Electrophoretic deposition of donor–acceptor nanostructures on electrodes for molecular photovoltaics
Hiroshi
Imahori
ab
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan
bFukui Institute for Fundamental Chemistry, Kyoto University, 34-4, Takano-Nishihiraki-cho, Sakyo-ku, Kyoto 606-8103, Japan. E-mail: imahori@scl.kyoto-u.ac.jp
First published on 19th September 2006
Abstract
Electrophoretic deposition of donor–acceptor nanostructures onto electrodes is reviewed in terms of light-to-electrical energy conversion. Various donors and acceptors and their composites have been deposited electrophoretically onto a nanostructured SnO2 or TiO2 electrode which exhibits photocurrent generation. In particular, bottom-up self-organization of porphyrin and fullerene molecules onto the nanostructured SnO2 electrodes has led to highly efficient photocurrent generation with an incident photon-to-current efficiency (IPCE) of up to ∼60%. Such examples will give us many valuable insights into the design of organic molecular electronics including solar cells, organic transistors, and light-emitting devices.
 Hiroshi Imahori | Hiroshi Imahori was born in Kyoto, Japan in 1961. He completed his doctorate on organic chemistry at Kyoto University. From 1990–1992 he was a post-doctoral fellow at the Salk Institute for Biological Studies, USA. In 1992 he became an Assistant Professor at ISIR, Osaka University. In 1999 he moved to the Graduate School of Engineering, Osaka University, as an Associate Professor. Since 2002 he has been Professor of Chemistry, Graduate School of Engineering, Kyoto University. |
1. Introduction
Recently, ecological considerations linked with the CO2/global warming problem have prompted us to utilize photovoltaic solar energy. However, the present cost of electricity from silicon-based photovoltaics is much higher than the current commercial prices of electricity generated by hydraulic power or nuclear and fossil fuels. Therefore, it is necessary to develop low-cost solar cells with a high power conversion efficiency (η). Organic solar cells would be promising candidates if they fulfil these requirements.1,2 It should be noted here that they bear unique advantages over inorganic solar cells (i.e., flexibility, lightness, and colorfulness). Since the beginning of the 1990s, substantial advances in power conversion efficiency have been made in molecular photovoltaics including dye-sensitized solar cells (up to η = 7–11%)3–6 and bulk heterojunction solar cells (up to η = 3–6%).7–14In this context, extensive efforts have been made in recent years to explore the photovoltaic and photoelectrochemical properties of electrodes modified with various donor and acceptor components toward the realization of highly efficient organic solar cells. In the design of organic solar cells we consider the following criteria: (i) extensive light-harvesting in the visible region, (ii) efficient energy transfer to the interface of the heterojunction (if necessary) and subsequent charge separation, and (iii) efficient injection of separated electrons and holes into their respective electrodes, minimizing undesirable charge recombination. Accordingly, it is of vital importance to organize suitable donor and acceptor molecules on the electrode surface in the nanometer scale for fulfilling these requirements. Versatile methods such as Langmuir–Blodgett (LB) films,15–22 self-assembled monolayers (SAMs),23–47 layer-by-layer deposition,48–59 vacuum deposition,8,14 chemical adsorption3–6,60,61 and spin coating7,9,10–13,61–64 have been adopted to construct such nanostructures on electrodes for photoelectrochemical devices and solar cells.
The electrophoretic deposition technique has been widely employed, particularly in film deposition from colloidal dispersions. Electrophoretic deposition is essentially a two-step process. First, particles such as macromolecules and colloids are charged in a polar solution and forced to move toward one of two electrodes inserted into the solution by applying an electric field to the suspension. Then, the particles are deposited onto the electrode gradually, leading to the formation of an organic thin film. Thus, it is a fast and economical process which makes it possible to control nano- and micro-structures on the electrode surface. In this review article, we focus on the electrophoretic deposition method, which allows donor and acceptor molecules to be assembled onto electrodes for molecular photoelectrochemical devices. Specifically, we emphasize the utilization of porphyrins as an electron donor and fullerenes as an electron acceptor, because they exhibit small reorganization energies of electron transfer,65–70 which makes it possible to generate a long-lived charge-separated state with a high quantum yield.
2. Electrophoretic deposition of clusters of single component
2.1 Fullerenes and other acceptors
Kamat et al. applied the electrophoretic deposition technique for C60 clusters [denoted as (C60)m] to obtain the C60 films on electrodes which absorb visible light intensively and exhibit a remarkable photoelectrochemical response.71,72 Namely, a toluene solution of C60 molecules is rapidly injected into acetonitrile to form C60 clusters with an average diameter of ∼300 nm due to the lyophobic nature of C60 in the mixed solvent (acetonitrile–toluene = 3 : 1, v/v), as illustrated in Fig. 1. Under application of a dc electric field (100–200 V), the C60 clusters in the mixed solvent become negatively charged and are deposited on a nanostructured SnO2 electrode as they are driven towards the positively charged electrode surface. The broad absorption of the C60 film in the visible and in the near-IR region, as well as the high molar absorptivity, makes the film suitable for harvesting solar energy. Under visible light irradiation (λ > 425 nm, input power (WIN) = 130 mW cm−2), using I−/I3− as the redox couple, the photoelectrochemical cell revealed a stable photocurrent of ∼0.14 mA cm−2 and an open circuit voltage (VOC) of ∼0.2 V. The maximum IPCE (incident photon-to-current efficiency) of ∼4% was observed at the excitation wavelength of 420 nm.71 The IPCE value is 2–3 orders of magnitude larger than previously reported values using functionalized C60 as photosensitizers.15–19,23,24 Photoinduced electron transfer between iodide ions (I−/I3− = 0.5 V vs NHE) and the excited states of C60 clusters (1C60*/C60•− = 1.7 V vs NHE; 3C60*/C60•− = 1.4 V vs NHE) is the primary step in the photocurrent generation (Fig. 2). The reduced C60 (C60/C60•− = −0.2 V vs NHE) then injects electrons into SnO2 nanocrystallites (ECB = 0 V vs NHE). The electrons transferred to the semiconductor nanocrystallites are driven to the counter electrode via external circuit to regenerate the redox couple.71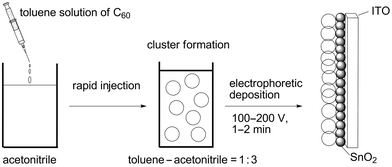 | ||
| Fig. 1 Schematic illustration of cluster formation of C60 by rapid injection and its electrophoretic deposition onto a nanostructured SnO2 electrode. | ||
 | ||
| Fig. 2 Energy diagram for photocurrent generation at an ITO/SnO2/(C60)m electrode. | ||
We designed two C60 derivatives with a phenylpyrrolidine moiety, 1 and 2, to examine the effect of the C60 substituents on the structure and photoelectrochemical properties of C60 clusters electrophoretically deposited on nanostructured SnO2 electrodes (Fig. 3).73 With introducing larger substituents into C60, the cluster size increases, whereas the IPCE decreases (2.3% for C60, 1.1% for 1, and 0.3% for 2 at 400 nm). The trend for photocurrent generation efficiency, as well as surface morphology on the electrode, can be rationalized by the steric bulkiness around the C60 molecules. It is noteworthy that the C60 molecule with two alkoxy chains 2 is suggested to give a bilayer vesicle structure in the mixed solvent, irrespective of the hydrophobic nature of both the C60 and alkoxy chain moieties.73 Such information will be valuable for the design of photoactive molecules, which are fabricated onto electrode surfaces to exhibit efficient photocurrent generation.
