Chemoresponsive viscosity switching of a metallo-supramolecular polymer network near the percolation threshold†
David M.
Loveless
,
Sung Lan
Jeon
and
Stephen L.
Craig
*
Department of Chemistry and Center for Biologically Inspired Materials and Material Systems, Duke University, Durham, NC 27708-0346, USA. E-mail: stephen.craig@duke.edu
First published on 3rd November 2006
Abstract
The precise manipulation of network percolation, combined with the previously reported effects of the kinetics of cross-linking interactions, provide a mechanism by which to optimize the stimulus-responsive mechanical properties of supramolecular gels. Specific metal–ligand coordinative bonds create cross-links between poly(4-vinylpyridine) in DMSO, and an abrupt change in mechanical properties is observed at a critical concentration of cross-linker. The change in mechanical properties is attributed to the onset of percolation within the network, and bulk mechanical properties are shown to be especially sensitive to external stimuli in the vicinity of the percolation threshold. The reversible control of bulk mechanics is demonstrated, and the magnitude of the response (changes of up to five orders of magnitude in modulus) is determined by the concentration and dissociation kinetics of the cross-linkers. Combinations of cross-linkers, individually present at concentrations below the percolation threshold, provide a related mechanism by which complex viscoelastic switching can be programmed at the small-molecule level.
Introduction
The reversible control of material properties represents an important and ongoing challenge in polymer and material science.1–3 For example, stimulus-responsive polymers undergo orders-of-magnitude changes in properties in response to light,4–6 ultrasound,7–10 electrochemistry, temperature11,12 and chemical signals. Paulusse and Sijbesma recently reviewed the importance and promise of molecular-based rheological switches, which might have applications in clutches, brakes, and vibration damping, among other fields.7 The use of chemically specific interactions as the critical entanglements in polymeric materials (“supramolecular” polymers) provides not only an avenue for the rational and predictive control of viscoelastic response in bulk materials, but also a productive mechanism by which to engineer stimulus-responsive behavior. One strength of this approach is how neatly it maps onto existing theoretical frameworks, for example transient network theories. In particular, classic work by Green and Tobolsky13 and Lodge14 and subsequent advances by others,15,16 have related the number and lifetime of entanglements to polymer mechanical properties. Often these entanglements are, at least in part, phenomenological, but in supramolecular polymers they are clearly defined by very specific and reversible chemical interactions. Environmental factors that impact the strength or kinetic response of those interactions, therefore, have a direct and predictable impact on polymer mechanics.The impact of the addition or loss of entanglements is often greatest in the vicinity of transitions, for example from sol to gel. Chambon and Winter,17,18 Rubinstein et al.,19 and Lang and Burchard,20 among others, have noted both the environmental sensitivity and complexity of material structure and properties around the percolation threshold, or gel point, at which a pseudo-infinite continuous network is created by the minimum number of physical or chemical connections between components. A thorough understanding of structure and properties in the area of the gel point remains an active and important area of research.21 Not surprisingly, Nature employs the percolation region productively, to create survival advantages through tissue whose mechanical properties switch rapidly and reversibly in response to chemical signals.22–24 As is the case in Nature's examples, the supramolecular strategy, as defined above, potentially allows the concentration of entanglements to be fine-tuned in such a way that this region of sensitivity can be accessed easily. Here, we explore the sensitivity of a previously reported family of metallo-supramolecular polymer networks in the vicinity of the transition from discrete polymer aggregates to a percolated network. In this work, we focus on the functional mechanical properties, particularly the viscosities, rather than the finer structural aspects of the polymer networks, and very sensitive, chemo- and thermoresponsive mechanical properties are realized.
We recently reported that supramolecular polymer networks (SPNs) formed from the coordinative cross-linking of poly(4-vinylpyridine) (PVP) by bis(PdII) or bis(PtII) pincer complexes provide exquisite, molecular control over bulk mechanical properties (Fig. 1).25 The bulk viscoelastic properties are controlled by the kinetics of the individual metal–pyridine coordination bonds, and relatively complex dynamic mechanical responses can be programmed through a “mix and match” strategy using combinations of cross-linkers.26 The control of mechanical properties extends over several orders of magnitude, reflecting the range of dissociation kinetics in the employed cross-linkers.
