).Synthesis
The synthetic routes for 1a–e and 2a–e are outlined in Scheme 1.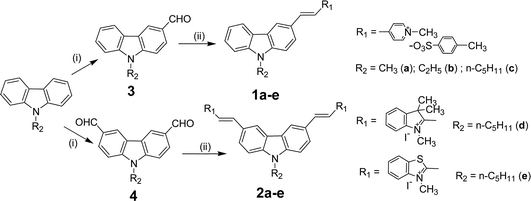 | ||
| Scheme 1 Synthetic route to 1a–e and 2a–e. Reagents and conditions: (i) POCl3/DMF, ClCH2CH2Cl, reflux; (ii) salts 5–7, piperidine (catalytic amount), ethanol, reflux. | ||
1,4-Dimethyl-pyridinium tosylate (5), 1,2,3,3-tetramethyl- indolium iodide (6), 1,2-dimethyl benzothiazolium iodide (7), N-alkyl-carbazole, N-alkyl-carbazolyl-3-aldehyde (3), N-alkyl-carbazolyl-3,6-dialdehydes (4) were synthesized according to literature methods.26–28 The compounds 1a–e and 2a–e are obtained by Knoevenagel condensation.27 The typical procedure for 1a–e and 2a–e is: salt 5 (10 mmol) and aldehyde 3 (10 mmol) were added to a 50 mL flask with 25 mL ethanol, followed by catalytic piperidine (1.0 mL). The resulting mixture was allowed to stir overnight. After concentration, the residue was recrystallized from acetonitrile–diethyl ether, further recrystallization gives the desired product. When a pentyl group was introduced at the 9-position, the solubility increased and further recrystallization is needed to afford purified products.
Results and discussion
One-photon optical properties
The normalized one-photon absorption and fluorescence spectra of 1c–e and 2c–e are shown in Fig. 1. The absorption spectra of 2c–e exhibit one weaker shoulder peak but the spectra of 1c–e do not. The lowest excited band and the second excited band of 2c–e observed are derived from the strong interaction between two branched components similar to 1c–e. The absorption maximum of 1D-ICT system is red shifted from 437 to 495 nm as the acceptor changed from pyridinium (1c) to indolium (1d). The same phenomenon is observed for the 2D system, with an absorption maximum of 458 nm for 2c, 495 nm for 2e, and 510 nm for 2d. | ||
| Fig. 1 Normalized Vis-UV spectra (a) and fluorescence spectra (b) of 1c–e and 2c–e. Excited wavelength at 500 nm for 1c/2c, 530 nm for 1d/2d and 1e/2e. | ||
As shown in Fig. 1(b), the fluorescence of indolium compounds show a red shift compared to pyridinium compounds. It is worth noting that the fluorescence spectrum of 2d is red shifted 5 nm compared to 1d for indolium derivatives, however another two couples exhibit nearly the same fluorescence spectra. With regard to the indolium cation, the acceptor is so strong that it affects the degree of intramolecular charge transfer of the 2D compound in its excited state. The energy of the 2D-ICTs is in the order of 2c > 2e > 2d, which means that indolium is the strongest acceptor and pyridinium is the weakest among these three cations.
The fluorescence quantum yields were measured at concentrations of ∼1–2 × 10−6 M according to the following expression with fluorescein in 0.1 M aqueous NaOH solution as a reference standard (Φ = 0.90) (eqn (1)).29,30
 | (1) |
All the one-photon photophysical data are listed in Table 1. The excitation and emission show no significant dependence on chain length at the substituted carbazolyl nitrogen. The quantum yields of 1D compounds are larger than those of the corresponding 2D compounds in methanol. The quantum yields can be improved using high viscosity solvents,31 those of 2c and 2d in glycerol increase to 7.7 and 7.6% from 1.3 and 0.7% in methanol, respectively.