 | ||
| Fig. 3 Fullerene derivatives 1 and 2 and perylene diimide 3 for the cluster formation. | ||
Kamat et al. assembled alkanethiol-tethered C60 onto a gold nanoparticle to form a photoelectrochemical device.74 By incorporating the alkanethiol-tethered C60 into dodecanethiol-capped gold nanoparticles during the gold reduction step, it is possible to bind ca. 90 C60 moieties per gold nanocore. The C60-modified gold nanoparticles in toluene are deposited electrophoretically onto a nanostructured SnO2 electrode. The maximum IPCE value of 0.8% was obtained at an excitation wavelength of 450 nm.74
The interaction between the sensitizer and the redox couple in dye-sensitized solar cells is an important factor that controls the power conversion efficiency.3–6 Kamat et al. employed C60 clusters to separate ruthenium dye and I−/I3− couple to minimize the undesirable sensitizer–redox couple interaction.75 The ruthenium dye device with the C60 layer exhibited the maximum IPCE of 64%, which is larger by 15% than that in the ruthenium dye device without the C60 layer.75
On the other hand, it is generally difficult to find suitable conditions for forming the clusters of other acceptors in the mixed solvent to deposit them electrophoretically onto the nanostructured SnO2 electrode. For instance, we could not deposit the clusters of perylene diimide 3 (Fig. 3) electrophoretically onto the ITO/SnO2 electrode probably due to their small size (13 nm) in the mixed solvent.76
2.2 Porphyrins and phthalocyanines
Electron donors such as porphyrins and phthalocyanines can make their clusters in an acetonitrile–toluene mixture. We deposited the clusters of porphyrins and phthalocyanines electrophoretically onto the semiconducting electrodes for photoelectrochemical devices.76,77 When a concentrated solution of 5,15-bis(3,5-di-tert-butylphenyl)porphyrin] 4a (Fig. 4) in toluene is mixed with acetonitrile by fast injection method (acetonitrile–toluene = 9 : 1, v/v), the molecules aggregate and form stable clusters [denoted as (4a)m].77 Under application of a high dc electric field (500 V), the porphyrin clusters which become negatively charged in the mixed solvent are deposited on the nanostructured TiO2 electrode as they are driven towards the positively charged electrode surface. Photoexcitation of the porphyrin film on the electrode in a photoelectrochemical device with visible light leads to relatively high photocurrent generation. The maximum photocurrent of 0.15 mA cm−2 and photovoltage of 0.25 V were attained using the I−/I3− redox couple. The IPCE value of 2.0% was achieved at an applied bias potential of 0.2 V vs SCE. Electron injection from the porphyrin excited singlet state (1H2P*/H2P•+ = −0.7 V vs NHE) to the TiO2 conduction band (ECB = −0.5 V vs NHE) is the primary step in the photocurrent generation (Fig. 5). Then, iodide (I−/I3− = 0.5 V vs NHE) gives electrons to the oxidized porphyrin (H2P/H2P•+ = 1.2 V vs NHE) to regenerate the redox couple.77 The broad photoresponse of these crystallites throughout the visible range, as well as the ease of assembling them on the electrode surface, paves the way for harvesting a wide wavelength range of solar light.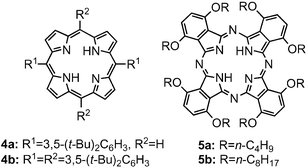 | ||
| Fig. 4 Porphyrins 4 and phthalocyanines 5 for the cluster formation. | ||
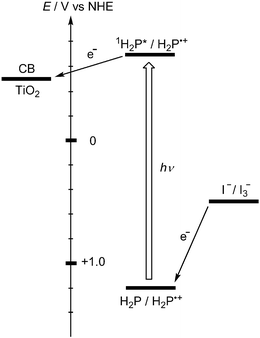 | ||
| Fig. 5 Energy diagram for photocurrent generation at ITO/TiO2/(4a)m electrode. | ||
Phthalocyanines 5a (R = n-C4H9) and 5b (R = n-C8H17) (Fig. 4) form optically transparent clusters in a mixture of toluene–acetonitrile after rapid injection of the toluene solution into acetonitrile [denoted as (5)m].76 The clusters of 5b in the toluene–acetonitrile mixture exhibit a structureless broad absorption in the 300–500 and 700–800 nm range with lower molar coefficients, compared to the monomeric form in toluene. These results confirm that the phthalocyanine molecules aggregate and form clusters in the mixed solvents as in the case of porphyrins and/or fullerenes. In contrast, phthalocyanine 5a does not reveal such a spectral change under the same conditions. This suggests that the long alkyl chains of 5b relative to those of 5a have a large impact on the formation of clusters in the mixed solvents. Under application of a high dc electric field (250 V for 2 min), the clusters (5b)m, which are positively charged in the toluene–acetonitrile, are driven towards the negatively charged electrode surface [denoted as ITO/SnO2/(5b)m].76 This behavior is in sharp contrast with porphyrins and fullerenes in which they are negatively charged under application of a dc voltage, leading to deposition on the positively charged electrode (vide supra). Photoelectrochemical measurements were performed under the standard three electrode system (0.15 V vs SCE). The IPCE value of the phthalocyanine photoelectrochemical device (0.1–1%)76 is rather smaller than those of the porphyrin devices using 4a (∼2.0%)77,78 and 4b (∼1%) (vide infra).79 In the case of the porphyrins, bulky t-butyl groups are introduced into the meta positions of the meso-phenyl groups of the porphyrins to avoid the undesirable aggregation that leads to the self-quenching of the porphyrin excited singlet state. On the other hand, the present phthalocyanine 5b possesses relatively long alkoxy chains which induce π–π stacking of the phthalocyanine rings, resulting in the relatively strong self-quenching of the excited singlet state. Such difference in the photophysics of the phthalocyanine and the porphyrins may be responsible for the difference in the IPCE values. This matches the strong tendency of phthalocyanines to aggregate on the nanostructured SnO2 or TiO2 electrodes, leading to low IPCE of phthalocyanine-sensitized solar cells (<4%).76
3. Electrophoretic deposition of clusters of donor and acceptor composites
3.1. Donor–acceptor linked systems