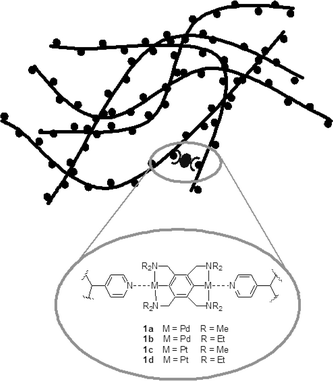 | ||
| Fig. 1 Schematic picture of PVP cross-linked with bimetallic compounds 1a–d. Triflate counter ions are omitted for clarity. | ||
It is desirable to combine the control of mechanical properties with the responsiveness afforded by the chemical specificity of the cross-linkers. In this paper, we report that combining the chemistry of the intermolecular cross-links with details of the percolation behavior provides a useful mechanism for engineering dramatic and reversible, chemically-induced changes in mechanical properties. Gel to sol transitions are induced by a range of chemical signals, including competing ligands and acids. Reversible gel to sol to gel transitions are reported using a variety of acids and bases, and the chemical acid–base signals can be coupled to thermal processes. Relaxation mechanisms below the sol-to-gel transition are independent of the kinetics of the cross-linking interactions, and, as previously reported, relaxations above the sol-to-gel transition are determined by the cross-linking kinetics. The magnitude of the change in properties is therefore determined by the cross-linking kinetics, and changes in viscosity of several orders of magnitude are demonstrated. Gel-to-gel transitions can be similarly engineered by a strategy using mixtures of cross-linkers. The onset of percolation, as defined by the concentration of cross-linkers, depends on the total weight percentage of the SPN and provides another degree of control in the systems. Although the fine details of the behavior are system-specific, the strategy to exploit sol-to-gel or other phase transitions as highly sensitive regions of stimulus-responsive behavior in supramolecular polymer systems is general.
Experimental
Materials
Dimethylsulfoxide (DMSO), poly(4-vinylpyridine) Mw = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (PVP), sodium bicarbonate, sodium triflate, triflic acid, sulfuric acid, dimethylaminopyridine (DMAP) and NaCl were used as received from Aldrich, Acros, or Fisher Scientific. The synthesis of 1a–1d can be found in the ESI.† The details of the SPN preparation have been reported previously.26 The cross-linker is reported as functional group equivalents relative to the amount of pyridine residues (2 × [1]/[pyridine]), where [pyridine] = (mass of PVP/molar weight of vinyl pyridine monomer (105.14 g mol−1))/volume. For example: 5 mg of 1d is added to 190 mg of PVP to create a 0.5% cross-linked sample. All samples were annealed at 70 °C for at least 4 h after chemical modification prior to mechanical characterization.
000 (PVP), sodium bicarbonate, sodium triflate, triflic acid, sulfuric acid, dimethylaminopyridine (DMAP) and NaCl were used as received from Aldrich, Acros, or Fisher Scientific. The synthesis of 1a–1d can be found in the ESI.† The details of the SPN preparation have been reported previously.26 The cross-linker is reported as functional group equivalents relative to the amount of pyridine residues (2 × [1]/[pyridine]), where [pyridine] = (mass of PVP/molar weight of vinyl pyridine monomer (105.14 g mol−1))/volume. For example: 5 mg of 1d is added to 190 mg of PVP to create a 0.5% cross-linked sample. All samples were annealed at 70 °C for at least 4 h after chemical modification prior to mechanical characterization.
