Novel microfibrous composite bed reactor: high efficiency H2 production from NH3 with potential for portable fuel cell power supplies†
Yong
Lu
*,
Hong
Wang
,
Ye
Liu
,
Qingsong
Xue
,
Li
Chen
and
Mingyuan
He
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, East China Normal University, Shanghai, China. E-mail: ylu@chem.ecnu.edu.cn; Fax: (86) 21 6223 3424; Tel: (86) 21 6223 3424
First published on 21st September 2006
Abstract
A novel microfibrous composite bed reactor was developed and was demonstrated for high efficiency hydrogen production by the decomposition of ammonia at moderate temperatures in portable fuel cell power system applications. By using a high-speed and low-cost papermaking technology combined with a subsequent sintering process, sinter-locked three-dimensional microfibrous networks consisting of ∼3 vol% 8 µm (dia.) nickel microfibers were utilized to entrap ∼35 vol% 100–200 µm dia. porous Al2O3 support particulates. A CeO2 promoter and active Ni component were then dispersed onto the pore surface of the entrapped Al2O3 support particulates by a stepwise incipient wetness impregnation method. The microfibrous structure took advantage of a large void volume, entirely open structure, high heat/mass transfer, high permeability, good thermal stability, and unique form factors. Addition of ceria significantly promoted the low-temperature activity of Ni/Al2O3 catalyst particulates incorporated into the micorfibrous structure. The use of fine particles of catalyst significantly attenuated the intraparticle mass transport limitations. As a result, the present novel microfibrous composite bed reactor provided excellent activity and structure stability in ammonia decomposition, as well as low pressure drop and high efficiency reactor design. At a 90% conversion of a 145 sccm ammonia feed rate, the microfibrous entrapped Ni/CeO2–Al2O3 catalyst composite bed could provide a 4-fold reduction of catalytic bed volume and a 5-fold reduction of catalytic bed weight (or 9-fold reduction of catalyst dosage), while leading to a reduction of reaction temperature of 100 °C, compared to a packed bed with 2 mm dia. Ni/CeO2–Al2O3 catalyst pellets. This composite bed was capable of producing roughly 22 W of hydrogen power, with an ammonia conversion of 99% at 600 °C in a bed volume of 0.5 cm3 throughout a 100 h continuous test. These initial and promising results established that the microfibrous nickel-based catalyst composites were effective for high efficiency production of hydrogen by ammonia decomposition, while achieving a significant reduction of overall catalytic bed weight and volume. We anticipate our assay to be a new point for small-scale hydrogen production, where the microfibrous catalytic reactors considered in isolation can satisfy several of the most fundamental criteria needed for useful operation.
Introduction
Microreaction technology, with its unprecedented heat and mass transfer advantages as well as uniform residence times and flow patterns, is one of the few technologies with the potential to develop efficient, environmentally benign, and compact processes.1,2 Microsystems are now available in many devices for applications including: micromixers and microreactors as alternative to batch production in the pharmaceutical and fine chemical industry, lab-on-chip devices, microsensors, advanced rapid throughput chemical and catalyst screening tools (e.g. combi), and distributed heating and cooling. For example, the chemical analysis devices are commonly referred to as micro total analytical systems (μTAS) and have been reported for a diverse range applications;3,4 there remains tremendous interest in miniaturized systems for chemical syntheses and catalysis,1,5 including highly exothermic reactions, e.g. the direct nitration or fluorination of aromatic compounds, the in situ production of hazardous compounds, e.g. phosgene, on chip catalysis, e.g. the screening of new catalysts and reactions devices, as well the gaining of kinetic and thermodynamic data for reactions.However, recent interest in powering fuel cells for small, portable electronic devices has provided particular impetus for research on microreactors for high efficiency hydrogen production.1,6 One of the strategies in this effort is to develop novel microreactor technologies to meet the fundamental criteria needed for a miniature fuel cell power system,7–10 and meanwhile, to employ a simple CO-free hydrogen production process. The decomposition of ammonia offers by far the simplest process for on-site production of COx-free hydrogen,7–9 compared to the production of hydrogen using steam reforming of hydrocarbons or oxygenated hydrocarbons11 that requires a complex combination of multiple processes12 to achieve the required low CO levels.13 This process allows a single feed stream and low overall device weight and volume,7–9 thus making it particularly preferred as an attractive source of hydrogen to power fuel cells for micro/portable power applications. Additionally, ammonia as a fuel has many other important aspects including its higher energy density (1.7 times as much hydrogen as liquid hydrogen per volume), its easier storage and distribution, and its low toxicity, low flammability and its assured purity which results from the method of manufacturing.7 Unfortunately, most reactor designs for ammonia decomposition used so far are the traditional catalyst particulates packed into microreactors that normally suffer from poor intraparticle mass/heat transfer, low contacting efficiency, high pressure drop, mechanical attrition, and catalyst clumping in a way that leads to fluid bypassing.1,14 Although the microchannel technologies combined with the catalyst washcoats can be used to avoid one or more of the frustrating problems encountered in the packed beds, the surface areas per unit reactor volume have remained unacceptably low.1,7,15 Hence, it is important to render novel microstructured materials for developing a miniature hydrogen generator for micro/portable fuel cell power system supplies. A monolithic anodized aluminum microreactor, with an increase in surface area, has been developed for ammonia decomposition.7 Regardless of the use of expensive Ru for achieving good performance, the fatal disadvantage of this reactor stems from the low melting point (661 °C) of the aluminum body. Kenis et al. recently reported a more interesting solution of a microreactor composed of an inverted beaded silicon carbide monolith with interconnected micronic pores (0.75–7.5 µm) as supports and Ru as catalyst to enable ammonia decomposition up to 1000 °C.8 Furthermore, microburners or microcombustors have also been developed for use as microscale heat sources for the microreactors, making the microreaction system of ammonia decomposition advantageous for portable power applications.8,9
Herein, we demonstrate a novel microfibrous composite bed reactor for the high efficiency production of hydrogen from ammonia in portable fuel cell power system applications. This approach permits high efficiency microprocesses and advanced design of microreactors, with many beneficial properties that avoid the frustrating problems encountered in the conventional approaches, e.g. conventional packed beds. Our previous microfibrous composite reactors, with entrapment of preferential oxidation (PROX) CO catalyst particulates and with entrapment of H2S sorbent particulates for hydrogen fuel cleanup in proton exchange membrane fuel cell (PEMFC) applications do both provide 3-fold or more promotion of bed utilization efficiency, while leading to a significant reduction of overall reaction bed weight and volume compared to packed beds, with 1–2 mm dia. catalyst per sorbent pellet.16,17 In the present study, sinter-locked microfibrous carriers consisting of ∼3.0 vol% of 8 µm dia. nickel fibers were utilized to entrap ∼35 vol% of 100–200 µm dia. CeO2-promoted Ni/Al2O3 catalyst particulates. The microfibrous composite bed reactors demonstrated effective and efficient ammonia decomposition to CO-free hydrogen, with significantly promoted low-temperature reactivity and multi-fold reduction of overall reactor weight and volume.
Experimental
Preparation of microfibrous structure with Ni-based catalysts
Sintered microfibrous entrapped 100–200 µm dia. active Al2O3 support particulates (SA: 270 m2 g−1) using nickel fibers (8 µm in dia. by 2–3 mm length, manufactured by Western Metal Material Co., Ltd., Xi'an, Shanxi province, China) were prepared through wet layup papermaking, followed by sintering in a hydrogen atmosphere. In this process, 0.7 g of cellulose (20–40 µm dia. by 100–1000 µm length) and 3.0 g of metal fibers were added into the container of a commercial blender that was filled with 1.5 L water, and blended vigorously to produce a uniform suspension. The suspension and 5.0 g of Al2O3 support particulates were transferred into the headbox of a 159 mm dia. circular sheet former (ZCX-159A, made in China) filled with 8.5 L water under manual mixing. The 159 mm circular preform was then formed by draining, pressing at ∼400 kN m−1, and drying in air at ∼110 °C. Pre-oxidation of the paper in air at 500 °C removed the cellulosic binders, and subsequent sintering in hydrogen at 950 °C entrapped the Al2O3 support particulates. The void volume fraction of the resulting microfibrous carrier product was defined as the ratio of its void volume to its total volume. One 5 cm × 5 cm square piece was cut from the large sheet; subsequently, the total volume of as-cut piece could be calculated by accurately scaling its thickness while its void volume could be estimated by weighing it before and after immersion in deionized water at room temperature.Rare earth oxide (REO) additives (i.e., CeO2 and La2O3) and Ni were then highly dispersed onto the pore surface of the entrapped Al2O3 support particulates by a stepwise incipient wetness impregnation (IWI) method, using their nitrate salts as precursors. Loadings of REO additives as well as Ni active components were varied from 5–15 wt%, without including the mass of nickel microfibers. The impregnated sample in each step was calcined in air at a temperature ranged from 250–550 °C for 2 h. In the case of preparation of a microfibrous entrapped Ni/CeO2–Al2O3 catalyst composed of 10 wt% Ni, 10 wt% CeO2 and 80 wt% Al2O3, for example, a piece of 5.0 g microfibrous carrier consisting of 2.9 g entrapped 100–200 µm dia. Al2O3 support particulates was incipiently impregnated with 2.1 cm3 of an aqueous solution of Ce(NO3)3·6H2O containing 0.29 g CeO2, followed by overnight drying and 2 h calcination at 450 °C, and then re-impregnated with another 2.1 cm3 aqueous solution of Ni(NO3)2·6H2O containing 0.29 g Ni, followed by overnight drying and 2 h calcination at 250 °C. By weighing the sample before and after each step of impregnation, it was found that 0.283 g of CeO2 and 0.289 g of Ni were placed onto the entrapped Al2O3 support particulates.
Microreactor evaluation
Catalyst reactivity was measured in an 11.9 mm inner dia. (i.d.) quartz tube heated by a temperature-controlled tube furnace. For each experiment, 5 pieces of 12 mm (dia.) by 0.9 mm (thick) circular chips of microfibrous catalyst sample were cut from a large microfibrous catalyst sheet and packed carefully into the tube reactor. The slightly larger dia. of the catalyst chip than the i.d. of the reactor was used to prevent the appearance of a gap between the reactor wall and the edge of the catalyst chip thereby avoiding the gas bypassing. Prior to the reaction, all catalysts were reduced with hydrogen (at a flow rate of 30 sccm) at 500 °C for 2 h. The product nitrogen and unconverted ammonia in the effluent gas was analyzed by an online GC equipped with a TCD and a 3 m Poropak Q packed column at 100 °C in an auxiliary box, using a hydrogen carrier gas. The ammonia conversion was calculated by a normalized method on nitrogen atom basis. The reactant stream of anhydrous ammonia (99.99%) was controlled with a calibrated mass flow controller. All experiments were carried out at atmospheric pressure. Control experiments throughout the temperature range of interest showed that the reactor induced negligible conversion even in the presence of an equivalent amount of nickel microfibers as in the microfibrous composites. The real reaction temperatures were measured using a thermocouple, the head of which was located at the center point of the thin microfibrous catalytic bed in touching manner or buried inside the packed bed.Characterizations
Photographs of microfibrous structure were recorded by an Olympus zoom stereomicroscope (SZ61) and by a FE-SEM instrument (Hitach S-4800). The surface area was determined using the BET method with a commercial unit (Quatronm Autosorb 3B), with nitrogen physisorption at −196 °C. Powder XRD experiments were conducted on a Bruker diffractometer (Bruker D8 Advance) with monochromatized Cu Kα radiation (λ = 0.15406 nm) at a setting of 40 kV and 40 mA in 2θ setup of 0.02°. Before the XRD analyses, the catalyst samples were reduced with hydrogen at 500 °C for 2 h. The Ni particle size (d) of each catalyst sample was estimated by the Scherrer formula on the basis of the Ni(111) diffraction line, and therefore the estimate of the Ni dispersion (D) could be approximated by assuming d (nm) = 1/D.18Pressure drop measurements
Pressure drop through the microfibrous composite bed was measured using a sample cell consisting of a 19 mm (i.d.) tube fitting with 14 mm (i.d.) transparent plastic tubing inserted into the ends of the fitting. The cell was equipped with spacers inside the fitting to hold the sample of circular microfibrous material properly. Two O-ring rubber gaskets were placed at the front and back of the sample for avoiding gas bypass. The cell was connected to the outlet of mass flow controller. The pressure drop was measured by a Magnehelica 2000–250 PA differential pressure gauge (Dwyer, USA), at various values of the inlet flow rate.Results and discussion
Microstructure and feature of microfibrous matrix
A microfibrous matrix with 100–200 µm dia. Al2O3 support particulates using 8 µm dia. nickel microfibers was prepared through regular wet layup papermaking with a subsequent sintering process. Fig. 1 shows the optical photographs for the typical microstructure of the microfibrous matrix with micronic particulates using nickel microfibers. Fig. 1a shows the pressed and dried paper preform before sintering. A three-dimensional open porous structure of nickel microfibers was trussed up with cellulose fibers as binders while fixing the micronic Al2O3 support particulates into the void space. Fig. 1b shows the sintered products of microfibrous structure with Al2O3 support particulates. Clearly, the cellulosic binders observed in Fig. 1a were completely removed by pre-oxidation prior to sintering since no carbon fibers with diameter similar to the cellulose fibers could be observed in Fig. 1b. The junctures of metal fibers were well sintered together to cause a locked three-dimensional network, thereby uniformly entrapping micronic particulates present in the paper preform (see Fig. 1b and Fig. 2a). | ||
| Fig. 1 Optical photographs of microfibrous structure prepared using 8 µm Ni fibers with 100–200 µm Al2O3 particulates. (a) preform paper, (b) after pre-oxidizing in air at 500 °C for cellulose removal and subsequently sintering in H2 at 950 °C for 45 min. | ||
 | ||
| Fig. 2 SEM images of morphology of (a) neat microfibrous entrapped Al2O3 particulates (same sample as in Fig. 1b), (b) after placing CeO2 additives and Ni species onto the sample a (same sample as that packed into the bed C). | ||
The feature of the microfibrous matrix with Al2O3 support particulates is described in Table 1. The sintered microfibrous network consisting of ∼3 vol% of 8 µm dia. nickel microfibers offered a very large capacity to load ∼35 vol% (∼58 wt%) of the micronic particulates. This new approach increased the particulates loading by a factor of 3 or more on a gravimetric basis and by a factor of 1000 or more on a volumetric basis, compared to traditional microchannel and honeycomb monoliths with the washcoats no more than a few microns thick.1,15 The microfibrous matrix provided a large void volume (∼62 vol%), and provided a much higher surface-to-volume ratio compared to the traditional microchannel and honeycomb monoliths (generally less than 10 m2 cm−3 (ref. 7)). The microfibrous matrix here gave a surface area of 69 m2 g−1, equivalent to a surface-to-volume ratio of ∼48 m2 cm−3 (surface area multiplied by apparent density of the microfibrous matrix).
| Composition | |
|---|---|
| Nickel fiber (vol%/wt%) | 3.3/42 |
| Al2O3 particulates (vol%/wt%) | 35/58 |
| Void volume fraction (vol%) | 61.7 |
| Density/g cm−3 | 0.7 |
| Surface-to-volume ratio/m2 cm−3 | 45 |
Reactivity of microfibrous composite bed with entrapment of REO-promoted Ni/Al2O3 for NH3 decomposition
Table 2 summarizes the characteristics of each of the seven catalytic reaction beds studied. Composite bed A represented the base case: microfibrous matrix just impregnated with nickel nitrate. Composite beds B to D were modified ones with CeO2 and La2O3. Beds E to G were the traditional packed beds, being used for comparison with bed C to distinguish the benefit of the microfibrous composite structure.| Reaction bed | A | B | C | D | E | F | G |
|---|---|---|---|---|---|---|---|
| Bed type | compositeb | compositec | compositec | composited | packede | packedf | packedd |
| a Each reaction condition was run for 2 h, during which the experimental data were collected. b Prepared by IWI and calcined at 250 °C. c Prepared by stepwise IWI (REO additives first with calcination at 450 °C and then Ni species with calcination at 250 °C). d Prepared by co-IWI with a mixture solution containing required amount of Ce(NO3)3·H2O and Ni(NO3)·H2O, followed by calcination at 350 °C. e Packed with equivalent amount of catalysts as those entrapped into bed C, diluted with 100–200 µm quartz beads to 0.5 ml). f Sandwiched equivalent amount of catalysts as those entrapped into bed C with bottom and top pieces of microfibrous mesh. g The value in parentheses corresponds to a reaction temperature of 650 °C | |||||||
| Bed features | |||||||
| Nickel fiber (vol%) | 3.3 | 3.3 | 3.3 | 3.3 | 0 | 50 | 0 |
| Catalyst loading (vol%) | 35 | 35 | 35 | 35 | 35 | 40 | 75 |
| Voidage (vol%) | 61.7 | 61.7 | 61.7 | 61.7 | — | — | 25 |
| Catalyst particle size/mm | 0.1–0.2 | 0.1–0.2 | 0.1–0.2 | 0.1–0.2 | 0.1–0.2 | 0.1–0.2 | 2 |
| Bed density | 0.74 | 0.78 | 0.78 | 0.78 | — | — | 1.0 |
| Catalyst composition | |||||||
| Support | Al2O3 | Al2O3 | Al2O3 | Al2O3 | Al2O3 | Al2O3 | Al2O3 |
| Catalyst additive | — | La2O3 | CeO2 | CeO2 | CeO2 | CeO2 | CeO2 |
| Additive loading (wt%) | 0 | 10 | 10 | 10 | 10 | 10 | 10 |
| Ni loading (wt%) | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Features for 90% conversion of 72 sccm NH3 feed rate | |||||||
| Bed volume/cm3 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 2.5 |
| Bed weight/g | 0.37 | 0.39 | 0.39 | 0.39 | — | — | 2.5 |
| Temperature/°C | 600 | 580 | 550 | 580 | 600 | 600 | 650 |
| Performance of 145 sccm NH3 feed at 600 °C (650 °C) in a bed of 0.5 ml g | |||||||
| NH3 conversion (mol%) | 83 (99.5) | 93 (99.9) | 99 (99.999) | 95 (99.9) | 84 | 83 | 35 (67) |
| H2 production rate/sccm | 180 (216) | 202 (217) | 215 (217) | 206 (217) | 196 | 193 | −(141) |
| NH3 residue (ppmv) | — (2500) | — (500) | 5000 (5) | — (500) | — | — | — (—) |
| TOFNH3 at 600 °C/s−1 | 4.8 | 5.6 | 6.4 | — | — | — | 1.1 |
(1) Promotive effect of rear earth oxides
Among Ru-, Ir-, and Ni-based catalysts, the hydrogen production activity of the non-precious Ni catalyst for ammonia decomposition, though lower, is not significantly different, making it suitable from an economic standpoint.11 It is worth noting that recent efforts result in promotion of the activity of Ni/Al2O3,19,20 in particular, achieving significantly promoted activity through using La2O3 additives.20 Hence, novel monolithic microfibrous composite beds were developed by highly dispersing Ni with REOs as promoters onto the pore surface of Al2O3 support particulates entrapped into the microfibrous mesh by an IWI method. Fig. 2 shows the SEM images of the neat microfibrous matrix (as in Fig. 1b) and the composite catalyst packed in bed C. By comparison, almost no observable coats of NiO and/or CeO2 appeared on both the fibers or the external surface of the porous support particulates. The average Ni particle sizes of the catalyst samples, determined from the mid-height width of the Ni(111) X-ray diffraction line, were in the range 15–37 nm, depending strongly on the Ni loading but slightly on the REO additives; Ni dispersion (D)18 ranged from 2.7% to 6.7% and could be approximated by assuming d (nm) = 1/D (see Tables S1 and S2 in ESI for detailed illustration†). For example, at constant Ni loading of 10 wt%, the average Ni particle size (d) and Ni dispersion (D) were: 21 nm and 4.7% for Ni/Al2O3 (bed A), 23 nm and 4.3% for Ni/La2O3–Al2O3 (bed B), and 25 nm and 4.0% for Ni/CeO2–Al2O3 (beds C and G). Moreover, XRD peaks of CeO2 for the entrapped CeO2–Al2O3 support samples were weak and broad, even at high CeO2 loading up to 15 wt% (see supplementary Fig. S1 in ESI for detailed illustration†). The above results suggested that the Ni species and CeO2 additives were highly dispersed onto the internal surface of the porous Al2O3 support particulates.Beds A (base), B (with La2O3) and C (with CeO2) in Table 2 were designed to have a constant Ni loading, using the same microfibrous matrix as shown in Fig. 1b. For beds B and C, REO additives and Ni were placed onto the pore surface of the entrapped Al2O3 support particulates by stepwise IWI method, but by co-IWI method for bed D. Comparing bed C with bed D, it is clear that the stepwise IWI method was superior to the co-IWI method. A loading of 10 wt% was preferred for both the CeO2 additive and the Ni; and the optimal calcination temperature was 450 °C in step (1) for loading CeO2, but was 250 °C in the subsequent step (2) for loading Ni (see supplementary Tables S1 to S4 in ESI for detailed illustration†). As shown in Table 2, at 600 °C the REO additives provided ∼12–19% higher activities (or ∼17–33% higher TOFs) in beds B and C compared to bed A, while the promotive effect of CeO2 was higher than that of La2O3, as indicated by the TOFs: 6.4 s−1vs. 5.6 s−1 (see Table 2 and supplementary Fig. S2 in ESI for detailed illustration†).
(2) Intensification effectiveness of microfibrous structure
A large void volume and high surface-to-volume ratio as well as the open structure and excellent heat conductivity of the microfibrous network are central to the notion of increasing the steady-state volumetric reaction rate. Intraparticle mass transport can be increased by decreasing the size of the individual particles, while intrabed mass transport can be increased by the ability of the microfibrous network to separate the particles in the absence of particle–particle contact or the use of traditional binders.21 Not surprisingly, as shown in Table 2 and Fig. S3 (in ESI†), bed C exhibited much higher activity for ammonia decomposition while achieving ultrahigh bed utilization efficiency, compared to beds E (packed with the equivalent amount of catalysts as those entrapped into bed C, diluted with quartz beads to 0.5 ml) and F (sandwiched equivalent catalyst with bottom and top pieces of microfibrous mesh). Note that the catalyst used in the cases of beds E and F was collected by disclosing the network of the same samples as those packed in bed C. At a 145 sccm ammonia feed rate, bed C provided much higher conversion than beds E and F in a temperature range from 550–650 °C (see supplementary Fig. S3 in ESI for detailed illustration†), while leading to a 50 °C reduction of the reaction temperature for 90% ammonia conversion (see Table 2). This novel microfibrous structure not only intensified intrabed heat transfer, thereby making the reaction bed temperature isothermal, but also might act as micromixer thereby mitigating the diffusion resistance in gas phase that appeared in microchannel reactors.7In comparison with bed G packed with 2 mm of catalyst pellets, at 90% conversion of a 72 sccm ammonia feed gas rate, bed C provided a 4-fold reduction of catalytic bed volume and a 5-fold reduction of catalytic bed weight (or 9-fold reduction of catalyst usage without including the mass of nickel microfibers), while leading to a reduction of reaction temperature of up to 100 °C (see Table 2), by taking advantage of the use of both the microfibrous structure and small particulate size. In the equivalent bed volume, at 600 °C and a 145 sccm ammonia feed rate, the conversion almost tripled in bed C compared to bed G (see Table 2). In the case of bed C (0.5 cm3), roughly 216 sccm of hydrogen was produced with an ammonia conversion of 99% at 600 °C or 99.999% at 650 °C (see Table 2), corresponding to an equivalent power of ∼22 W for a fuel cell. This power is significant for a laptop computer, yet the catalyst bed was three fifth the area of a US penny (19 mm (dia.) by 1.4 mm thick) and two times thicker.
The Thiele modulus for the particles in beds C and G were estimated according to the Weisz criterion22 (ϕs = (dN/dt)(1/C0)(R2/Deff), where dN/dt is the reaction rate (mol cm−3 s−1), C0 reactant concentration (mol cm−3), R particle radius (cm) and Deff effective diffusivity (cm2 s−1)). The calculation for the particles in bed C (temperature 550 °C, conversion 90%, ammonia flow rate 145 sccm, particle radius 7.5 × 10−3 cm) provided a value of ϕs of ca. 0.64, which is slightly below the regime for mass-transfer effects (ϕs > 1). However, an estimate of ϕs for the particles in bed G (temperature 600 °C, conversion 35%, ammonia flow rate 145 sccm, particle radius 0.1 cm) was approximated to a value of ca. 134, suggesting the presence of high mass transport limitations, thereby reducing the catalytic effectiveness of the large particle catalyst.
Besides the above advance, the microfibrous composite, with the unique form factors, can be made into thin sheets (from submillimeter to several millimetres in thickness) of large area and/or pleated to control pressure drop and contacting efficiency in a beneficial manner different from other traditionally employed contacting schemes, including packed beds, fluid beds, monoliths, or wovens (see supplementary Fig. S4 in ESI for detailed illustration†).
(3) Comparison with a state-of-the-art microreactor using Ru
A promising small ammonium cracker microsystem for hydrogen generation is an aluminium-anodized alumina microchannel reactor with 3.5 wt% Ru and 0.8 wt% K (including the mass of aluminium body).7 That reactor, with a 140 µm channel and a 300 µm thick fin, was 9.2 mm wide, 12.6 mm long, and 4 mm thick (equivalent to 0.46 cm3, including 0.11 cm3 void volume contributed by the channels), while having a weight of 0.95 g (calculated by assuming that the volume of 0.35 cm3, except for the void volume of 0.11 cm3, in the microchannel reactor was aluminium with a density of 2.7 g cm−3). The fractional conversions of a 145 sccm ammonia feed to bed C and the microchannel reactor with Ru in ref. 7versus the reaction temperatures were plotted for comparison (see supplementary Fig. S5 in ESI for detailed illustration†). Our microfibrous composite bed C demonstrated ammonia conversions similar to the literature microreactor with Ru when the temperature increased to 600 °C, whereas the low-temperature activity of our microfibrous composite bed was not as good as that literature microreactor: 81% vs. 88% at 550 °C and 45% vs. 63% at 500 °C. By comparison, in equivalent bed volume our microfibrous composite bed provided an almost 1.5-fold reduction of the bed weight at 99% ammonia conversion of a 145 sccm feed gas rate at 600 °C. In contrast to the disadvantage of the aluminium-anodized alumina microchannel reactor, stemming from the low melting point of its aluminium body, the use of a nickel microfibrous structure with a higher melting temperature permitted high temperature operation, thereby being able to yield fast rates or approach complete ammonia conversion (see Fig. 5 in posterior section).Stability of activity and microfibrous structure
A long time test was carried out on the composite bed C at 600 °C and a 145 sccm ammonia feed gas rate. Fig. 3 shows the ammonia conversion versus time on stream and the SEM image of the microfibrous catalyst composites after the test for 100 h. Ammonia conversion was sustained at around 99% throughout the test of 100 h. The microfibrous catalyst composite circle chip kept its monolithic shape as perfect as the fresh sample even after this 100 h test, while the sinter-locked network microstructure remained robust (see Fig. 3). The above results suggested that the microfibrous catalyst composites had both excellent activity and structure stability. | ||
| Fig. 3 NH3 conversion of a 145 sccm feed rate vs. time on stream at 600 °C and SEM image of the microstructure of the composite sample packed into the bed C after 100 h test. | ||
Benefit of low pressure drop from high premeability
In addition, the pressure drop is also an important portable fuel cell system design consideration. Actually, very low pressure drop was produced in our microfibrous composite bed due to its high permeability. The same sample as that packed into bed C was used to measure a pressure drop through the microfibrous composite bed in a 14 mm (i.d.) tube at room temperature. The thickness of the microfibrous bed was 2.7 mm and the cross-sectional area of the microfibrous bed was 1.54 cm2. Gas flow rate was increased from 200 to 1500 sccm to vary the face velocity from 2.16 to 16.23 cm s−1. Fig. 4 shows the pressure drop versus face velocity of N2 across the microfibrous bed.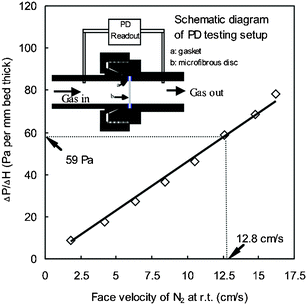 | ||
| Fig. 4 Profile of pressure drop (PD) vs. face velocity of N2 over per millimeter thickness of microfibrous composite with 100–200 µm Ni/CeO2–Al2O3 using 8 µm nickel fibers (same sample as packed into bed C). | ||
In our present case of ammonia decomposition at 600 °C, 99% conversion of 145 sccm feed rate would result in a gas flow rate of ∼850 cm3 min−1 (at 600 °C) in this hot reaction bed region. The corresponding face velocity of 12.8 cm s−1 was calculated by dividing the gas flow rate by the cross-sectional area of the tube reactor (i.d., 11.9 mm). At this face velocity, as indicated in Fig. 4, the pressure drop of 59 Pa was produced per millimeter of our microfibrous composite bed under the reaction conditions.
Promising applications
In solid oxide fuel cell (SOFC) applications, the product stream (H2 and N2) with thousand-ppm NH3 residue can be directly fueled to the anode without degradation of the performance of SOFC, while completely converting the trace ammonia to N2 and H2O with zero emission of NOx.23 Recently, a micro-SOFC stack with high power output and rapid start-up (within 1 min) has been reported to be suitable for portable power applications.24 Electrical power might thus be generated uninterruptedly to drive portable electronic devices by an integrated micro-SOFC/microfibrous ammonia cracker system fed with just ammonia. In PEMFC applications, however, the trace amount of ammonia in the product stream needs to be reduced to ppb-level to protect the membrane electrolyte assembly.25 As seen in Fig. 5, although a higher ammonia conversion could be achievable at a relatively low temperature at the expense of faster rates, the operation window seems very small to achieve a 99.9998% ammonia conversion, for reducing ammonia residue to less than 1 ppm, in reasonable reaction temperature ranges at an acceptable hydrogen production rate. For example, increasing the reaction temperature from 650 °C to 700 °C increased ammonia conversion from 99.999% (corresponding to 5 ppm ammonia residue) to 99.9998% (corresponding to 1 ppm ammonia residue) at a 145 sccm ammonia feed rate in bed C; ammonia conversion of 99.9998% could just be achieved at a feed rate as low as 2 sccm in this bed at 600 °C. Actually, the trace ammonia can be reduced to ppb levels by passing the product stream through an adsorber.6 Even so, ammonia conversion approaching to 100% is quite helpful for either longer gas life or smaller size of the adsorber.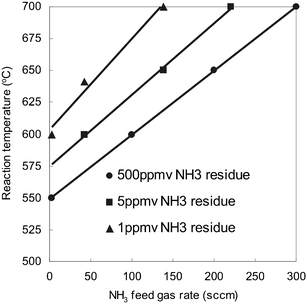 | ||
| Fig. 5 Reaction temperature vs. NH3 feed gas rate at three levels of NH3 residue in product gas stream in bed C (0.5 cm3). See footnotes in Table 2 for the description of bed C. | ||
Recently, reports of an intermediate temperature direct ammonia fuel cell using a proton conducting solid electrolyte have been appeared.26–28 Until this new approach reaches the high technology reliability level comparable to the SOFCs and PEMFCs, it is worthwhile to develop a on-site hydrogen generator using ammonia decomposition because it is uncontested that hydrogen is the ideal fuel for present fuel cells.
Of course, a reactor operating in the real world requires a compact heating source as the part of a microreaction system. Considering the unique form factors of our micorfibrous composite, we proposed a small plate-type reactor with integrated microcombustor design (see supplementary Fig. S6 in ESI for detailed illustration†). The work along this line is in progress in our group.
Conclusions
We demonstrated a novel microsystem process that utilizes a sintered-locked microfibrous carrier consisting of ∼3.0 vol% of 8 µm dia. nickel microfibers to entrap ∼35 vol% of 100–200 µm dia. CeO2-promoted Ni/Al2O3 catalyst particulates for high efficiency CO-free hydrogen production by ammonia decomposition. The resulting microfibrous catalyst composites possess unique physical properties in terms of void volume, pore structure, surface-to-volume ratio, permeability, thermal conductivity, particle size, and form factor. The use of a large void volume metallic microfibrous structure might facilitate interlayer heat and mass transfer, while the entrapment of very small catalyst particulates might significantly reduce the intraparticle mass transfer. By taking the above beneficial properties, together with the promotive effect of CeO2, the microfibrous composite beds provided a 4-fold reduction of catalytic bed volume and a 5-fold reduction of catalytic bed weight (or 9-fold reduction of catalyst dosage), while reducing the reaction temperature by up to 100 °C compared to the packed bed of 2 mm dia. Ni/CeO2–Al2O3 catalyst pellets at a 90% conversion of a 72 sccm ammonia feed rate. This approach permitted the production of ∼22 W of hydrogen power by ammonia decomposition, with a conversion of 99% at 600 °C—all in a volume of 0.5 cm3 throughout a 100 h continuous test. The microfibrous structure remained robust even after this 100 h test. The thermal stability of the microfibrous structure also allowed high temperature operation for yielding fast hydrogen production rate or achieving ultra-low ammonia residue in the hydrogen stream. In addition, the microfibrous composite provided high permeability, thereby leading to a low pressure drop across the microfibrous composite bed. Whereas the microfibrous composite bed reactor considered in isolation can satisfy several of the most fundamental criteria needed for useful operation, the commercialization of a small hydrogen generator for miniature fuel cell power system remains challenging, as a reactor operating in the real world requires a compact heating source and must meet other performance and reliability criteria.Acknowledgements
This work is supported by grants to Y. Lu from National Natural Science Foundation of China (No. 20590366, No. 20570360), and the Science & Technology Commission of Shanghai Municipality (No. 05QMX1418, No. 05DJ14002).References
- J. D. Holladay, Y. Wang and E. Jones, Chem. Rev., 2004, 104, 4767 CrossRef CAS.
- A. J. Tüdos, G. A. J. Besselink and R. B. M. Schasfoort, Lab Chip, 2001, 1, 83 RSC.
- A. G. Hadd, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1999, 71, 5206 CrossRef CAS; Y. H. Chen, W. C. Wang, K. C. Young, T. T. Chang and S. H. Chen, Clin. Chem., 1999, 45, 1938 CAS; G. M. Greenway, S. J. Haswell and P. H. Petsul, Anal. Chim. Acta, 1999, 382, 1 CrossRef CAS.
- A. Valero, F. Merino, F. Wolbers, R. Luttge, I. Vermes, H. Andersson and A. Berg, Lab Chip, 2005, 5, 49 RSC.
- N. G. Wilson and T. McCreedy, Chem. Commun., 2000, 733 RSC.
- R. Neale, Electron. Eng., 2000, 72(883), 9 Search PubMed.
- J. C. Ganley, E. G. Seebauer and R. I. Masel, J. Power Sources, 2004, 137, 53 CAS.
- Christian, M. Mitchell, D.-P. Kimb and P. J. A. Kenis, J. Catal., 2006, 241, 235 CrossRef.
- L. R. Arana, S. B. Schaevitz, A. J. Franz, M. A. Schmidt and K. F. Jensen, J. Microelectromech. Syst., 2003, 12, 600 CrossRef CAS.
- A. S. Chellappa, C. M. Fischer and W. J. Thomson, Appl. Catal., A, 2002, 227, 231 CrossRef CAS.
- V. Hacker and K. Kordesch, in Handbook of Fuel Cells – Fundamentals, Technology and Applications, ed. W. Vielstich, A. Lamm and H. Gasteiger, John Wiley & Sons, Ltd, Chichester, 2003, vol. 3, pp.121–127 Search PubMed.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964 CrossRef CAS; G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075 CrossRef CAS; M. Krumpelt, T. R. Krause, J. D. Carter, J. P. Kopasz and S. Ahmed, Catal. Today, 2002, 77, 3 CrossRef CAS; C. S. Song, Catal. Today, 2002, 77, 17 CrossRef CAS.
- T. McCreedy, Trends Anal. Chem., 2000, 19, 396 CrossRef CAS; J. Kobayashi, Y. Mori, K. Okamoto, R. Akiyama, M. Ueno, T. Kitamori and S. A. Kobayashi, Science, 2004, 304, 1305 CrossRef CAS.
- J. C. Ganley, E. G. Seebauer and R. I. Masel, AIChE J., 2004, 50(4), 829 CrossRef CAS.
- K. Watanabe, M. Sakairi, H. Takahashi, K. Takahiro, S. Nagata and S. Hirai, J. Electrochem. Soc., 2001, 148, B473 CrossRef CAS; X. Wu, D. Weng, L. Xu and H. Li, Surf. Coat. Technol., 2001, 145, 226 CrossRef CAS.
- B. K. Chang, Y. Lu and B. J. Tatarchuk, Chem. Eng. J., 2006, 115, 195 CrossRef CAS.
- Y. Lu, N. Sathitsukasnoh, A. Queen and B. J. Tatarchuk, in Microreactor Technology and Process Intensification, ed. Yong Wang and Jamelyn D. Holladay, American Chemical Society Publications Division, Washington, DC, USA, distributed by Oxford University Press, 2005, ch. 25, pp 406–423 Search PubMed.
- M. Boudart and H. Hwang, J. Catal., 1975, 39, 44 CrossRef CAS.
- S. F. Yin, B. Q. Xu, X. P. Zhou and C. T. Au, Appl. Catal., A, 2004, 277, 1 CrossRef CAS.
- J. Zhang, H. Y. Xu, X. L. Jin, Q. J. Ge and W. Z. Li, Appl. Catal., A, 2005, 290, 87 CrossRef CAS.
- D. K. Harris, D. R. Cahela and B. J. Tatarchuk, Composites, Part A, 2001, 32, 1117 Search PubMed; D. R. Cahela and B. J. Tatarchuk, Catal. Today, 2001, 69, 33 CrossRef CAS.
- P. B. Weisz, Chem. Eng. Prog. Symp. Ser., 1959, 55, 29 Search PubMed.
- J. Staniforth and R. M. Ormerod, Green Chem., 2003, 5, 606 RSC.
- Z. P. Shao, S. M. Haile, J. Ahn, P. D. Ronney, Z. L. Zhan and S. A. Barnett, Nature, 2005, 435, 795 CrossRef CAS.
- F. A. Uribe, T. Zawodzinski, Jr. and S. Gottesfeld, J. Electrochem. Soc., 2002, 149, A293 CrossRef CAS.
- N. Maffei, L. Pelletier, J. P. Charland and A. McFarlan, J. Power Sources, 2006, 140, 264.
- A. McFarlan, L. Pelletier and N. Maffei, J. Electochem. Soc., 2004, 151, A930 Search PubMed.
- A. Wojcik, H. Middleton, I. Damopoulos and J. Van Herler, J. Power Sources, 2003, 118, 342 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 to S4, Fig. S1 to S6. See DOI: 10.1039/b608555e |
| This journal is © The Royal Society of Chemistry 2007 |