Kamat et al. deposited clusters of C60-aniline linked dyad 6 as thin films on a nanostructured SnO2 electrode under the influence of an electric field (Fig. 6).80 Photoelectrochemical measurements were carried out under the standard two electrode arrangement. The maximum IPCE of ∼4% is noted at 450 nm,80 which is comparable to that (∼4%) of similar photoelectrochemical device with C60 itself.71 Thus, there is no substantial effect of C60 as an electron mediator on photocurrent enhancement in this system. | ||
| Fig. 6 Donor–acceptor linked molecules 6–9 for the cluster formation. | ||
In a collaboration with Kamat et al., we applied the electrophoretic deposition method to porphyrin–fullerene linked dyads 7 (M = H2, Zn) to assemble the dyad clusters on a nanostructured SnO2 electrode (Fig. 6).81 The cluster films of 7, when electrophoretically deposited on the nanostructured SnO2 electrodes, are photoactive and generate anodic photocurrent under standard three electrode arrangement (0 V vs Ag/AgCl) [denoted as ITO/SnO2/(7)m/LiI + I2/Pt device]. Although the light-harvesting efficiency (maximal absorbance = 1.6) is improved by a factor of 11 compared to the monolayer system (∼0.04), the maximum IPCE values are found to be still low (0.36% for ITO/SnO2/(7(M = H2))m/LiI + I2/Pt device and 0.42% for ITO/SnO2/(7(M = Zn))m/LiI + I2/Pt device).81 The maximum IPCE values are smaller than those of photoelectrochemical devices (∼9%) in which a similar porphyrin–C60 dyad is physically adsorbed onto a SnO2 electrode.60 Although such donor–acceptor linked molecules are designed to exhibit a long-lived charge-separated state with a high quantum yield in solutions,65–70 the linkage between the donor and the acceptor would not be optimized for the desirable molecular packing of the dyads in the thin film, so that separated hole and electron are not injected efficiently into the respective electrodes.
Fukuzumi and Kamat et al. employed formanilide–anthraquinone dyad 8a and ferrocene (Fc)–formanilide–anthraquinone dyad 8b (Fig. 6) as components of photoelectrochemical devices where composite clusters of 8a or 8b with C60 are assembled onto a SnO2 electrode using the electrophoretic deposition method.82 Photocurrent measurements were performed under the standard three electrode arrangement [denoted as ITO/SnO2/(8a + C60)m or (8b + C60)m/NaI + I2/Pt device]. The maximum IPCE value (9.7% at 470 nm) in ITO/SnO2/(8a + C60)m/NaI + I2/Pt device is 10 times as large as that (0.98% at 465 nm) in ITO/SnO2/(8b + C60)m/NaI + I2/Pt device (0 V vs SCE). Such enhancement in photocurrent generation can be ascribed to the dramatic difference in the lifetimes of the charge-separated states of 8a (900 µs) and 8b (20 ps) in DMSO.82
Fukuzumi and Imahori et al. reported photovoltaic cell comprising of 9-mesityl-10-carboxymethylacridinium ion 9 and C60 clusters (Fig. 6).83 9-Mesityl-10-carboxymethylacridinium ion 9 is adsorbed chemically onto a SnO2 electrode, followed by the electrophoretic deposition of the C60 cluster onto the SnO2 electrode. Photoelectrochemical measurements were performed using the standard three electrode system [denoted as ITO/SnO2/9 + (C60)m/NaI + I2/Pt device]. The IPCE value in ITO/SnO2/9 + (C60)m/NaI + I2/Pt device reaches 25% at 480 nm at an applied potential of 0.2 V vs SCE, whereas the maximum IPCE value of ITO/SnO2/9/NaI + I2/Pt device is 2% at 460 nm.83
Fukuzumi and Kamat et al. deposited electrophoretically the composite clusters of C60 and TiO2 nanoparticles modified with 9 (TiO2–9) in acetonitrile–toluene (3 : 1, v/v) onto a nanostructured SnO2 electrode.84 The TiO2 nanoparticles act as scaffold to organize 9 and C60. The IPCE value in ITO/SnO2/(TiO2–9 + C60)m/NaI + I2/Pt device reaches 37% at 480 nm at an applied potential of 0.2 V vs SCE, whereas the maximum IPCE value of ITO/SnO2/(TiO2–9)m/NaI + I2/Pt device is 5% at 480 nm.84 Although 9 is reported to exhibit an extremely long-lived charge-separated state with a high quantum yield,85 C60 as an electron mediator between the charge-separated state and the SnO2 electrode is necessary to achieve efficient photocurrent generation.
3.2. Donor and acceptor composite systems
Composites of donor and acceptor in the form of clusters have been assembled as three-dimensional network on an electrode surface for attaining efficient photocurrent generation. Kamat et al. prepared composite clusters of various donors (N,N-dimethylaniline, N,N-dimethyl-p-toluidine, ferrocene, N-methylphenothiazine, and N,N-dimethyl-p-anisidine) and 1,2,5-triphenylpyrrolidinofullerene which are deposited electrophoretically onto a nanostructured SnO2 electrode.86 The maximum IPCE of ∼0.05% is obtained at 420 nm,86 which is much smaller than those of a similar photoelectrochemical device with C60 (1.6–4% at 420 nm)71 and fullerene derivatives (0.2–0.8% at 400 nm).73In a collaboration with Kamat et al., we applied bottom-up self-assembly to porphyrin and fullerene single components and the composites to construct a novel organic solar cell (dye-sensitized bulk heterojuntion solar cell), possessing both the dye-sensitized and bulk heterojunction characteristics.78,87,88 We have already found, for the first time, that there is π–π interaction between porphyrin and fullerene in the ground and excited states.89–93 The specific interaction between porphyrin and fullerene is suitable for electrophoretic deposition of the composite clusters. Bearing this in mind, we make a supramolecular complex of porphyrin and fullerene in toluene due to the π–π interaction (step 1).78,87,88 Then, the supramolecular complex is self-assembled into larger clusters in a mixture of toluene and acetonitrile due to lyophobic interaction between the complex and the mixed solvent (step 2). Finally, the larger clusters can be further associated onto a nanostructured SnO2 electrode using the electrophoretic deposition technique (step 3).