Rheological measurements
Rheological data were obtained using a Bohlin VOR rheometer with a concentric cylinder geometry (fixed bob and rotating cup). The sample (∼2 mL) was loaded into the cup, with heating when necessary to allow the sample to flow into the geometry, and the bob was lowered into the cup. The sample was allowed to equilibrate to 23 °C. Steady shear and low deformation oscillatory data were obtained and processed with the Bohlin VOR software.Results and discussion
Percolation and the sol-to-gel transition
Transient SPNs PVP·1 (Fig. 1) were formed at 10% by total weight in DMSO by mixing and annealing appropriate ratios of PVP and cross-linkers 1a–d. The mechanical properties of the mixtures were investigated as a function of cross-linking concentration (reported as stoichiometric functional group equivalents) using a combination of steady and oscillatory shear rheology in a cup and bob geometry. The effect of cross-linking concentration on viscosity is shown in Fig. 2. Below a critical concentration of cross-linker, Ccr(ca. 0.8%, reported as functional group equivalents of metal to pyridine), the four mixtures behave as free-flowing sols and their viscosities are very similar to that of a comparable 10 weight% solution of PVP in DMSO (approaching 0.02 Pa s); there is a negligible difference in viscosity between the mixtures formed from the four different cross-linkers.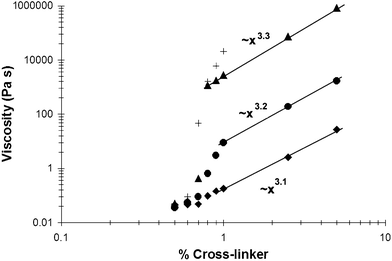 | ||
| Fig. 2 The effect of cross-linking percentage on viscosity for the different transient networks. Each network was made at a total SPN weight percentage of 10% in DMSO. The viscosity was measured by either steady or oscillatory shear rheology. (◆) PVP·1a, (●) PVP·1b, (▲) PVP·1c, and (+) PVP·1d. Scaling laws fit to the high cross-linking regime are shown for PVP·1a–c. Dynamic viscosities are reported for the lowest accessible frequency (0.001 s−1), at which viscosity is independent of decreasing frequency for all samples except PVP·1d. | ||
Above Ccr, however, the absolute and relative behaviors of the mixtures are qualitatively and quantitatively quite different. As seen in Fig. 2, the viscosity increases––dramatically in some cases—over a relatively narrow range of cross-linking content to produce materials that behave like weak gels. We attribute the change in behavior to a sol-to-gel transition brought about by percolation to form a pseudo-infinite transient network. Above Ccr, the relative viscosities of the samples are proportional to the lifetime of the coordinative metal–pyridine bonds that define the cross-links. For the three SPNs for which a complete data set could be obtained (low frequency measurements on the most viscous sample, PVP·1d, are beyond the limits of our rheometer), the cross-linking dependence of the viscosity obeys a constant power law of ∼x3.1 from completed percolation through 5% cross-linking, in agreement with previous measurements on a related set of SPNs from our laboratory.25
The transition threshold, expressed in terms of the percentage of cross-linker, depends on the total concentration of PVP·1 in DMSO (Fig. 3). The shaded regions 0.001–0.01 and 0.01–0.1 Pa s in Fig. 3 roughly correspond to sols below the percolation threshold and the 10–100 Pa s region is the region above percolation, where the zero-shear viscosity is dependent on the dissociation kinetics of the cross-link. The percolation threshold varies from 0.6% cross-linker at 15 total weight%, to ∼0.8% at 10 total weight%, to 1.0% at 7.5 total weight%. The lighter shaded regions corresponding to 0.1–1 and 1–10 Pa s are the area of greatest sensitivity as the network transitions from a non-percolated to a fully percolated state with very slight changes in the cross-linking or the weight percentage. The percolation threshold expected for random placement of cross-linkers between linear polymers is given by eqn (1):27
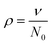 | (1) |
000). The theoretical Ccr for percolation occurs when each chain is connected to two other chains, a condition calculated from eqn (1) to occur at 0.35% cross-linker (again in functional group equivalents) for the system employed here. All of the measured values of Ccr are greater than the theoretical value of 0.35%, but the observed Ccr moves slowly toward the theoretical value as the total concentration of PVP·1 increases. At the highest concentration of network (15% by weight), Ccr is between 0.5% and 0.6%. The percolation behavior is consistent with the existence of an equilibrium between intramolecular and intermolecular cross-linking (see Discussion, below).
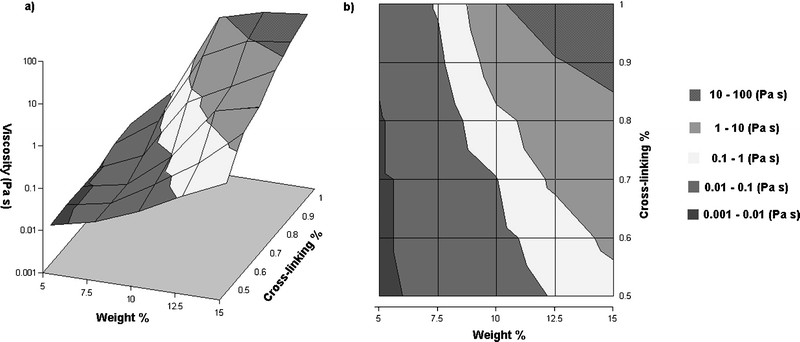 | ||
| Fig. 3 Zero-shear viscosity plotted versus the weight percentage and cross-linking percentage (reported as metal functional group equivalents) for a series of transient networks of PVP (Mw = 60000) and cross-linker 1b in DMSO at 23 °C. | ||
Response to chemical stimuli
Of particular interest in Fig. 2 and 3 is that the transition from sol-like behavior to gel-like behavior occurs over a very narrow range of cross-linking percentage. The change in cross-linker concentration across the “transition zone” from sol to gel is only 0.1%–0.2%, which corresponds to 0.7–1.8 mg of actual material added to a 2 g sample. As seen in Fig. 2, it is over this concentration range, and not in the percolated network, that the mechanical properties are most sensitive to the chemical composition of the SPN. For example, a chemical change in 0.09% of the total weight of the material effects a change in mechanical properties of nearly 5 orders of magnitude (0.05 Pa s to 1300 Pa s) for a 10% by weight PVP·1c network (and presumably over 6 orders of magnitude for PVP·1d). Once an effectively infinite network is formed, its relaxation is dominated by the relaxation timescale of the individual cross-links, rather than diffusion of smaller, discrete structures.25 The magnitude of the property change therefore also depends on the cross-linking kinetics (Fig. 2), and so the viscosity change in these transitions would be greater still if one used a cross-linking agent with a much slower dissociation rate.The sensitivity suggested by Fig. 2 is manifested in a series of chemically induced mechanical responses. For example, chloride binds much more tightly to the pincer complexes than does pyridine, and the addition of just 0.004 weight% NaCl (0.081 mg) to a 10% by weight network of PVP·1c at a cross-linking concentration of 0.8% (by functional group), theoretically enough to disable 15% of the cross-linkers, results in a 215-fold reduction in viscosity (1300 to 6 Pa s). Another 100-fold reduction is realized upon the addition of more NaCl (0.081 mg) (from 6 to 0.06 Pa s, Fig. 4a). The magnitude of the response is, as expected from Fig. 2, greatly diminished for SPNs with cross-linker densities further removed from the percolation threshold. For example, when the cross-linker content is increased to 1% 1c, the addition of 0.004 weight% NaCl has a far more modest impact on viscosity (2800 to 1500 Pa s).
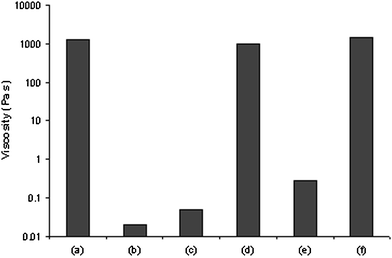 | ||
| Fig. 4 Chemoresponsive SPN (0.8% cross-linked PVP·1c at 10% total weight) and its response to certain stimuli. (a) Initial zero-shear viscosity, (b) a + NaCl (0.008 wgt%), (c) a + sulfuric acid, (d) c + NaHCO3, (e) a + triflic acid, (f) e + NaHCO3. | ||
Similar transitions to a free-flowing sol are observed upon addition of DMAP, which also outcompetes PVP for the metal coordination sites (data not shown). By chemically targeting the bismetallic cross-linkers, it is possible to ‘disable’ them and influence the nature of the network they define. When the amount of active cross-linker drops below the Ccr, the SPN changes from a pseudo-infinite structure to a solution of discrete, freely diffusing, aggregates, resulting in a dramatic dip in its moduli and viscosity.
Other chemical signals are equally effective. A range of acids, including HCl, H2SO4, and triflic acid, lead to similar reductions in viscosity upon addition to a minimally percolated network. The example of triflic acid is noteworthy, because the triflate counter-ion is a sufficiently weak ligand that it does not compete with DMSO for the metal center,25 and so the mechanical response can be attributed to the action of the protons, presumably on the pyridine side chains. Addition of triflic acid (4.5 mmol) to 0.8% network of PVP·1c at 10 total weight% leads to a 6500-fold reduction in viscosity (1300 to 0.2 Pa s). The change in viscosity is fully reversed by the addition of stoichiometric NaHCO3 to the sol and subsequent annealing (Fig. 4f). Because the viscosity returns to a value that is almost identical to that of the original sample, and because the addition of NaHCO3 to the SPN in the absence of acid has a negligible effect on viscosity, we attribute the effects of acid and base to reversible protonation and deprotonation of the pyridine residues and concomitant shifts in the metal–ligand coordination equilibrium. This acid-induced gel-to-sol transition and subsequent reversal can be repeated multiple times, although eventually the viscosity of the gel state decreases as the total weight% of the sample decreases. Upon addition, NaHCO3 reacts with the acid, restores the basicity of the pyridine residues, and allows again binding between the platinum center and the pyridine residues on the polymer chains.
The pH-sensitive phase behavior of the SPNs can be coupled to thermal triggers, for example the heat-induced decomposition of urea to ammonia recently used by Song et al. to trigger pH-responsive properties of hydrogels.28 Here, the urea decomposition reverses the effect of added acid, described above. A mixture of 0.8% 1c and PVP (10 weight% in DMSO) is normally above percolation, but the addition of 20 weight% H2SO4 and 15 weight% urea turns the viscous network (η = 1300 Pa s) into a free flowing sol. The reduced viscosity is due to the presence of acid; the effect of urea alone on viscosity is too small to measure. Heating for 16 h at 90 °C releases ammonia and neutralizes the acid, raising the viscosity back to nearly that of its initial state (η = 1100 Pa s).
In the above examples, the sensitive response is by necessity coupled to a gel-to-sol transition, but for many potential applications a less fluid lower endpoint is desired. The sharp chemoresponsiveness can be coupled to what is effectively a gel-to-gel transition by exploiting a mixture of cross-linkers. When two cross-linkers are mixed, each at concentrations above the percolation threshold, we have previously reported that the zero-shear viscosity is determined by the kinetics of the slower cross-linker; the faster cross-linkers are not flow-limiting, and so they are effectively invisible except under high-frequency oscillatory shear.26
The viscosity of the heterogeneously cross-linked SPNs differs qualitatively from that reported previously when the total cross-linker content is above the percolation threshold, but that of the slower cross-linker alone is not. In this case, all cross-links are flow-limiting, and dissociation of the faster cross-links determines the viscosity. For example, consider the viscosity of 10% by weight PVP·(1b + 1d). If 1b and 1d are each present at 1%, above the percolation threshold, the dynamic viscosity at 0.001 Hz is 2700 Pa s (the limits of our rheometer prevent the determination of an absolute zero-shear viscosity for samples containing 1d because of its extremely slow kinetics, but scaling from PVP·1c gives an expected value of 48000 Pa s), which is essentially identical to that of PVP·1d at 1% cross-linker. If, however, 1b and 1d are each present at 0.5%, individually below––but in combination above––the percolation threshold, the measured viscosity approaches that of 1% PVP·1b.
This behavior provides an intriguing mechanism for programming chemoresponsive switching. When 1.5 mg of DMAP is added to 2.01 g of PVP·(1b + 1d) (10% by weight in DMSO) with a cross-linking concentration of 1% in each, the DMAP disables portions of both cross-linkers (competition of DMAP vs. pyridine is almost identical for 1b and 1d29) of a percolated 1% network. The chemical signal drops the individual cross-linking components below the percolation threshold, and the viscosity changes from that of a percolated 1% 1d network to a viscosity approaching that of a percolated 1% 1b network (2700 Pa s to 7 Pa s) (empirically, from a fairly stiff gel to a very weak gel). In addition, the storage modulus, G′, shows the transition from an intact network controlled by the slow 1d cross-links towards a network whose dynamic mechanical properties are dominated the relaxations due to 1b (Fig. 5).
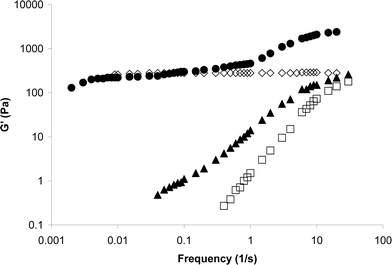 | ||
| Fig. 5 Storage modulus, G′, vs. frequency for (◇) 1% PVP·1d, (□) 1% PVP·1b, (●) 2% PVP·(1b·1d); 1% of each cross-link, and (▲) 2% PVP·(1b·1d); 1% of each cross-link, after the addition of 1.5 mg DMAP to 2.01 g of the SPN. All networks are 10% by total weight in DMSO at 23 °C. | ||
Discussion
Dramatic sensitivity in the viscoelastic properties of the SPN PVP·1 is observed only over a narrow range of cross-linker concentration. The critical range of cross-linker concentration corresponds to a transition between discrete aggregates of PVP·1 and a continuous networked structure. Below a critical concentration of cross-linker, the relaxations are dominated by the hydrodynamics of discrete aggregates that are effectively intact on the timescale of the relaxation, as evidenced by the independence of the viscosity on cross-linker dissociation kinetics. Above the critical concentration, however, the relaxations within a continuous network require dissociation of the cross-linkers, and the mechanical properties are dominated by the cross-linker kinetics: PVP·1d is more viscous than PVP·1c, which is more viscous than PVP·1b, which is more viscous than PVP·1a.The percolation threshold, expressed in terms of the number of added cross-linkers, is greater than that expected from theoretical models.27 The absence of apparent cross-links is not due to weaker-than-expected thermodynamics of the cross-linking interactions within the network, because the percolation threshold is effectively independent of thermodynamics: the Pt-based cross-linkers 1c and 1d have association constants ∼2 orders of magnitude greater than those of the Pd-based cross-linkers 1a and 1b, but the onset of percolation occurs at the same concentration. In any cross-linked network, there is a possibility of chemically intact but mechanically inactive cross-links. These inactive cross-links might include, for example, those that create what are effectively isolated cyclic sub-structures within the network. The fraction of cross-linkers that are “intermolecular” should increase with PVP concentration, and in fact the concentration of added cross-linker necessary for percolation, Ccr, does decrease as the total concentration of PVP increases.
The transition around the percolation threshold is the region in which the changes in viscoelastic properties are most sensitive. In the case of SPN PVP·1c, for example, the change in the viscosity from 0.6%–0.8% cross-linker was 19000-fold (0.062 and 1300 Pa s for 0.6% and 0.8% respectively), while the change in viscosity from 0.8%–1.0% is roughly a factor of two (1300 to 2800 Pa s). In these SPNs, the cross-links are very well defined and they control directly the mechanical properties of the networks.25 It is possible, therefore, to manipulate the cross-links in such a manner as to control the resulting mechanical properties of the SPN. For the pincer cross-linkers 1, the metal binding sites are preferentially occupied by better ligands, and in fact the addition of the stronger ligands Cl− or DMAP has an effect on material properties that is consistent with expectations based on the known coordination chemistry of the metal complexes. Similarly, added acid will protonate pyridine ligands and shift the metal–ligand coordination equilibrium towards unbound metal. This relatively simple chemical insight also has direct and dramatic consequences for material properties; the addition of small amounts of acid reduces the SPN to a free-flowing solution. Unlike the addition of chloride, however, the acid-induced changes in viscosity are easily reversed; either by the simple addition of stoichiometric NaHCO3 base, or by the in situ thermal generation of ammonia from urea.
In the examples of gel-to-sol transitions, changes in viscosity of up to five orders of magnitude are demonstrated, but in practice the sensitivity could be much greater. The lower viscosity limit is determined largely by the independent diffusion of PVP aggregates, and it therefore does not depend on the kinetics of the cross-linking interactions. On the other hand, the viscosity at the upper end of the percolation transition is directly proportional to the lifetime of those interactions. As a result, there exists very precise control over the viscoelastic behavior of the intact SPN, including relatively complex frequency responses. We note that, in these systems, the chemistry of a single unit––the pincer-pyridine complex––can be adjusted to control both the sensitivity and the properties of the “on” (high viscosity percolated) state.
Control of the low-viscosity “off” state is achieved by using a combination of cross-linkers. The flow limiting cross-linkers are determined by the minimal combination of slowest components that is necessary to exceed the percolation threshold. Sharp transitions from one percolated limit to another (or multiple others) are easily engineered. Because combinations of cross-linkers can also be used to program complex viscoelastic profiles,26 reasonably specific mechanical states can be accessed within the percolation-driven switches. Such approaches might be especially fruitful in damping applications, although a successful system would require switching mechanisms that are more rapid than those afforded by molecular diffusion.7 Of course, the SPNs are not limited to “two-state” systems, and the ability to rationally and rapidly access properties from across the entire range of behaviors in the percolation regime might often be desirable. To that end, the details of the gel point and its behavior are still an area of ongoing research interest,30 and while the current investigation has focused on viscosity switching as a functional property, well-defined SPNs such as those employed here might prove useful in understanding the nature of the sol-to-gel transition, especially in combination with recent transient network theories. Finally, one can also imagine that combinations of supramolecular interactions, with very different dependences on a range of stimuli,31,32 could be used to generate very responsive and finely tuned materials.
Conclusions
The chemical responsiveness of the viscosities of a family of supramolecular polymer networks formed from poly(4-vinylpyridine) and pincer-Pd and pincer-Pt cross-linkers is reported. As the concentration of cross-linker increases, a critical concentration, Ccr, is reached. The behavior above and below Ccr is consistent with a transition from a sol of discrete polymer aggregates to a percolated polymer network. The transition region of the viscosity around Ccr is sharp, and conversion from sol to gel occurs over a very narrow cross-linker percentage. The Ccr depends on the total weight% of network, and suggests an equilibrium between intra- and inter-molecular cross-linkers. The sharp transition region can be exploited to obtain polymer networks with sensitive chemical responsiveness. Changes in viscosity of several orders of magnitude are demonstrated, and the magnitude of the response depends directly on the lifetime of the cross-linking interaction. Reversible control of the metal–ligand cross-link, through relatively simple chemical signals, allows one to toggle between percolated and non-percolated states whose viscoelastic properties differ dramatically.The use of well-defined SPNs has several advantages as stimulus-responsive polymers. First, in contrast to covalent polymers, the interaction is reversible. Second, in contrast to other physical gels, the nature of the interaction is very specific. An understanding of interactions between molecular components allows the rational engineering of property endpoints and mechanisms (stimuli) to toggle between them. The combination of that molecular understanding with the details of the sol-to-gel transitions in SPNs offers a promising avenue by which to maximize the sensitivity of a mechanical response to external stimuli.
Acknowledgements
This research was funded by NIH (EB001037). DML was supported by NSF IGERT (DGE-0221632) and a Burroughs Wellcome Fellowship. We thank program participants T. Chance, S. Groome, S. Hoffman, and W. McCloud, for sample preparation and initial observations during a high school teacher outreach program funded by NSF (CHE-00-94224 to M. Fitzgerald). We thank S. Rowan for pointing out the example of the sea cucumber, cited in refs. 22–24.References
- B. Jeong and A. Gutowska, Trends Biotechnol., 2002, 20, 305–311 CrossRef CAS.
- B. Kippelen, S. R. Marder, E. Hendrickx, J. L. Maldonado, G. Guillemet, B. L. Volodin, D. D. Steele, Y. Enami, Sandalphon, Y. J. Yao, J. F. Wang, H. Rockel, L. Erskine and N. Peyghambarian, Science, 1998, 279, 54–57 CrossRef CAS.
- P. S. Stayton, T. Shimboji, C. Long, A. Chilkoti, G. Chen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472–474 CrossRef CAS.
- T. Suzuki, S. Shinkai and K. Sada, Adv. Mater., 2006, 18, 1043–1046 CrossRef CAS.
- Z. Hu, X. Lu and J. Gao, Adv. Mater., 2001, 13, 1708–1711 CrossRef CAS.
- H. Sakai, Y. Orihara, H. Kodashima, A. Matsumura, T. Ohkubo, K. Tsuchiya and M. Abe, J. Am. Chem. Soc., 2005, 127, 13454–13455 CrossRef CAS.
- J. M. J. Paulusse and R. P. Sijbesma, Angew. Chem., Int. Ed., 2006, 45, 2334–2337 CrossRef CAS.
- T. Naota and H. Koori, J. Am. Chem. Soc., 2005, 127, 9324–9325 CrossRef CAS.
- J. M. J. Paulusse and R. P. Sijbesma, Angew. Chem., Int. Ed., 2004, 43, 4460–4462 CrossRef CAS.
- J. M. J. Paulusse, J. P. J. Huijbers and R. P. Sijbesma, Chem.–Eur. J., 2006, 12, 4928–4934 CrossRef CAS.
- S. Sivakova, D. A. Bohnsack, M. E. Mackay, P. Suwanmala and S. J. Rowan, J. Am. Chem. Soc., 2005, 127, 18202–18211 CrossRef CAS.
- C.-L. Lin and W.-Y. Chiu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5923–5934 CrossRef CAS.
- M. S. Green and A. V. Tobolsky, J. Chem. Phys., 1946, 14, 80–89 CrossRef CAS.
- A. S. Lodge, Trans. Faraday Soc., 1956, 52, 120–130 RSC.
- F. Tanaka and S. F. Edwards, J. Non-Newtonian Fluid Mech., 1992, 43, 247–271 Search PubMed.
- M. Rubinstein and A. N. Semenov, Macromolecules, 1998, 31, 1386–1397 CrossRef CAS; R. H. W. Wientjes, R. J. J. Jongschaap, M. H. G. Duits and J. Mellema, J. Rheol., 1999, 43, 375–391 CrossRef CAS; T. Annable, R. Buscall, R. Ettelaie and D. Whittlestone, J. Rheol., 1993, 37, 695–726 CrossRef CAS.
- F. Chambon and H. H. Winter, Polym. Bull., 1985, 13, 499–503 CAS.
- H. H. Winter and F. Chambon, J. Rheol., 1986, 30, 367–382 CrossRef CAS.
- L. M. Leibler, M. Rubinstein and R. H. Colby, Macromolecules, 1991, 24, 4701–4707 CrossRef.
- P. Lang and W. Burchard, Macromolecules, 1991, 24, 814–815 CrossRef CAS.
- E. D. Gado, L. d. Arcangelis and A. Coniglio, J. Phys.: Condens. Matter, 2002, 14, 2133–2139 CrossRef.
- J. A. Trotter, J. Tipper, G. Lyons-Levy, K. Chino, A. H. Heuer, Z. Liu, M. Mrksich, C. Hodneland, W. S. Dillmore, T. J. Koob, M. M. Koob-Emunds, K. Kadler and D. Holmes, Biochem. Soc. Trans., 2000, 28, 357–362 CrossRef CAS.
- G. K. Szulgit and R. E. Shadwick, J. Exp. Biol., 2000, 203, 1539–1550 Search PubMed.
- T. Motokawa and A. Tsuchi, Biol. Bull., 2003, 205, 261–275 Search PubMed.
- W. C. Yount, D. M. Loveless and S. L. Craig, J. Am. Chem. Soc., 2005, 127, 14488–14498 CrossRef CAS.
- D. M. Loveless, S. L. Jeon and S. L. Craig, Macromolecules, 2005, 38, 10171–10177 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry,Cornell University Press,Ithaca, NY, 1969 Search PubMed.
- J. Song, V. Malathong and C. R. Bertozzi, J. Am. Chem. Soc., 2005, 127, 3366–3372 CrossRef CAS.
- S. L. Jeon, D. M. Loveless, W. C. Yount and S. L. Craig, Inorg. Chem., in press.
- A. T. t. Cate and R. P. Sijbesma, Macromol. Rapid Commun., 2002, 23, 1094–1112 CrossRef CAS.
- K. P. Nair, J. M. Pollino and M. Weck, Macromolecules, 2006, 931–940 CrossRef CAS.
- S. J. Rowan and J. B. Beck, Faraday Discuss., 2005, 128, 43–53 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: full experimental details. See DOI: 10.1039/b614026b |
| This journal is © The Royal Society of Chemistry 2007 |