| λ abs/nm | ε max a/M−1 cm−1 | λ em/nm | ST b/cm−1 | Φ c | λ TPAmax d/nm | Φ δ e | δ f/GM | |
|---|---|---|---|---|---|---|---|---|
| a Molar absorption coefficient. b Stokes shift ST = (1/λabs) − (1/λem). c Fluorescence quantum yield in methanol determined relative to fluorescein in 0.1 M NaOH. d The wavelength of the TPA maximum in the range 790–880 nm. e Two photon emission cross section. f TPA cross section, 1 GM = 10−50 cm4 s photon−1. g Measured in glycerol. ND not detected. | ||||||||
| 1a | 435 | 27 800 | 568 | 5383 | 0.11 | 880 | 52.6 | 470 |
| 1b | 435 | 26 100 | 568 | 5383 | 0.096 | 880 | 47.0 | 490 |
| 1c | 437 | 25 800 | 568 | 5278 | 0.090 | 880 | 41.0 | 456 |
| 1d | 495 | 42 300 | 575 | 2811 | 0.019 | — | — | — |
| 0.034g | 880 | 6.9g | 203g | |||||
| 1e | 474 | 23 700 | 569 | 3522 | 0.061 | ND | ND | ND |
| 2a | 456 | 57 550 | 568 | 4324 | 0.011 | 800 | 19.1 | 1737 |
| 2b | 456 | 55 430 | 568 | 4324 | 0.013 | 810 | 22.6 | 1740 |
| 2c | 458 | 57 500 | 569 | 4259 | 0.013 | 800 | 21.5 | 1650 |
| 0.077g | 800 | 115g | 1495g | |||||
| 2d | 510 | 40 300 | 581 | 2396 | 0.007 | — | — | — |
| 0.076g | 880 | 36g | 474g | |||||
| 2e | 495 | 87 100 | 570 | 2658 | 0.039 | 840 | 5.9 | 151 |
Two-photon absorption properties
The two-photon induced fluorescence emission spectra of 1c–d and 2c–e are presented in Fig. 2. Compare to their one photon excited fluorescence, all TPEF of these compounds are red shifted 14–20 nm which may be due to the effect of reabsorption in the higher concentration of 10−4 M. Fig. 3 presents the relationship of input power and output fluorescence intensity under the two-photon induced excitation. The fluorescence intensity is nearly linear to input power I2in with very good fixing (R = 0.9999), which provides clear evidence to confirm that the fluorescence is derived from two-photon induced excitation of these compounds. | ||
| Fig. 2 The normalized two-photon induced fluorescence emission spectra of 1c–d and 2c–e excited at 820 nm. | ||
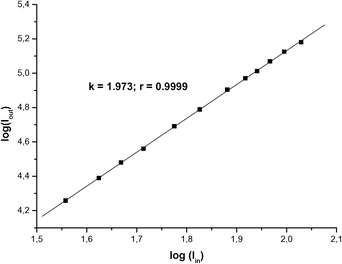 | ||
| Fig. 3 The relationship of input intensity and output intensity during the TPA cross section measurements for 1c. The solid squares are experimental spots and solid line is the fitting line. | ||
TPA cross sections were determined by the two-photon-induced excited fluorescence (TPEF) method.32 It is assumed that the quantum efficiencies after two-photon excitation are the same as those after one-photon excitation. The TPA cross sections are obtained via the TPEF method with calibration against fluorescein with a known Φδ value in aqueous NaOH solution (pH 11) at concentrations of 1.0 × 10−4 M for the measurement by using femtosecond pulse laser as the excitation resource. The samples were dissolved in solvents at concentrations of ∼1–2 × 10−4 M. To ensure that the measured signals were solely due to TPA, the dependence of TPEF on the incident intensity was verified in each case to be quadratic.
Then the TPA cross section δ were calculated on the basis of the following expression (eqn (2))
 | (2) |
The shifts of TPA cross section of 1c–d and 2c–e upon excitation by wavelengths from 790–880 nm were presented in Fig. 4. The TPA maxima below 880 nm appear at around 800 nm for 2a–c with the TPA cross sections δ = 1737, 1740, 1650 GM, respectively; 880 nm for 2d with δ = 474 GM and 840 nm for 2e with δ = 151 GM, which are contrasted to the shoulder peak as the second intramolecular charge transfer state. It is clearly shown that the second ICT state gives great contributions to TPA spectrum for 2D-ICT molecules. The TPA excited emission maximium and the TPA cross section maximium of these compounds were summarized in Table 1.