The electrophoretic deposition method is adopted to prepare a film of the composite clusters of porphyrin 4a and C60 [denoted as (4a + C60)m] on an ITO/SnO2 electrode.78 Photocurrent measurements were performed under the standard two electrode system [denoted as ITO/SnO2/(4a + C60)m/NaI + I2/Pt device]. The short circuit photocurrent density (JSC) of 0.18 µA cm−2, open circuit voltage (VOC) of 0.21 V, fill factor (ff) of 0.35, and η of 0.012% (input power (WIN) = 110 mW cm−2) are obtained for ITO/SnO2/(4a + C60)m/NaI + I2/Pt device. The maximum IPCE value of 4.5% at 430 nm for ITO/SnO2/(4a + C60)m/NaI + I2/Pt device is larger than the sum (∼2%) of the IPCE value (0.6%) for ITO/SnO2/(4a)m/NaI + I2/Pt and that (1.6%) for ITO/SnO2/(C60)m/NaI + I2/Pt reference devices. Note that the maximum IPCE value of 17% is obtained at 460 nm for ITO/SnO2/(4a + C60)m/NaI + I2/Pt device at an applied potential of 0.2 V vs SCE. Such enhancement in the photocurrent generation of the composite cluster devices from 4a and C60 demonstrates that charge separation between the excited porphyrin and C60 in the supramolecular complex is a dominating factor for efficient photocurrent generation. Namely, ultrafast photoinduced electron transfer occurs from the porphyrin singlet excited state (1H2P*/H2P•+ = −0.7 V vs NHE) to C60 (C60/C60•− = −0.2 V vs NHE) in the complex. The reduced C60 transfers electrons to the conduction band of SnO2 nanocrystallites (ECB = 0 V vs NHE) by electron hopping through large excess of C60 molecules. The regeneration of H2P clusters (H2P/H2P•+ = 1.2 V vs NHE) is achieved by the iodide/triiodide couple (I−/I3− = 0.5 V vs NHE) present in the electrolyte system (Fig. 7).78
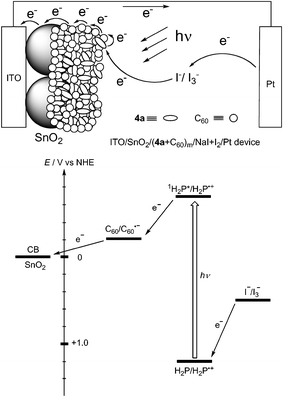 | ||
| Fig. 7 Schematic photocurrent generation and energy diagram in dye-sensitized bulk heterojunction solar cells consisting of 4a and C60. | ||
The present organic solar cell, “dye-sensitized bulk heterojunction solar cell,” is unique in that it possesses both characteristics of dye-sensitized and bulk heterojunction solar cells. Namely, initial charge separation occurs at the interface of the donor and the acceptor, which is typical characteristic of bulk heterojunction solar cells. Nevertheless, the following other processes are similar to those in dye-sensitized solar cells. It is noteworthy that the composite film reveals the multilayer structure on ITO/SnO2, which presents a striking contrast to monolayer structure of adsorbed dyes on TiO2 electrodes in dye-sensitized solar cells. Therefore, we can expect improvement of the photovoltaic properties by modulating both the structures of electrode surfaces and donor–acceptor multilayers.
A drawback of the electrophoretic deposition of donor–acceptor composites is the difficulty of codeposition of positively charged donors and negatively charged acceptors or vice versa, which are electrophoretically deposited onto respective electrodes. For instance, under application of a high dc electric field, the clusters of C60 and C60 derivatives, which tend to be negatively charged in toluene–acetonitrile, are driven towards the positively charged electrode surface. On the other hand, the clusters of phthalocyanines, which tend to be positively charged in toluene–acetonitrile, are driven toward the negatively charged electrode surface. Thus, the composite clusters of phthalocyanine and C60 cannot be deposited onto the same electrode. Nevertheless, the composite clusters of phthalocyanine 5b and perylene diimide 3 [denoted as (5b + 3)m] can be attached onto ITO/SnO2 electrodes by the electrophoretic deposition [denoted as ITO/SnO2/(5b + 3)m].76 Under application of a high dc electric field (250 V for 2 min), the clusters (5b + 3)m (which are positively charged in toluene–acetonitrile) are driven towards the negatively charged electrode surface. Although the IPCE value of the ITO/SnO2/(5b + 3)m device (<1%)76 is rather low compared to similar porphyrin systems,77–79 the enhancement of the photocurrent generation efficiency of the composite system is achieved relative to the phthalocyanine reference system without the perylene diimide. This is an example for dye-sensitized bulk heterojunction solar cells using different combination of donor and acceptor (i.e., phthalocyanine and perylene diimide).76
3.3. Pre-organized porphyrin systems
Supramolecular assembly of donor–acceptor molecules is a potential approach to create a desirable phase-separated, interpenetrating network involving molecular-based nanostructured electron and hole highways in the blend films of donor and acceptor for bulk heterojunction solar cells. However, different, complex hierarchies of self-organization going from simple molecules to devices have limited improvement of the device performance. To construct such complex hierarchies comprising of donor and acceptor molecules on electrode surfaces, pre-organized molecular systems are excellent candidates for achieving the molecular architectures. In particular, porphyrins have been three-dimensionally organized using dendrimers,79,94 oligomers,95 and nano- and micro- particles96–104 as nanoscaffold to combine with fullerenes for organic solar cells. | ||
| Fig. 8 Molecular structures of multiporphyrin dendrimers 10 (n) (n: number of the porphyrins in the dendrimer (n = 4, 8, 16)) for dye-sensitized bulk heterojunction solar cells. | ||

 | ||
| Fig. 10 Schematic diagram for step-by-step organization of porphyrin–alkanethiol 12 and C60 onto a nanostructured ITO/SnO2 electrode via porphyrin-modified gold nanoparticle 13 (n = 5, 11, 15). | ||
We further continued this novel approach to construct a light energy conversion system using the supramolecular incorporation of C60 molecules (guest) into tailored, large and bucket-shaped surface holes (host) on porphyrin-modified gold nanoparticles 14 in the mixed solvent, followed by the electrophoretic deposition on a nanostructured SnO2 electrode (Fig. 11).99,100 First, some of porphyrin molecules on a gold nanoparticle 13 (n = 11) are exchanged with short alkanethiol molecules to yield 14 (n = 11) with large and bucket-shaped surface holes (step 1). The SnO2 electrodes modified with the composite clusters of 14 (n = 11) with C60 are prepared as in the case of 13 and C60 composite system. The difference in the porphyrin–C60 ratio is found to affect the structures and photoelectrochemical properties of the composite clusters in the mixed solvents as well as on the SnO2 electrodes. Photoelectrochemical measurements were performed with the standard three electrodes system [denoted as ITO/SnO2/(14(n = 11) + C60)m/LiI + I2/Pt device]. The maximum IPCE value (42%) of ITO/SnO2/(14(n = 11) + C60)m/LiI + I2/Pt device with large, bucket-shaped holes is larger by a factor of 3 than that (16%) of ITO/SnO2/(13(n = 11) + C60)m/LiI + I2/Pt device with small, wedge-shaped surface holes on 13 (n = 11) at an applied potential of +0.06 V vs SCE. This clearly demonstrates that the shape and size of the host holes on the three-dimensional porphyrin-modified gold nanoparticles have a large impact on the photoelectrochemical properties. Remarkable enhancement in the photoelectrochemical performance as well as broader photoresponse in the visible and infrared relative to the reference systems demonstrates that the bottom-up self-organization approach provides a novel perspective for the development of efficient organic solar cells.99,100
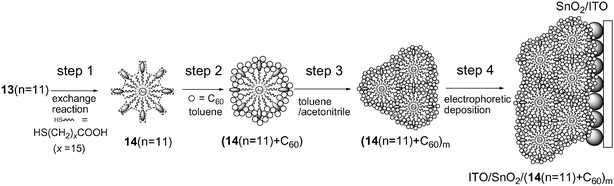 | ||
| Fig. 11 Schematic diagram for step-by-step organization of porphyrin-modified gold nanoparticle 13 (n = 11) and C60 onto a nanostructured ITO/SnO2 electrode via porphyrin-modified gold nanoparticle 14 (n = 11) with a large, bucket-shaped hole on the surface for the binding of C60. | ||
The drawbacks of utilizing gold nanoparticles as nanoscaffolds are their expensive cost and the relatively strong energy transfer quenching of the porphyrin excited singlet state by the gold surface. Thus, inexpensive nanoparticles as a scaffold of porphyrin molecules without such energy transfer quenching would be required to improve photocurrent generation efficiency in the composite cluster systems with C60. Silica nanoparticles are potential candidates because of their low cost, non-photoactive characteristics, and facile functionalization. A silica micro- and nano-particle 15 has been successfully employed as a scaffold to self-organize porphyrin and C60 molecules on a nanostructured SnO2 electrode (Fig. 12).101,102 The quenching of the porphyrin excited singlet state on the silica nanoparticle is suppressed significantly, showing that silica nanoparticles are promising scaffolds for organizing photoactive molecules three-dimensionally in nanometer scale. Marked enhancement of the photocurrent generation (10–17%) was achieved in the porphyrin-modified silica particle devices with C60 compared to the reference device (4% for 13 (n = 5)) with a similar length of spacer between the porphyrin and the particle in which gold core was employed as a scaffold of porphyrins instead of silica particles. The rather small IPCE value (10%) of the silica nanoparticle device relative to that using silica microparticles (17%) may result from poor electron and hole mobility in the composite film due to poor connection between the composite clusters of porphyrin-modified silica nanoparticle and C60 in micrometer scale.101,102

We examined the substituent effect of meso-tetraphenylporphyrin 16 on the nanostructure and photoelectrochemical properties of SnO2 electrodes modified electrophoretically with the composite clusters of 16 and C60 (Fig. 13).107 To facilitate the supramolecular complexation between the porphyrin and C60, electron-donating substituents (i.e., methoxy group) are introduced into the 3,5-positions (16a), 3,4,5-positions (16b), and 2,6-positions (16c) of the meso-phenyl groups at the porphyrin ring. The maximum IPCE value (59% at 425 nm) of ITO/SnO2/(16a + C60)m device is much larger than those of ITO/SnO2/(16b + C60)m (10%), ITO/SnO2/(16c + C60)m (5.7%), and other related porphyrin and C60 single component composite devices (1–17%).78,79 It should be emphasized here that the simple substituent of methoxy groups onto the meta-positions of the meso-phenyl groups at the porphyrin ring is responsible for the efficient photocurrent generation, which is much superior to the systems from the more complex, time-consuming pre-organized porphyrin molecular assemblies and C60.94–104 Various spectroscopic and surface analyses revealed that the molecular arrangement of 16a and C60 composite clusters on the SnO2 electrode involves a similar specific molecular packing of single crystal 16a·2C60·toluene in which the porphyrin and C60 make an alternative layer structure where the closest porphyrin moieties (center-to-center distance (Rcc) = 14.2 Å) are arranged in a one-dimensional chain with a dihedral angle of 66°, while the closest C60 moieties (Rcc = 10.2 Å) in a two-dimensional sheet with sandwiching 16a between two C60 molecules (Fig. 14). The segregated nanoarrays of porphyrin and fullerene on the SnO2 electrode allow the system to undergo ultrafast electron transfer or charge transfer (<100 fs) between the supramolecular complex of the porphyrin and C60 molecules, followed by hole and electron relay through the nanostructured one-dimensional porphyrin chains and two-dimensional C60 sheets, leading to efficient photocurrent generation.107 In bulk heterojunction solar cells, many researchers have suggested the importance of nanostructured electron and hole transporting highways which have never been confirmed experimentally.1,7–14 Our finding is the first experimental demonstration for this hypothesis. Such results provide many valuable insights into the design of molecular photovoltaics in the nanoscale.
 | ||
| Fig. 13 Molecular structures of self-assembling porphyrins 16 used in this study. | ||
 | ||
| Fig. 14 Molecular packing of 16a·(C60)2·(toluene). Parts of the C60 and toluene molecules are omitted for clarity. | ||
4. Summary
This review article has focused on recent advances in electrophoretic deposition for photoelectrochemical devices and organic solar cells. Electron transfer reactions between donors and acceptors have been well understood on the basis of electron transfer parameters including the driving force, electronic coupling matrix elements, reorganization energy, and temperature, in the light of Marcus theory of electron transfer. In the next stage, chemists must apply such principle to donor–acceptor assemblies in a solid-state which exhibits specific functions of interest. However, it is still difficult to predict molecular packing of the solid-state donor–acceptor composites and its resultant function from the molecular structures of the donor and acceptor. Recent advances in electrophoretic deposition of donor–acceptor nanostructures on electrodes will give us a deep insight into the design of molecular devices. In such cases, utilization of weak intermolecular interactions between donor–donor, acceptor–acceptor, and donor–acceptor for forming the nanostructures on the electrode plays a key role in the construction of organic molecular electronics such as organic transistors, solar cells, and light-emitting devices. Preprogrammed, self-assembling simple molecules will allow us to achieve such functions without difficulty in terms of synthesis and fabrication. Moreover, the search for novel and better nanomaterials such as carbon nanotubes108,109 and nanoparticles110 should continue to combine with electrophoretic deposition methods for the development of organic molecular electronics.Acknowledgements
The authors are deeply indebted to the work of all collaborators and co-workers whose names are listed in the references (in particular, Prof. S. Fukuzumi, Prof. H. Lemmetyinen, Prof. N. V. Tkachenko, Prof. P. V. Kamat, and Prof. M. Crossley). H. I. thanks Grants-in-Aid from MEXT, Japan (Priority Areas of Chemistry of Coordination Space (No. 18033025) and of Super-Hierarchical Structures (No. 18039019), and 21st Century COE on Kyoto University Alliance for Chemistry), NEDO, and Kurata and Sekisui foundations for financial support.References
- Organic Photovoltaics, ed. S. S. Sun and R. S. Sariciftci, CRC, Boca Raton, 2005 Search PubMed.
- B. A. Gregg, J. Phys. Chem. B, 2003, 107, 4688 CrossRef CAS.
- A. Hagfeldt and M. Grätzel, Acc. Chem. Res., 2000, 33, 269 CrossRef CAS.
- N. S. Lewis, Inorg. Chem., 2005, 44, 6900 CrossRef CAS.
- M. Grätzel, J. Photochem. Photobiol., A, 2004, 163, 3 CrossRef.
- M. Wei, Y. Konishi, H. Zhou, M. Yanagida, H. Sugihara and H. Arakawa, J. Mater. Chem., 2006, 16, 1287 RSC.
- F. Padinger, R. S. Rittberger and N. S. Sariciftci, Adv. Funct. Mater., 2003, 13, 85 CrossRef CAS.
- M. Hiramoto, H. Fujiwara and M. Yokoyama, Appl. Phys. Lett., 1991, 58, 1062 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CrossRef CAS.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS.
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425 CrossRef CAS.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Ind. Ed., 2003, 42, 3371 Search PubMed.
- J. Xue, S. Uchida, B. P. Rand and S. R. Forrest, Appl. Phys. Lett., 2004, 84, 3013 CrossRef CAS.
- J. Jin, L. S. Li, Y. Li, Y. J. Zhang, X. Chen, D. Wang, S. Jiang and T. J. Li, Langmuir, 1999, 15, 4565 CrossRef CAS.
- C. Luo, C. Huang, L. Gan, D. Zhou, W. Xia, Q. Zhuang, Y. Zhao and Y. Huang, J. Phys. Chem., 1996, 100, 16685 CrossRef CAS.
- W. Zhang, Y. Shi, L. Gan, C. Huang, H. Luo, D. Wu and N. Li, J. Phys. Chem. B, 1999, 103, 675 CrossRef CAS.
- D. Zhou, L. Gan, C. Luo, H. Tan, C. Huang, G. Yao, X. Zhao, Z. Liu, X. Xia and B. Zhang, J. Phys. Chem., 1996, 100, 3150 CrossRef CAS.
- W. Zhang, L. Gan and C. Huang, J. Mater. Chem., 1998, 8, 1731 RSC.
- N. V. Tkachenko, E. Vuorimaa, T. Kesti, A. S. Alekseev, A. Y. Tauber, P. H. Hynninen and H. Lemmetyinen, J. Phys. Chem. B, 2000, 104, 6371 CrossRef CAS.
- N. V. Tkachenko, V. Vehmanen, J.-P. Nikkanen, H. Yamada, H. Imahori, S. Fukuzumi and H. Lemmetyinen, Chem. Phys. Lett., 2002, 366, 245 CrossRef CAS.
- T. Vuorinen, K. Kaunisto, N. V. Tkachenko, A. Efimov and H. Lemmetyinen, Langmuir, 2005, 21, 5383 CrossRef CAS.
- H. Imahori, T. Azuma, S. Ozawa, H. Yamada, K. Ushida, A. Ajavakom, H. Norieda and Y. Sakata, Chem. Commun., 1999, 557 RSC.
- H. Imahori, T. Azuma, A. Ajavakom, H. Norieda, H. Yamada and Y. Sakata, J. Phys. Chem. B, 1999, 103, 7233 CrossRef CAS.
- O. Enger, F. Nuesch, M. Fibbioli, L. Echegoyen, E. Pretsch and F. Diederich, J. Mater. Chem., 2000, 10, 2231 RSC.
- T. Akiyama, H. Imahori, A. Ajavakom and Y. Sakata, Chem. Lett., 1996, 907 CAS.
- H. Imahori, S. Ozawa, K. Ushida, M. Takahashi, T. Azuma, A. Ajavakom, T. Akiyama, M. Hasegawa, S. Taniguchi, T. Okada and Y. Sakata, Bull. Chem. Soc. Jpn., 1999, 72, 485 CrossRef CAS.
- H. Yamada, H. Imahori and S. Fukuzumi, J. Mater. Chem., 2002, 12, 2034 RSC.
- D. Hirayama, T. Yamashiro, K. Takimiya, Y. Aso, T. Otsubo, H. Norieda, H. Imahori and Y. Sakata, Chem. Lett., 2000, 570 CrossRef CAS.
- D. Hirayama, K. Takimiya, Y. Aso, T. Otsubo, T. Hasobe, H. Yamada, H. Imahori, S. Fukuzumi and Y. Sakata, J. Am. Chem. Soc., 2002, 124, 532 CrossRef CAS.
- H. Imahori, H. Yamada, S. Ozawa, K. Ushida and Y. Sakata, Chem. Commun., 1999, 1165 RSC.
- H. Imahori, H. Yamada, Y. Nishimura, I. Yamazaki and Y. Sakata, J. Phys. Chem. B, 2000, 104, 2099 CrossRef CAS.
- H. Imahori, H. Norieda, H. Yamada, Y. Nishimura, I. Yamazaki, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 100 CrossRef CAS.
- S. Saha, L. E. Johansson, A. H. Flood, H.-R. Tseng, J. I. Zink and J. F. Stoddart, Small, 2005, 1, 87 CrossRef CAS.
- S. Saha, E. Johansson, A. H. Flood, H.-R. Tseng, J. I. Zink and J. F. Stoddart, Chem.—Eur. J., 2005, 11, 6846 CrossRef CAS.
- K.-S. Kim, M.-S. Kang, H. Ma and A. K.-Y. Jen, Chem. Mater., 2004, 16, 5058 CrossRef CAS.
- M. Morisue, S. Yamatsu, N. Haruta and Y. Kobuke, Chem.—Eur. J., 2005, 11, 5563 CrossRef CAS.
- H. Yamada, H. Imahori, Y. Nishimura, I. Yamazaki and S. Fukuzumi, Adv. Mater., 2002, 14, 892 CrossRef CAS.
- H. Yamada, H. Imahori, Y. Nishimura, I. Yamazaki, T. K. Ahn, S. K. Kim, D. Kim and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 9129 CrossRef CAS.
- H. Imahori, M. Kimura, K. Hosomizu and S. Fukuzumi, J. Photochem. Photobiol., A, 2004, 166, 57 CrossRef CAS.
- H. Imahori, M. Kimura, K. Hosomizu, T. Sato, T. K. Ahn, S. K. Kim, D. Kim, Y. Nishimura, I. Yamazaki, Y. Araki, O. Ito and S. Fukuzumi, Chem.—Eur. J., 2004, 10, 5111 CrossRef CAS.
- V. Chukharev, T. Vuorinen, A. Efimov, N. V. Tkachenko, M. Kimura, S. Fukuzumi, H. Imahori and H. Lemmetyinen, Langmuir, 2005, 21, 6385 CrossRef CAS.
- M. Isosomppi, N. V. Tkachenko, A. Efimov, K. Kaunisto, K. Hosomizu, H. Imahori and H. Lemmetyinen, J. Mater. Chem., 2005, 15, 4546 RSC.
- Y.-J. Cho, T. K. Ahn, H. Song, K. S. Kim, C. Y. Lee, W. S. Seo, K. Lee, S. K. Kim, D. Kim and J. T. Park, J. Am. Chem. Soc., 2005, 127, 2380 CrossRef CAS.
- A. Ikeda, T. Hatano, S. Shinkai, T. Akiyama and S. Yamada, J. Am. Chem. Soc., 2001, 123, 4855 CrossRef CAS.
- T. Konishi, A. Ikeda and S. Shinkai, Tetrahedron, 2005, 61, 4881 CrossRef CAS.
- E. Bustos, J. Manriquez, L. Echegoyen and L. A. Godínez, Chem. Commun., 2005, 1613 RSC.
- M. Lahav, V. Heleg-Shabtai, J. Wasserman, E. Katz, I. Willner, H. Durr, Y.-Z. Hu and S. H. Bossmann, J. Am. Chem. Soc., 2000, 122, 11480 CrossRef CAS.
- C. Luo, D. M. Guldi, M. Maggini, E. Menna, S. Mondini, N. A. Kotov and M. Prato, Angew. Chem., Int. Ed., 2000, 39, 3905 CrossRef CAS.
- D. M. Guldi, F. Pellarini, M. Prato, C. Granito and L. Troisi, Nano Lett., 2002, 2, 965 CrossRef CAS.
- F. B. Abdelrazzaq, R. C. Kwong and M. E. Thompson, J. Am. Chem. Soc., 2002, 124, 4796 CrossRef CAS.
- D. M. Guldi, I. Zilbermann, G. A. Anderson, K. Kordatos, M. Parato, R. Tafuro and L. Valli, J. Mater. Chem., 2004, 14, 303 RSC.
- I. Zilbermann, G. A. Anderson, D. M. Guldi, H. Yamada, H. Imahori and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2003, 7, 357 CrossRef CAS.
- D. M. Guldi, I. Zilbermann, G. A. Anderson, A. Li, D. Balbinot, N. Jux, M. Hatzimarinaki, A. Hirsch and M. Prato, Chem. Commun., 2004, 726 RSC.
- D. M. Guldi, I. Zilbermann, A. Lin, M. Braun and A. Hirsch, Chem. Commun., 2004, 96 RSC.
- S. Conoci, D. M. Guldi, S. Nardis, R. Paolesse, K. Kordatos, M. Prato, G. Ricciardi, M. G. H. Vicente, I. Zilbermann and L. Valli, Chem.—Eur. J., 2004, 10, 6523 CrossRef CAS.
- D. M. Guldi and M. Prato, Chem. Commun., 2004, 2517 RSC.
- D. M. Guldi, J. Phys. Chem. B, 2005, 109, 11432 CrossRef CAS.
- P. G. Hoertz and T. E. Mallouk, Inorg. Chem., 2005, 44, 6828 CrossRef CAS.
- F. Fungo, L. Otero, C. D. Borsarelli, E. N. Durantini, J. J. Silber and L. Sereno, J. Phys. Chem. B, 2002, 106, 4070 CrossRef CAS.
- H. Imahori, J.-C. Liu, H. Hotta, A. Kira, T. Umeyama, Y. Matano, G. Li, S. Ye, M. Isosomppi, N. V. Tkachenko and H. Lemmetyinen, J. Phys. Chem. B., 2005, 109, 18465 CrossRef CAS.
- H. Imahori, J.-C. Liu, K. Hosomizu, T. Sato, Y. Mori, H. Hotta, Y. Matano, Y. Araki, O. Ito, N. Maruyama and S. Fujita, Chem. Commun., 2004, 2066 RSC.
- C.-H. Huang, N. D. McCenaghan, A. Kuhn, J. W. Hofstraat and D. M. Bassani, Org. Lett., 2005, 7, 3409 CrossRef CAS.
- Y. Liu, S. Xiao, H. Li, Y. Li, H. Liu, F. Lu, J. Zhuang and D. Zhu, J. Phys. Chem. B, 2004, 108, 6256 CrossRef CAS.
- H. Imahori, K. Hagiwara, T. Akiyama, M. Aoki, S. Taniguchi, T. Okada, M. Shirakawa and Y. Sakata, Chem. Phys. Lett., 1996, 263, 545 CrossRef CAS.
- H. Imahori and Y. Sakata, Adv. Mater., 1997, 9, 537 CAS.
- H. Imahori and Y. Sakata, Eur. J. Org. Chem., 1999, 2445 CrossRef CAS.
- H. Imahori and S. Fukuzumi, Adv. Mater., 2001, 13, 1197 CrossRef CAS.
- H. Imahori, Y. Mori and Y. Matano, J. Photochem. Photobiol., C, 2003, 4, 51 CrossRef CAS.
- H. Imahori, Org. Biomol. Chem., 2004, 2, 1425 RSC.
- P. V. Kamat, S. Barazzouk, K. G. Thomas and S. Hotchandani, J. Phys. Chem. B, 2000, 104, 4014 CrossRef CAS.
- S. Barazzouk, S. Hotchandani and P. V. Kamat, Adv. Mater., 2001, 13, 1614 CrossRef.
- H. Hotta, S. Kang, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda and H. Imahori, J. Phys. Chem. B, 2005, 109, 5700 CrossRef CAS.
- P. K. Sudeep, B. I. Ipe, K. G. Thomas, M. V. George, S. Barazzouk, S. Hotchandani and P. V. Kamat, Nano Lett., 2002, 2, 29 CrossRef CAS.
- P. V. Kamat, M. Haria and S. Hotchandani, J. Phys. Chem., 2004, 108, 5166 Search PubMed.
- A. Kira, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda, M. Isosomppi, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, Langmuir, 2006, 22, 5497 CrossRef CAS.
- T. Hasobe, H. Imahori, S. Fukuzumi and P. V. Kamat, J. Mater. Chem., 2003, 13, 2515 RSC.
- T. Hasobe, H. Imahori, S. Fukuzumi and P. V. Kamat, J. Phys. Chem. B, 2003, 107, 12105 CrossRef CAS.
- T. Hasobe, P. V. Kamat, M. A. Absalom, Y. Kashiwagi, J. Sly, M. J. Crossley, K. Hosomizu, H. Imahori and S. Fukuzumi, J. Phys. Chem. B, 2004, 108, 12865 CrossRef CAS.
- P. V. Kamat, S. Barazzouk, S. Hotchandani and K. G. Thomas, Chem.—Eur. J., 2000, 6, 3914 CrossRef CAS.
- H. Imahori, T. Hasobe, H. Yamada, P. V. Kamat, S. Barazzouk, M. Fujitsuka, O. Ito and S. Fukuzumi, Chem. Lett., 2001, 784 CrossRef CAS.
- K. Okamoto, T. Hasobe, N. V. Tkachenko, H. Lemmetyinen, P. V. Kamat and S. Fukuzumi, J. Phys. Chem. A, 2005, 109, 4662 CrossRef CAS.
- T. Hasobe, S. Hattori, H. Kotani, K. Ohkubo, K. Hosomizu, H. Imahori, P. V. Kamat and S. Fukuzumi, Org. Lett., 2004, 6, 3103 CrossRef CAS.
- T. Hasobe, S. Hattori, P. V. Kamat, Y. Wada and S. Fukuzumi, J. Mater. Chem., 2005, 15, 372 RSC.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS.
- V. Biju, S. Barazzouk, K. G. Thomas, M. V. George and P. V. Kamat, Langmuir, 2001, 17, 2930 CrossRef CAS.
- H. Imahori, J. Phys. Chem. B, 2004, 108, 6130 CrossRef CAS.
- H. Imahori and S. Fukuuzmi, Adv. Funct. Mater., 2004, 14, 525 CrossRef CAS.
- H. Imahori, K. Hagiwara, M. Aoki, T. Akiyama, S. Taniguchi, T. Okada, M. Shirakawa and Y. Sakata, J. Am. Chem. Soc., 1996, 118, 11771 CrossRef CAS.
- N. V. Tkachenko, C. Guenther, H. Imahori, K. Tamaki, Y. Sakata, S. Fukuzumi and H. Lemmetyinen, Chem. Phys. Lett., 2000, 326, 344 CrossRef CAS.
- H. Imahori, N. V. Tkachenko, V. Vehmanen, K. Tamaki, H. Lemmetyinen, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2001, 105, 1750 CrossRef CAS.
- V. Vehmanen, N. V. Tkachenko, H. Imahori, S. Fukuzumi and H. Lemmetyinen, Spectrochim. Acta, Sect. A, 2001, 57, 2229 CrossRef CAS.
- P. D. W. Boyd and C. A. Reed, Acc. Chem. Res., 2005, 38, 235 CrossRef CAS.
- T. Hasobe, Y. Kashiwagi, M. Absalom, K. Hosomizu, M. J. Crossley, H. Imahori, P. V. Kamat and S. Fukuzumi, Adv. Mater., 2004, 16, 975 CrossRef CAS.
- T. Hasobe, P. V. Kamat, V. Troiani, N. Solladié, T. K. Ahn, S. K. Kim, D. Kim, A. Kongkanand, S. Kuwabata and S. Fukuzumi, J. Phys. Chem. B, 2005, 109, 19 CrossRef CAS.
- T. Hasobe, H. Imahori, S. Fukuzumi and P. V. Kamat, J. Am. Chem. Soc., 2003, 125, 14962 CrossRef CAS.
- T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, S. K. Kim, D. Kim, A. Fujimoto, T. Hirakawa and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216 CrossRef CAS.
- H. Imahori, A. Fujimoto, S. Kang, H. Hotta, K. Yoshida, T. Umeyama, Y. Matano and S. Isoda, Tetrahedron, 2006, 62, 1955 CrossRef CAS.
- H. Imahori, A. Fujimoto, S. Kang, H. Hotta, K. Yoshida, T. Umeyama, Y. Matano and S. Isoda, Adv. Mater., 2005, 17, 1727 CrossRef CAS.
- H. Imahori, A. Fujimoto, S. Kang, H. Hotta, K. Yoshida, T. Umeyama, Y. Matano, S. Isoda, M. Isosomppi, N. V. Tkachenko and H. Lemmetyinen, Chem.—Eur. J., 2005, 11, 7265 CrossRef CAS.
- H. Imahori, K. Mitamura, T. Umeyama, K. Hosomizu, Y. Matano, K. Yoshida and S. Isoda, Chem. Commun., 2006, 406 RSC.
- H. Imahori, K. Mitamura, Y. Shibano, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda, Y. Araki and O. Ito, J. Phys. Chem. B, 2006, 110, 11399 CrossRef CAS.
- T. Hasobe, S. Hattori, P. V. Kamat, Y. Urano, N. Umezawa, T. Nagano and S. Fukuzumi, Chem. Phys., 2005, 319, 243 CrossRef CAS.
- T. Hasobe, S. Hattori, P. V. Kamat and S. Fukuzumi, Tetrahedron, 2006, 62, 1937 CrossRef CAS.
- H. Imahori, M. Arimura, T. Hanada, Y. Nishimura, I. Yamazaki, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 335 CrossRef CAS.
- H. Imahori, Y. Kashiwagi, Y. Endo, T. Hanada, Y. Nishimura, I. Yamazaki, Y. Araki, O. Ito and S. Fukuzumi, Langmuir, 2004, 20, 73 CrossRef CAS.
- S. Kang, T. Umeyama, M. Ueda, Y. Matano, H. Hotta, K. Yoshida, S. Isoda and H. Imahori, Adv. Mater., 2006, 18 DOI:10.1002/adma.200600312.
- S. Barazzouk, S. Hotchandani, K. Vinodgopal and P. V. Kamat, J. Phys. Chem. B, 2004, 108, 17015 CrossRef CAS.
- T. Hasobe, S. Fukuzumi and P. V. Kamat, Angew. Chem., Int. Ed., 2006, 45, 755 CrossRef CAS.
- I. Robel, B. A. Bunker and P. V. Kamat, Adv. Mater., 2005, 17, 2458 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |