Solvent resistant microfluidic DNA synthesizer†
Yanyi Huang‡§a, Piero Castrataro‡ab, Cheng-Chung Lee‡ac and Stephen R. Quake*a
aDepartment of Bioengineering, Stanford University and Howard Hughes Medical Institute, California 94305, USA. E-mail: quake@stanford.edu
bCRIM Laboratory, Scuola Superiore Sant'Anna, Viale R. Piaggio 34, 56025, Pontedera (PI), Italy
cDepartment of Bioengineering, California Institute of Technology, Pasadena, California 91125, USA
First published on 30th November 2006
Abstract
We fabricated a microfluidic DNA synthesizer out of perfluoropolyether (PFPE), an elastomer with excellent chemical compatibility which makes it possible to perform organic chemical reactions, and synthesized 20-mer oligonucleotides on chip.
Synthetic chemistry presents numerous instances in which it would be useful to automate and miniaturize reactions. Many synthesis problems require trial and error effort in order to optimize yield and a miniaturized automated chemical synthesizer would reduce manpower, reagent consumption, and cost. There are other cases, such as DNA synthesis, where the process has been optimized over decades of work and one desires to take advantage of these efficient reaction chemistries. Reducing reagent consumption for chemical synthesis also offers the possibility of reducing waste proportionately and is thus environmentally friendly.
Microfluidic devices have shown great potential towards realizing this goal.1 However, because chemical synthesis requires the use of a wide variety of solvents, most microfluidic work in this area has been limited to continuous flow reactors fabricated in glass or silicon, which limits the workable complexity and reagent savings.2,3 In special cases where the solvents are mild, it has been possible to use integrated elastomeric micromechanical valves to perform batch synthesis at the nanogram scale of radiopharmaceuticals4 and ‘click-chemistry’ protein ligands.5 Hua et al. have attempted DNA synthesis in a hybrid silicon–elastomer microfluidic valve system, but with limited chemical characterization of the products.6 We previously reported a photocurable perfluoropolyether (PFPE) elastomer, which is compatible with a wide variety of solvents7 and is a suitable material with which to fabricate micromechanical valves.8
Here we report a microfluidic DNA oligonucleotide synthesizer made of PFPE which performs reaction cycles adopted from the widely used phosphoramidite method.9,10 The synthetic procedure is shown in Scheme 1. The device (Fig. 1) is capable of synthesizing 60 pmol of DNA oligonucleotides while consuming less than 500 nL of 0.1 mol L−1 phosphoramidite solution in each reaction cycle. The reduction of reagent consumption is significant: a 60 fold reduction over conventional automation. This approach demonstrates the usefulness of integrated micromechanical valves for complicated multi-step organic synthetic reactions and enables automated chemical experiments with a wide variety of solvents.
 | ||
| Fig. 1 Optical micrograph of the PFPE DNA synthesizer chip. The channels have been filled with food dyes to indicate the different functional parts of the chip. | ||
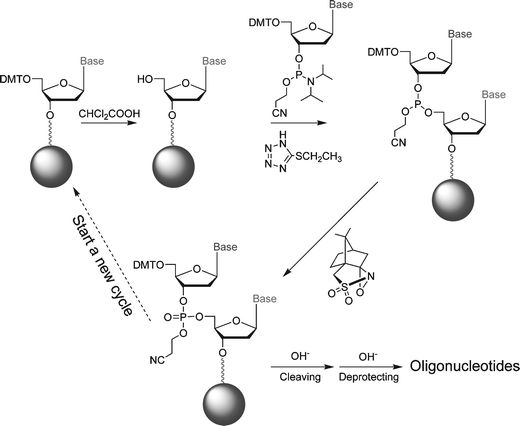 | ||
| Scheme 1 DNA synthesis procedure. | ||
PFPE was synthesized according to ref. 7. Microfluidic molds were prepared using standard photolithographic methods. The uncured PFPE was bubbled with oxygen-free nitrogen before UV exposure and then poured or spin-coated onto the molds to form layers with different thicknesses. The three layered architecture of the synthesizer is based on valve structures used in other elastomeric devices.11,12 The layers were partially cured by UV light and then bonded together with further UV exposure. All of the PFPE fabrication steps were carried out under nitrogen atmosphere inside a glove-box.
The first nucleotide (dT in our experiments) was pre-attached to the porous silica beads (pore size 200 nm, Sepax Technologies, DE, USA) with a base-cleavable succinyl linker at the 3′-end. The 5′-position was protected with a dimethoxytrityl (DMT) group. The cross-section of the column is 300 × 15 µm, and the typical length of the column is 4 mm. All the reaction reagents were delivered through Teflon tubes from vials pressurized by argon (10 psi). The column valves were actuated with 40 psi pressure to prevent the beads from escaping.
We used 3% dichloroacetic acid in dichloromethane (DCM) as the deblocking reagent. DCM is a solvent that poses severe challenges with conventional elastomers such as polydimethyl siloxane (PDMS).13 Microfluidic channels made in PDMS will swell and become clogged within seconds when exposed to DCM, but PFPE is resistant to DCM and the chip is fully functional for hours.
We substituted iodine with (1S)-(+)-(10-camphorsulfonyl)oxaziridine (CSO),14 as the oxidizing reagent because in our experiments CSO led to purer target products. The coupling step requires two reagents, phosphoramidite and 5-ethylthio-1H-tetrazole as activators. The two reagents were piped into the synthetic column in alternate fashion. The total time of each synthetic cycle is 9 min, including 2 min of deblocking, 2 min of coupling, 2 min of oxidizing and three washing steps, 1 min each.
We synthesized 20-mer DNA oligonucleotides with the sequence: 5′-CCG ACC TGG ATA CTG GCA TT-3′. After the last cycle, we used deblocking solution to deprotect the DMT group at the 5′-end. Finally the beads were washed using acetonitrile before they were flushed out of the column into a micro-vial. The beads were then lyophilized and cleaved using concentrated ammonium hydroxide for 1 h. The resulting solution was collected and kept at room temperature for 4 h to completely remove the side-protecting group of each base. The solution was lyophilized again to eliminate residual NH3 and water. We re-suspended the synthesized oligonucleotide samples into Tris-EDTA buffer (pH 7.5) to carry out HPLC/MS detection and electrophoresis, without further purification. Electrospray ionization (ESI) MS confirms the molecular weight of 6092.5 (calculated: 6093) of the final product and shows a yield of 67.7%, with most of the impurities coming from single deletions (23%) and single insertions (4%). The gel image (Fig. 2(B)) also shows that the synthesized product had the same mobility as the commercially ordered HPLC-purified sample with the same sequence.
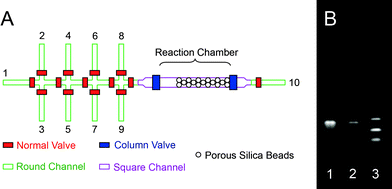 | ||
| Fig. 2 Schematic of microfluidic oligonucleotides synthesizer. (A) The fluidic channels consist of both rounded and squared profiles. The first eight channels are assigned to specific reagents: (1) acetonitrile, (2) deblocking reagent, (3) oxidizing reagent, (4) activator, (5) dT-CE phosphoramidite, (6) Pac-dA-CE phosphoramidite, (7) iPr-Pac-dG-CE phosphoramidite, and (8) Ac-dC-CE phosphoramidite. The ninth channel serves two functions. During experimental setup it is used as an inlet for silica beads. During the experiment it is used as an outlet for unwanted reagents that are left in the main channel. A solid-phase reaction column is formed in situ4 using partially closed column valves to trap the silica beads (5 µm in diameters). (B) Gel image of (1) oligonucleotides synthesized using our microfluidic device, (2) HPLC-purified oligonucleotides standard purchased from IDT, and (3) HPLC-purified (dT)10-(dT)15-(dT)20 oligonucleotide size standard purchased from IDT. The motility of the synthesized product is identical to that of commercially ordered HPLC-purified sample. However, trace byproduct can be seen in the unpurified sample. | ||
To further test that our synthesized oligonucleotide had the correct sequence, we measured the melting curve of the sample with complementary strands and oligonucleotides containing a single-base mismatch. Our synthesized DNA was labeled at the 5′-end with Cy3-phosphoramidite. The two HPLC purified strands, complementary and single-mismatched, are purchased, with carboxyfluorescein-labels (FAM) at the 3′-ends.
When the two strands are hybridized, the fluorescent intensity of FAM is significantly reduced through FRET interaction between FAM and Cy3 fluorophores. When the temperature exceeds the melting temperature, the two strands separate and the FAM signal recovers. By monitoring the fluorescent intensity of the FAM versus temperature, we measured the melting curve of the oligonucleotides (Fig. 3). The measurement was carried on a commercial microfluidic chip (Digital Isolation and Detection Chip, Fluidigm, South San Francisco, CA) which takes 12 parallel measurements at a time and each measurement is performed with 1200 replicates. Compared with the standard samples, our synthesized oligonucleotides show similar melting temperature but higher fluorescent intensity at lower temperatures, indicating that there is faint impurity in the product. This observation coincides with the results of electrophoresis and mass spectrometry.
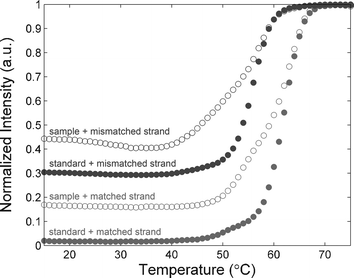 | ||
| Fig. 3 Melting curve measurements of single strand (ss)-DNA 20-mer with complementary strand and oligonucleotides with a single mismatch, respectively. | ||
In conclusion, we have demonstrated that microfluidics can be used for batch synthesis of DNA using phosphoramidite reagents and conventional solvents. We envision a number of direct applications, in spite of the fact that only picomoles of product are produced. First, DNA is special in that it can be biochemically amplified and it has been shown that one can perform whole gene synthesis with as little as femtomoles of the source oligonucleotides.15 Second, if the assay is to be performed on the chip, it is possible to elute the product DNA in nanolitre volumes, thus creating concentrations that are comparable to what are used at the benchtop. This may be useful, for example, in screening siRNA sequences,16 creating DNA nanostructures,17 and for DNA computing.18,19
This work was supported by the NIH Director's Pioneer Award and the DARPA Optofluidic Center. The authors thank S. Maerkl and J. Liu for helpful discussions.
Notes and references
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- A. de Mello and R. Wootton, Lab Chip, 2002, 2, 7N–13N RSC.
- K. Jahnisch, V. Hessel, H. Lowe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef.
- C. C. Lee, G. Sui, A. Elizarov, C. J. Shu, Y. S. Shin, A. N. Dooley, J. Huang, A. Daridon, P. Wyatt, D. Stout, H. C. Kolb, O. N. Witte, N. Satyamurthy, J. R. Heath, M. E. Phelps, S. R. Quake and H. R. Tseng, Science, 2005, 310, 1793–1796 CrossRef CAS.
- J. Wang, G. Sui, V. P. Mocharla, R. J. Lin, M. E. Phelps, H. C. Kolb and H. R. Tseng, Angew. Chem., Int. Ed., 2006, 45, 5276–5281 CrossRef CAS.
- A. Hua, Y. Xia, O. Srivannavit, J.-M. Rouillard, X. Zhou, X. Gao and E. Gulari, J. Micromech. Microeng., 2006, 16, 1433–1443 CrossRef.
- R. M. Van Dam, PhD thesis, California Institute of Technology, 2005.
- J. P. Rolland, R. M. Van Dam, D. A. Schorzman, S. R. Quake and J. M. DeSimone, J. Am. Chem. Soc., 2004, 126, 2322–2323 CrossRef CAS.
- R. L. Letsinger and W. B. Lunsford, J. Am. Chem. Soc., 1976, 98, 3655–3661 CrossRef CAS.
- M. D. Matteucci and M. H. Caruthers, J. Am. Chem. Soc., 1981, 103, 3186–3191.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS.
- I. Ugi, P. Jacob, B. Landgraf, C. Rupp, P. Lemmen and U. Verfurth, Nucleosides Nucleotides, 1988, 7, 605–608 CAS.
- J. Tian, H. Gong, N. Sheng, X. Zhou, E. Gulari, X. Gao and G. Church, Nature, 2004, 432, 1050–1054 CrossRef CAS.
- Y. Dorsett and T. Tuschl, Nat. Rev. Drug Discovery, 2004, 3, 318–329 CrossRef CAS.
- P. W. K. Rothermund, Nature, 2006, 440, 297–302 CrossRef CAS.
- N. C. Seeman, Science, 2003, 421, 427–431.
- A. Condon, Nat. Rev. Genet., 2006, 7, 565–575 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional characterization results. See DOI: 10.1039/b613923j |
| ‡ These authors contributed equally to this work. |
| § Current address: Department of Advanced Materials and Nanotechnology, College of Engineering, Peking University, China. |
| This journal is © The Royal Society of Chemistry 2007 |