 | ||
| Fig. 4 The TPA cross sections of 1c–d and 2c–e at different wavelengths. | ||
Two-photon initiated photopolymerization
As shown above, compound 2c possesses a large TPA cross section in the range of 780–820 nm which is widely used in 3D laser microlithography. In order to investigate the two-photon photosensitivity of chromophore 2c, the threshold energy of polymerization was evaluated by analyzing the polymer spots produced by single-spot exposures. The threshold power for 2c in PR1 is 3.0 mW.The microstructure with high spatial resolution was successfully fabricated by using induced laser power of 10 mW with exposure time of 2 ms. A microstructure of diamond-lattice photonic crystal33 with a single-layer 8 × 8 lattice was successfully fabricated, the SEM image of this is illustrated in Fig. 5. Usually, 1–5wt% of photoinitiator in resins have been used for photopolymerization,34 however, only 0.1wt% of 2c was mixed in our experiments. This result clearly indicated that 2c is a photoinitiator with high TPP initiating efficiency. In our previous study on stilbazolium cations using semiempirical calculations,35 we found that stilbazolium cations are easily excited to obtain a diradical conformation in their excited state. The high TPP initiating efficiency of 2c should be due to the diradical form of the stilbazolium structure.
 | ||
| Fig. 5 SEM image of 3D diamond photonic crystal microstructure. | ||
Conclusions
In conclusion, C2v symmetric carbazole-based 2D hemicyanines compounds as well as their 1D analogues were synthesized, and their one photon absorption, steady state fluorescence, TPA and TPEF were investigated. The results showed that the 2D systems possessed larger TPA cross sections than the 1D systems although the 1D systems have higher fluorescence quantum yields. The threshold of TPP for the C2v symmetric carbazole-based 2D stilbazolium compound, 3,6-bis[2-(1-methylpyridinium) vinyl]-9-pentyl-carbazole ditosylate (2c) was determined to be 3.0 mW and it was successfully used as TPP photoinitiator for TPP microfabrication.Acknowledgements
This work was supported in part from the “One Hundred Overseas Talents Program” of the Chinese Academy of Sciences (CAS) and the “Nonlinear Nanophotonics” Project of Japan Science and Technology Agency (JST).References
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777 CrossRef CAS.
- K. D. Belfield, K. J. Schafer, Y. Liu, J. Liu, X. B. Ren and E. W. Van Stryland, J. Phys. Org. Chem., 2000, 13, 837 CrossRef CAS.
- Denk, W. Strickler, J. H. Webb and W. W., Science, 1990, 248, 73 CrossRef CAS.
- R. H. Kohler, J. Cao, W. R. Zipfel, W. W. Webb and M. R. Hansen, Science, 1997, 276, 2039 CrossRef CAS.
- M. J. Miller, S. H. Wei, I. Parker and M. D. Cahalan, Science, 2002, 296, 1869 CrossRef CAS.
- S. Kawata, H.-B. Sun, T. Tanaka and K. Takada, Nature, 2001, 412, 697 CrossRef CAS.
- B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I. Y. S. Lee, D. McCord-Maughon, J. Q. Qin, H. Rockel, M. Rumi, X. L. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398, 51 CrossRef CAS.
- T.-C. Lin, S. J. Chung, K.-S. Kim, X.-P. Wang, G.-S. He, J. Swiatkiewicz, H. E. Pudavar and P. N. Prasad, Adv. Polym. Sci., 2003, 161, 157 CAS.
- B. A. Reinhardt, L. L. Brott, S. J. Clarson, A. G. Dillard, J. C. Bhatt, R. Kannan, L. Yuan, G. S. He and P. N. Prasad, Chem. Mater., 1998, 10, 1863 CrossRef CAS.
- R. Kannan, G. S. He, L. Yuan, F. Xu, P. N. Prasad, A. G. Dombroskie, B. A. Reinhardt, J. W. Baur, R. A. Vaia and L.-S. Tan, Chem. Mater., 2001, 13, 1896 CrossRef CAS.
- O.-K. Kim, K.-S. Lee, H. Y. Woo, K.-S. Kim, G. S. He, J. Swiatkiewicz and P. N. Prasad, Chem. Mater., 2000, 12, 284 CrossRef CAS.
- M. Rumi, J. E. Ehrlich, A. A. Heikal, J. W. Perry, S. Barlow, Z. Hu, D. McCord-Maughon, T. C. Parker, H. Röckel, S. Thayumanavan, S. R. Marder, D. Beljonne and J.-L. Brédas, J. Am. Chem. Soc., 2000, 122, 9500 CrossRef CAS.
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Röckel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Science, 1998, 281, 1653 CrossRef CAS.
- P. C Ray and J. Leszczynski, J. Phys. Chem. A, 2005, 109, 6689 CrossRef.
- C. F. Zhao, G. S. He, J. D. Bhawalkar, C. K. Park and Paras N. Prasad, Chem. Mater., 1995, 7, 1979 CrossRef CAS.
- Y. F. Zhou, F. P. Meng, X. Zhao, S. Y. Feng and M. H. Jiang, Chem. Phys., 2001, 269, 441 CrossRef CAS.
- K. Kamada, K. Ohta, Y. Iwase and K. Kondo, Chem. Phy. Lett., 2003, 372, 386 Search PubMed.
- A. Abbotto, L. Beverina, R. Bozio, A. Facchetti, C. Ferrante, G. A. Pagani, D. Pedron and R. Signorini, Org. Lett., 2002, 4, 1495 CrossRef CAS.
- X. M. Wang, D. Wang, G. Y. Zhou, W. T. Yu, Y. F. Zhou, Q. Fang and M. H. Jiang, J. Mater. Chem., 2001, 11, 1600 RSC.
- L. Porres, O. Mongin, C. Katan, M. Charlot, T. Pons, J. Mertz and M. Blanchard-Desce, Org. Lett., 2004, 6, 47 CrossRef CAS.
- M. Drobizhev, A. Karotki, Y. Dzenis, A. Rebane, Z. Suo and C. W. Spangler, J. Phys. Chem. B, 2003, 107, 7540 CrossRef CAS.
- S.-J. Chung, K.-S. Kim, T.-C. Lin, G. S. He, J. Swiatkiewicz and P. N. Prasad, J. Phys. Chem. B, 1999, 103, 10741 CrossRef CAS.
- X.-M. Duan, H. Konami, S. Okada, H. Oikawa, H. Matsuda and H. Nakanishi, J. Phys. Chem., 1996, 100, 17780 CrossRef CAS.
- B. J. Coe, J. A. Harris, B. S. Brunschwig, J. Garin and J. Orduna, J. Am. Chem. Soc., 2005, 127, 3284 CrossRef CAS.
- B. J. Coe, J. A. Harris, L. A. Jones, B. S. Brunschwig, K. Song, K. Clays, J. Garín, J. Orduna, S. J. Coles and M. B. Hursthouse, J. Am. Chem. Soc., 2005, 127, 4845 CrossRef CAS.
- W. J. Kuo, G. H. Hsiue and R. J. Jeng, Macromolecules, 2001, 34, 2373 CrossRef CAS.
- B. J. Coe, J. A. Harris, I. Asselberghs, K. Clays, G. Olbrechts, A. Persoons, J. T. Hupp, R. C. Johnson, S. J. Coles, M. B. Hursthouse and K. Nakatani, Adv. Funct. Mater., 2002, 12, 110 CrossRef CAS.
- Y. Zhang, L. Wang, T. Wada and H. Sasabe, Macromolecules, 1996, 29, 1569 CrossRef CAS.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- R. A. Velapoldi1 and H. H. Tonnesen, J. Fluoresc., 2004, 14, 465 CrossRef.
- Y. Y. Huang, T. R. Cheng, F. Y. Li, C. P. Luo, C.-H. Huang, Z. G. Cai, X. R. Zeng and J. Y. Zhou, J. Phys. Chem. B, 2002, 106, 10031 CrossRef CAS.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481 Search PubMed.
- K. Kaneko, H. B. Sun, X.-M. Duan and S. Kawata, Appl. Phys. Lett., 2003, 83, 2091 CrossRef CAS.
- L. H. Nguyen, M. Straub and M. Gu, Adv. Funct. Mater., 2005, 15, 209 CrossRef CAS.
- X.-M. Duan, T. Wada, S. Okada, H. Oikawa, H. Sasabe and H. Nakanishi, MRS Symp. Proc., 2000, 581 Search PubMed , B.3.31-1-6.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |