Oxidative properties of FeO2+: electronic structure and solvation effects†‡
Manuel J.
Louwerse
and
Evert
Jan Baerends
*
Theoretical Chemistry, Vrije Universiteit Amsterdam, De Boelelaan 1083, 1081 HV, Amsterdam, The Netherlands
First published on 28th November 2006
Abstract
An electronic structure analysis is provided of the action of solvated FeO2+, [FeO(H2O)5]2+, as a hydroxylation catalyst . It is emphasized that the oxo end of FeO2+ does not form hydrogen bonds (as electron donor and H-bond acceptor) with H-bond donors nor with aliphatic C–H bonds, but it activates C–H bonds as an electron acceptor. It is extremely electrophilic, to the extent that it can activate even such poor electron donors as aliphatic C–H bonds, the C–H bond orbital acting as electron donor in a charge transfer type of interaction. Lower lying O–H bonding orbitals are less easily activated. The primary electron accepting orbital in a water environment is the 3σ*α orbital, an antibonding combination of Fe-3dz2 and O-2pz, which is very low-lying relative to the π*α compared with, for example, the σ* orbital in O2 relative to its π*. This is ascribed to relatively small Fe-3dz2 with O-2pz overlap, due to the nodal structure of the 3dz2.The H-abstraction barrier is very low in the gas phase, but it is considerably enhanced in water solvent. This is shown to be due to strong screening effects of the dielectric medium, leading to relative destabilization of the levels of the charged [FeO(H2O)5]2+ species compared to those of the neutral substrate molecules, making it a less effective electron acceptor. The solvent directly affects the orbital interactions responsible for the catalytic reaction.
1. Introduction
The oxoiron(IV) ion (ferryl ion) FeO2+ has long been known to be the active center in heme iron enzymes such as cytochrome P450.1,2 Its activity is generally ascribed to the H-abstraction/oxygen rebound mechanism proposed about 30 years ago by Groves and co-workers.3,4 There is also evidence for some non-heme iron enzymes,5,6 including bleomycin,7 and iron containing zeolites like FeZSM-5,8 that the active intermediates are ironoxo species. Nonetheless, in particular in the classical Fenton reaction9 (oxidation by H2O2 catalyzed by ferrous ions in aqueous solution), the presence and role of ferryl ion has been the subject of long debate.10–13 DFT calculations have indicated that in Fenton chemistry the production of an FeO2+ intermediate seems to be thermodynamically favorable over production of OH˙ radicals,14 which was later substantiated by Car–Parrinello molecular dynamics simulations in water solution.15–17 On the other hand, the occurrence of FeO2+ in Fenton chemistry has very recently been put in doubt.18 The issue appears far from settled.13Significantly, the presence of the FeO2+ moiety has over the past years been experimentally established in a number of non-heme enzymes and complexes. Que and co-workers have characterized and studied the catalytic activity of FeO2+ with tetradentate and pentadentate nitrogen lone pair ligands19,20 (see also Feringa et al.21). Very recently the pentaaquaferryl ion was unequivocally characterized experimentally,18 and its activity as an oxygen atom transfer reagent demonstrated. The electronic structure of the bare FeO2+ (and FeO+) have been the subject of several investigations,22–25 some in connection with gas phase studies of reactivity.23–27 Many discussions of the electronic structure of non-heme complexes of FeO2+ have appeared in recent years.28–35
The purpose of the present paper is twofold. First, we will give a detailed electronic structure explanation of the remarkable capability of the ferryl ion to hydroxylate even the strong aliphatic C–H bonds, in terms of the nature and energies of the participating molecular orbitals (MOs). Second, we will address the issue of the much higher barrier to reaction that has been found in aqueous solution for reaction of FeO2+ with methane36 and methanol,37 compared to the reaction with the penta-aqua complex ([FeO(H2O)5]2+) in the gas phase.
To set the stage, we display in Table 1 a sample of the interaction energies and reaction barriers we have obtained in earlier work for simple substrate molecules like methane,36methanol,37 and hydrogen peroxide28 and for the interaction with water, calculated in the gas phase and in solution. The reaction barriers are for hydrogen abstraction, the first step in the rebound mechanism for hydroxylation. In Fig. 1 some of the reactant complexes are depicted. In Fig. 2 the precise bonding distances and angles are given.
![Pictures of the complexes under study. (a) Methane interacts with [FeO(H2O)5]2+ in a linear configuration. (b), (c) Methanol can interact with [FeO(H2O)5]2+ either via its methyl group (b) or via its hydroxyl group (c), in both cases leading to a very strong interaction. (d) In the gas phase water rotates its oxygen atom towards the oxo-oxygen of FeO2+ to make an O–O interaction.](/image/article/2007/CP/b613182d/b613182d-f1.gif) | ||
| Fig. 1 Pictures of the complexes under study. (a) Methane interacts with [FeO(H2O)5]2+ in a linear configuration. (b), (c) Methanol can interact with [FeO(H2O)5]2+ either via its methyl group (b) or via its hydroxyl group (c), in both cases leading to a very strong interaction. (d) In the gas phase water rotates its oxygen atom towards the oxo-oxygen of FeO2+ to make an O–O interaction. | ||
![Distances and angles in the optimized complexes of [FeO(H2O)5]2+ with methane, methanol, and water, respectively.](/image/article/2007/CP/b613182d/b613182d-f2.gif) | ||
| Fig. 2 Distances and angles in the optimized complexes of [FeO(H2O)5]2+ with methane, methanol, and water, respectively. | ||
Table 1 includes results that demand an explanation. In the first place, except for methane, the interactions with FeO2+ are rather strong: the interactions of water, methanol, and hydrogen peroxide vary from 43 to 81 kJ mol–1, which is 2 to 4 times the strength of an average hydrogen bond. Remarkably, although methanol interacts rather strongly via its methyl group with the FeO2+ ion, the interaction of methane is much weaker.
Methanol may also bind with its hydroxyl group to FeO2+, in which case the bonding is 13 kJ mol–1 weaker than for bonding with the methyl group. However, the interaction is not by hydrogen bond donation to the oxo ligand of the [FeO(H2O)5]2+ complex, and neither is the case for bonding of water. This can be recognized in the orientation of the molecules: the interaction is primarily with the lone pairs on the oxygen atoms of water and methanol.
A very salient feature of Table 1 is the strong increase of the transition state barrier for the H-abstraction in water solution compared to the extremely small barriers in the gas phase. These barriers in solution have been obtained from Car–Parrinello molecular dynamics , using the technique of thermodynamic integration along the reaction path.36,37 They therefore represent free energy barriers.
We will rationalize these findings with the help of electronic structure considerations. In their seminal work, Schröder, Schwarz, Shaik, and co-workers have explained the electronic structure and the reactivity of the related FeO+ ion in the gas phase.23–25 They have stressed the analogy of the electronic structure and the reactivity of FeO+ to that of the O2 molecule: it is a π biradical , and in the optimal reaction path spin-crossings occur leading to a two-state-reactivity (TSR) mechanism.
However, for FeO2+ we will identify as the particular electronic structure feature that lends it its special role in oxygenation catalysis, the availability of an extremely low-lying empty acceptor orbital, the 3σ* orbital, an antibonding combination of the Fe-3dz2 and the O-2pz. In fact, this low-lying σ* LUMO makes FeO2+ more analogous to the F2 molecule than to O2, F2 also being highly reactive owing to a low-lying σ* acceptor orbital. FeO2+ is extremely electrophilic indeed. It activates C–H bonds by acting as an electron acceptor to the C–H σ bonding orbital, even though this orbital is low-lying and in principle a very poor donor orbital. The activation does not at all involve donation of electrons into the C–H σ* orbital; in fact, such donation does not even proceed into the O–H σ* orbital, which is the reason the oxo oxygen does not participate in hydrogen bonding (as H-bond acceptor).
The large solvent effect on the barrier height cannot be understood simply from different solvation stabilization of reactants and transition state . We will show that it is caused by modification of the electronic structure mechanism of the reaction by the solvent environment. The relevant orbital levels of [FeO(H2O5]2+ and of the substrate molecules undergo differential shifts in solution, which modifies interaction strengths and reaction barriers. This is another example of the important role the solvent may play in modifying orbital interaction patterns, as also recently observed in a calculation of pKa’s in water.38
2. Methods
All calculations have been performed with the ADF (Amsterdam density functional) package39–42 using the BLYP43,44 density functional. All electrons are included in the calculations and large STO basis sets are used, which are available in the ADF library of standard basis sets,39 namely the QZ4P set (a quadruple-ζ basis set with four sets of polarization functions) for iron and the TZ2P set (a triple-ζ basis set with two sets of polarization functions) for the other atoms. Additionally, the calculations were corrected for relativistic effects using the zero-order regular approximation (ZORA)45 approach.First, the calculations were performed in the gas phase with the first coordination shell of water molecules around the iron ion included in the calculations, resulting in [FeO(H2O)5]2+. Secondly, for the calculations in solution the same penta-aqua oxo complexes were used, and the solvent was modeled as a dielectric, using the conductor-like screening model (COSMO)46,47 as implemented48 in ADF. As parameters for the COSMO model, the following radii were used: 1.6 Å for Fe2+, 1.7784 Å for O, 1.3 Å for H, and 1.4 Å for the solvent molecules. The dielectric constant used was 78.4. We have verified that this model for the solvent yields the same effects as found earlier in Car–Parrinello dynamics36,37 work, where explicit solvent water molecules have been included which were treated fully quantum mechanically. The COSMO model is used to demonstrate that the observed solvent effects can be attributed to long-range dielectric screening effects.
We analyze the bonding of this [FeO(H2O)5]2+ complex with water, methane, and methanol, respectively, using the fragment orbital analysis as it is implemented in the ADF package.49 For each complex the geometry is first optimized, and in this optimized geometry each complex is divided into two fragments: the [FeO(H2O)5]2+ complex and the H2O, CH4, or CH3OH molecule. For these fragments the molecular orbitals are calculated, and then the total electronic structure is recalculated based on the fragment orbitals. In this way the contributions of the fragment orbitals to the complex molecular orbitals are found. Also, the interaction energy can be decomposed49–51 into Pauli repulsion, electrostatic interaction, and orbital interactions between the fragments plus the energy cost to deform the fragments from their optimized geometries in the isolated fragment to the geometry they obtain in the complex.
The fragment orbital analysis can only be performed with one set of orbitals on each fragment, i.e. the ones obtained in spin-restricted calculations on the fragments. It cannot use simultaneously two sets of orbitals, such as the α and β spinorbitals obtained in spin-unrestricted calculations. Therefore for the [FeO(H2O)5]2+ fragment the spin-restricted orbitals were determined. The proper S = 2 ground state was then prepared by assigning the correct spin-unrestricted electron occupations (excess α spin electrons, see next section). This ground state , however, lacks the energy lowering (“spin polarization”) from further self-consistent optimization of the spinorbitals. This leads to a systematic overestimation of the interaction energy by an amount of 28–29 kJ mol–1 (depending on the exact fragment geometry). We corrected the orbital interaction term for this overestimation, so that this energy term and the total bond energy properly reflect the energy with respect to the spin polarized [FeO(H2O)5]2+ complex. All reported values in this article for orbital interactions and total interactions have been corrected in this manner.
The energy levels of the substrate molecules are strongly influenced by the net charge of the ironoxo complex and shift down considerably when these molecules approach the charged complex. Even though orbital interaction diagrams are only qualitative, the orbital levels should convey an intuitive grasp of the orbital mixings, and therefore the abovementioned shift should at least qualitatively be taken into account. So in the orbital schemes (Fig. 7, 9, and S2–S5) the water, methane, and methanol orbitals are all shifted (in each case all levels by the same constant) from the isolated molecule values. The constant is determined as the average shift of the non-interacting orbitals of a substrate molecule as calculated in the complex. Note that the shift can differ for different molecules, presumably because the bond distances are different.
When such a shifted HOMO starts to mix with the FeO2+LUMO , the relative levels of these two orbitals change somewhat more, because of the charge transfer that occurs. Therefore, the abovementioned shift, as depicted in the figures and given in the tables, is only qualitative. In the final self-consistent field, the substrate HOMO will be slightly lower in energy and the FeO2+LUMO slightly higher.
3. Results: transition state barriers for H-abstraction in the gas phase
Electronic structure of gas phase FeO2+ and [FeO(H2O)5]2+
First we will discuss the FeO2+ species itself. In Fig. 3a a schematic orbital diagram is shown for the FeO2+ species in the gas phase. A detailed description of the composition of these orbitals can be found in Table 1 of ref. 28. The 1σ orbital is not involved in the bonding, it is practically 100% O-2s. The iron and the oxygen atoms form bonding and antibonding orbitals of σ symmetry, 2σ (dz2 + pz) and 3σ* (dz2 – pz) and of π symmetry, 1π (dxz,yz + px,y) and 2π* (dxz,yz – px,y), where we take the Fe–O bond as the z axis. Although in transition metal complexes the antibonding combinations are commonly denoted as the (formally) “d orbitals”, we note that in this case the orbitals are actually strong mixtures, indicating almost covalent bonds.28 In fact, the bonding combinations 2σ and 1π are the ones with the larger d contributions (53% dz2 and 67% dxz,yz, respectively). The dδ orbitals (dx2–y2 and dxy), on the other hand, are purely nonbonding.![Orbital level diagram for bare FeO2+ and for the FeO orbitals in [FeO(H2O)5]2+. Both molecules are in the S = 2 ground state. Because of the high spin state, there is a large energy difference between α- and β-spin orbitals. In both cases the lowest empty orbital that is involved in Fe–O bonding is the 3σ*α orbital.](/image/article/2007/CP/b613182d/b613182d-f3.gif) | ||
| Fig. 3 Orbital level diagram for bare FeO2+ and for the FeO orbitals in [FeO(H2O)5]2+. Both molecules are in the S = 2 ground state . Because of the high spin state, there is a large energy difference between α- and β-spin orbitals. In both cases the lowest empty orbital that is involved in Fe–O bonding is the 3σ*α orbital. | ||
The 2σ and 1π are doubly occupied, with both spin α and spin β electrons, but the 2π* is only occupied with spin α electrons, as in O2. Since the dδ orbitals are also only occupied with α electrons, there are four unpaired α spin electrons, i.e. S = 2. The surplus of α electrons causes a strong stabilizing field due to the exchange interaction, so all α levels are considerably lower than the corresponding β levels.
The 3σ*α is the lowest unoccupied orbital and the important frontier orbital of this moiety. The empty β spin dδ orbitals are actually at practically the same energy, but will not play a role since they are not involved in the Fe–O bonding, and will be shielded from an incoming substrate molecule by the equatorial ligands. The 2π*β orbitals and the 3σβ are also low-lying empty orbitals (the important orbitals for the case of FeO+), but these orbitals are less important as acceptor orbitals than the 3σ*α, since they are roughly 1 eV higher in energy. The 3σα orbital has considerable amplitude at the oxo end (it has 48% O-2pz character, versus 37% Fe-dz2). Its particularly low energy and the large amplitude at the oxo group cause it to act as an electron acceptor in strong charge transfer interactions, even with relatively low-lying donor orbitals such as the C–H bonding orbital. As we will see below, this gives FeO2+ its unique capability to activate such bonds.
Due to its occupation pattern (1π42π2) and TSR mechanism, FeO+ has been compared to O2.23–25 Because for FeO2+ there is no TSR mechanism, but instead the reactivity is driven by the very low-lying empty σ* orbital, FeO2+ is more analogous to the F2 molecule.
Of course, the interaction with a substrate molecule will be modified when the FeO2+ is surrounded by ligands. In Fig. 3b we give the level scheme for [FeO(H2O)5]2+, and in Fig. 4 the orbitals are depicted. A complete description of all molecular orbitals in this complex can be found in Table 2 of ref. 28. In the level scheme, two blocks with H2Olone pair levels are indicated, in-between the FeO2+-derived levels: the σ (2a1) lone pairs and the π (1b2) lone pairs.
![Pictures of the FeO orbitals in the [FeO(H2O)5]2+ complex. Depicted are the α orbitals; for the β orbitals the mixing with the water orbitals is somewhat different. The plane of drawing is the xz plane (Fe–O is along the z axis). For the 1δxy and 1δx2–y2 orbitals the plane of drawing is the equatorial (xy) plane and for 1πy and 2πy* it is the yz plane.](/image/article/2007/CP/b613182d/b613182d-f4.gif) | ||
| Fig. 4 Pictures of the FeO orbitals in the [FeO(H2O)5]2+ complex. Depicted are the α orbitals; for the β orbitals the mixing with the water orbitals is somewhat different. The plane of drawing is the xz plane (Fe–O is along the z axis). For the 1δxy and 1δx2–y2 orbitals the plane of drawing is the equatorial (xy) plane and for 1πy and 2πy* it is the yz plane. | ||
The main change in the oxoiron(IV) levels with respect to the bare FeO2+ is for the dδ levels. The dx2–y2, with lobes along the axes, is pushed up by antibonding interaction with the σ lone pairs of the equatorial H2Os, cf. the antibonding character displayed in Fig. 4 for the dx2–y2. As already highlighted in ref. 28, the higher lying π lone pairs of the equatorial H2O push the dxy orbitals down in a bonding interaction, as clearly visible in Fig. 4 for the dxyα orbital. As a consequence, there is a large splitting (2.7 eV) between the dx2–y2α and dxyα orbitals. The situation is different for the higher lying unoccupied dx2–y2β and dxyβ orbitals. These orbitals are both pushed up by antibonding interactions with the H2Olone pairs, the dxyβ somewhat less by the π interaction with the π lone pairs than the dx2–y2β by the σ interaction with the σ lone pairs. As a result, the dx2–y2β and dxyβ levels are split by only 1.1 eV, and the dxyβ is clearly the lowest lying empty orbital.
As noted before, the dδ orbitals, although important for the interaction with the water ligands, do not play a role in the interaction with substrate molecules. However, the effects we have been discussing are important for the spin state of the system. When more strongly σ donating equatorial ligands are used, the dx2–y2α may be pushed above the dxyβ, so that the lowest state would correspond to an empty dx2–y2α and occupied dxyβ. The dxy is then doubly occupied, and the total spin becomes S = 1 (only two unpaired 2π* electrons), with the consequence that there is less relative stabilization of the α spin levels. Because the dx2–y2α and dxyβ levels are not involved in the Fe–O bond, nor in the interaction with substrate molecules, one might assume that the spin-state of FeO2+ is not playing a crucial role in its reactivity.29 However, because the energy level of the 3σ*α orbital is of prime importance for the reactivity (as we will show), the decrease in the stabilization of the α levels when the spin-state changes will still have an important effect on the chemistry.
Another change induced by the H2O ligands, in which the axialwater ligand plays a role, is the relative destabilization of the 3σ*α with respect to the 2π*α and 2π*β orbitals. As the precise energy level of the 3σ*α orbital is very important, the axial ligand could play a major role in influencing the reactivity of FeO2+. In the case of water ligands the 3σ*α remains the most important acceptor orbital, but one could also conceive of ligand environments where the role of the 3σ*α and the 2π*β orbitals is reversed, leading again to the TSR mechanism as found for FeO+; this has been found, for instance, in the case of cytochrome P450.2
We will return to the role of ligands other than H2O elsewhere.52
We wish to emphasize that the interaction of substrate molecules with FeO2+ is not by way of hydrogen bonding to the oxo oxygen. To act as a H-bond acceptor, FeO2+ would have to be able to donate electrons into the σ* orbital of the O–H bond of the H-bond donor. The 2π*α orbital is the only available orbital which would be suitable for this type of interaction. It is, however, so low-lying that it cannot set up effective hydrogen bonds; moreover, the 2π* is only half occupied (with α spin electrons). All interactions with [FeO(H2O)5]2+ are by way of a charge transfer into the 3σ*α orbital.
In view of the importance of the 3σ*α orbital for the action of FeO2+ as strong electron acceptor, we give a contour plot in Fig. 5 of the 3σ*α orbital of [FeO(H2O)5]2+. The 3σ* orbital is an antibonding combination of the dz2 orbital of Fe and the pz orbital of the oxo-oxygen. There is some hybridization at the O which yields the 3σ* a pronounced outward lobe. However, the antibonding of the O-2pz with dz2 is mitigated by the positive overlap that the pz can obtain with the equatorial torus of the dz2. This bonding effect is clearly visible in Fig. 5. As a result, the overlap between the dz2 and the O-2pz is only 0.10 (e.g. in O2 the pz–pz overlap is 0.23), and consequently, the 3σ* orbital is lower in energy than one would generally expect for an antibonding pz–pz σ* orbital: the present 3σ*α orbital is only 2.0 eV above the 2π*α orbitals, while for O2, for instance, the gap is 9.2 eV, and for the very reactive F2 it is still 3.3 eV.
![Contour plot of the 3σ*α orbital of [FeO(H2O)5]2+ in the plane of the Fe–O bond. There is both bonding and antibonding overlap between the Fe-3dz2 and the O-2pz orbitals, explaining the low energy of this antibonding orbital.](/image/article/2007/CP/b613182d/b613182d-f5.gif) | ||
| Fig. 5 Contour plot of the 3σ*α orbital of [FeO(H2O)5]2+ in the plane of the Fe–O bond. There is both bonding and antibonding overlap between the Fe-3dz2 and the O-2pz orbitals, explaining the low energy of this antibonding orbital. | ||
The substrate molecules: methane, methanol and water
A few comments on the frontier orbitals of the substrate molecules water, methane, and methanol are in order. In the figures and tables, we use the orbital energies of these molecules in exactly the configurations they have in the complexes with [FeO(H2O)5]2+, see Fig. 1. This means, for instance, that for methane one of the C–H bonds is elongated, so that the degeneracy among the T2 set of C–H bonding orbitals is lifted, and the orbital most localized on the elongated C–H is at higher energy (–9.07 eV) compared to the other still degenerate orbitals (–9.35 eV). In water the frontier orbitals are the σ and π lone pairs, at –9.25 eV (σ lone pair) and at –7.15 eV (π lone pair).Methanol has an interesting HOMO . It is a π* antibonding combination of a π orbital (with respect to the H–O–C plane) at the OH group and a minus combination of C–H bonding orbitals at the CH3 group (see Fig. 6). On account of its antibonding character, this orbital is relatively high lying (–6.00 eV), and therefore more amenable to interaction with the LUMO of FeO2+ (both via the OH and the CH lobes) than the HOMOs of the other substrate molecules. Analogous to water, there is also a σ lone pair (in the H–O–C plane), which is the HOMO –1 at –7.83 eV. It also has considerable amplitude at the CH3 group.
 | ||
| Fig. 6 The HOMO of methanol is an antibonding combination of the C–H bonding orbitals and the O lone pair. As a result of the antibonding character, this is a relatively high-lying orbital with a C–H bond contribution. | ||
It is important to note that, due to the antibonding interaction between the C–H bonds and the π lone pair at oxygen, the HOMO of methanol is much higher in energy than the HOMO of methane, although both have partly C–H bond character. As a result, methanol has C–H bond character (20–25%, depending on the geometry) in a relatively high-lying orbital, which makes the C–H bonds of methanol easier to activate than the C–H bonds of methane.
Interaction of [FeO(H2O)5]2+ with methane, methanol, and water
The geometries of the optimized complexes are shown in Fig. 2. The bonding with the C–H bonds of methane and methanol leads to a practically linear Fe–O⋯H–CR3 arrangement, but the OH-bonded methanol and the water molecule interact at an angle with the Fe–O bond. However, for all four complexes we studied, the potential energy surfaces are very flat between linear and bent geometries, and the precise geometries are likely determined by subtle electrostatic effects; the overlap with the 3σ*α orbital of [FeO(H2O)5]2+ is hardly influenced by these angles.Note that both the water and the OH-bonded methanol are interacting via their oxygen π lone pairs (the HOMOs ). Water rotates to optimize the overlap with its lone pair, but even the interaction of methanol occurs via its O-2pπ orbital. Indeed, the hydrogen is more or less in-between the two oxygen atoms, but it does not point directly to the oxo-oxygen and the overlapping orbital is the O-2pπ orbital of methanol. When we force the methanol into an orientation analogous to the orientation of the water molecule, the energy is only marginally higher.
For complexes of [FeO(H2O)5]2+ with substrate molecules the bonding interaction is characterized by a charge transfer from the HOMO of the substrate molecule into the 3σ*α LUMO of FeO2+. We studied the interactions with water, methane, and methanol. The important difference between these molecules, causing the differences in interaction strengths seen in Table 1, is the energy of the HOMO : the HOMO can interact more strongly with the unoccupied acceptor orbital when it lies at higher energy. The activation of a C–H bond by electron donation out of the C–H bonding orbital is usually weak due to the low energy of this type of orbital. However, the 3σ*α LUMO of [FeO(H2O)5]2+ is particularly low-lying, as explained before, and is therefore capable of this feat.
The double positive charge of [FeO(H2O)5]2+ makes it difficult to compare the 3σ* energy to that of the HOMOs of the neutral substrate molecules. In Fig. 7 a schematic picture is shown of the orbital interaction diagrams for the interactions of [FeO(H2O)5]2+ with methanol, methane, and water. In this schematic picture, only the α-spin orbitals are drawn, and for the [FeO(H2O)5]2+–molecule complex only the molecular orbitals resulting from the HOMO –LUMO interactions are included. The net positive charge on the [FeO(H2O)5]2+ unit stabilizes the orbitals of the substrate molecules when they approach (see section 2), and this lowering is reproduced in the figure. Detailed orbital interaction diagrams and tables with the mixing percentages for all orbitals are given in the ESI.‡
![Schematic orbital interaction diagrams for interactions with [FeO(H2O)5]2+, with only α electrons shown. To the right the orbital energies of the isolated substrate molecules, and in the next columns the stabilized orbital energies due to the approach to the charged [FeO(H2O)5]2+, see text. Only the mixings of the substrate HOMOs with the [FeO(H2O)5]2+LUMO are indicated. The complete diagrams are given in the ESI.](/image/article/2007/CP/b613182d/b613182d-f7.gif) | ||
| Fig. 7 Schematic orbital interaction diagrams for interactions with [FeO(H2O)5]2+, with only α electrons shown. To the right the orbital energies of the isolated substrate molecules, and in the next columns the stabilized orbital energies due to the approach to the charged [FeO(H2O)5]2+, see text. Only the mixings of the substrate HOMOs with the [FeO(H2O)5]2+LUMO are indicated. The complete diagrams are given in the ESI.‡ | ||
The important quantities are the mixing percentages with which the fragment orbitals enter the complex orbitals. For CH4 the (shifted) HOMO lies sufficiently far below the FeO2+LUMO that only a small (but unmistakable) admixture (11%) of the 3σ*α into the C–H bond orbital occurs, corresponding to a complexation energy of only –9 kJ mol–1. For methanol, the HOMO is at considerably higher energy than the CH4HOMO , so a much stronger charge transfer into the 3σ*α may be expected. Indeed, both for complexation with a C–H bond (–70 kJ mol–1), and complexation with oxygen lone pairs (–57 kJ mol–1), the bonds are much stronger than for CH4. For water the shifted HOMO energy lies in-between those of CH4 and CH3OH, and the charge transfer interaction is similar to that of CH4, leading to a very comparable orbital interaction energy. The large difference in the strength of the complexation of water compared to methane (–43 vs. –9 kJ mol–1) is a result of the electrostatic interaction (see discussion below). We stress again that the water–oxo bond is not a hydrogen bond, but a charge transfer from the HOMO of water into the LUMO of FeO2+.
We now substantiate our picture of the bonding in these cases, in particular the importance of the donor–acceptor orbital interactions, with the data given in Tables 2 and 3. In Table 2, the total interaction energy is decomposed into deformation energy (the energy required to deform a free molecule to the geometry it has in the complex), Pauli repulsion, electrostatic interactions, and orbital interactions (here corresponding to the charge transfer interaction). In Table 3 the charge transfer is further analyzed. We have tried to quantify the effect of the 2+ charge of the FeO2+ on the HOMO orbital energies of methane, methanol, and water in the manner indicated earlier (the amount by which the orbitals shift is estimated by taking the average shift of the non-interacting orbitals for each molecule). Also, the overlap of the HOMO of the small molecule with the LUMO of [FeO(H2O)5]2+ has been calculated, as this influences the total strength of the interaction. The trends in the orbital interaction term in the interaction energy can be rationalized using these data.
| CH4 | CH3OH | HOCH3 | H2O | |
|---|---|---|---|---|
| HOMO (isolated molecule)/eV | –9.1 | –6.0 | –6.0 | –7.1 |
| Shifted HOMO in field of 2+ charge/eV | –14.1 | –13.0 | –12.8 | –13.7 |
| Overlap of HOMO with FeO LUMO | 0.057 | 0.045 | 0.017 | 0.035 |
| Admixing of FeO LUMO /% | 11 | 45 | 42 | 13 |
| Orbital interaction/kJ mol–1 | –31 | –155 | –100 | –32 |
| Total interaction/kJ mol–1 | –9 | –7 | –57 | –43 |
| H-abstraction barrier/kJ mol–1 | 23 | 2 | 22 | — |
When we compare methane to methanol (both interacting via their C–H bond), we see a huge difference in the strength of the orbital interaction. This is a direct result of the fact that the HOMO of methanol is at higher energy, also after the stabilization by the 2+ charge, resulting in an orbital interaction of 155 kJ mol–1 for methanol, versus 31 kJ mol–1 for methane. This effect is partly cancelled by the larger deformation energy and larger Pauli repulsion. This is quite typical: stronger orbital interactions that lead to stronger bonding also lead to shorter bond lengths. So the bonding distance is slightly shorter in the case of methanol, causing larger deformation energies and larger Pauli repulsion. This partly counterbalances the stronger orbital interaction, but methanol nevertheless binds many times stronger than methane, purely because of the difference in the relative orbital energies.
When methanol interacts with FeO2+via its OH group, the orbital interaction is some 55 kJ mol–1 weaker. This is because in this orientation the overlap of the π* HOMO of methanol and the 3σ* LUMO of FeO2+ is considerably smaller (0.017 versus 0.045). Naturally, the orbital levels are virtually the same as in the CH-bonding configuration, and since in the final self-consistent field there is near degeneracy between the 3σ* and the (stabilized) HOMO of methanol, in both cases the amount of mixing comes close to 50%. However, because of the smaller overlap, the strength of the orbital interaction is weaker in the OH-bonding complex. Note that due to the weaker bonding as a result of the weaker orbital interaction, the Pauli repulsion is also weaker and the deformation energy smaller, so the net difference with the CH-bonding complex is diminished to only 13 kJ mol–1.
Finally, we consider the interaction of [FeO(H2O)5]2+ with a water molecule. In this case, in the optimized geometry, the water molecule orients its π lone pair, the HOMO , towards the FeO2+. Comparing to methane, we note that the orbital energy of the waterHOMO is more favorable, but the overlap is less favorable, and the orbital interaction strength of 32 kJ mol–1 is very similar to the 31 kJ mol–1 of methane. However, since water has a dipole, the electrostatic interaction with the charged [FeO(H2O)5]2+ is much stronger, and the Pauli repulsion is smaller (smaller overlaps), so the total interaction energy (–43 kJ mol–1) is significantly larger than for methane.
Bond activation
We have proposed a model for the reactivity of FeO2+ based on its ability to accept electron charge in its low-lying LUMO . We were also able to explain differences in interaction energies with various substrates on the basis of the relative orbital energies of the species involved. We will now relate this result to the height of the reaction barriers for H-abstraction as given in Table 3. These barriers were calculated by optimizing the transition state for H-abstraction, and were found to be extremely low, ranging from only 2 to 23 kJ mol–1.The effectiveness of the C–H bond activation by FeO2+, by charge transfer from the HOMO of the substrate molecule into the LUMO of FeO2+, will depend on the relative orbital energies. For methanol, with its high-lying HOMO , the charge transfer is so large in the transition state that almost a full electron is transferred. As the HOMO of methanol has a large contribution from the C–H bond, this means that the C–H bond is considerably weakened. The O–H bond of methanol is much less activated by the charge transfer interaction with FeO2+ simply because the O–H bond is not contributing to the HOMO (see Fig. 6).
For methane it seems surprising that the C–H bond is activated at all, as the complexation energy and the charge transfer in the reactants complex is small. Nevertheless, the H-abstraction barrier is still as low as 23 kJ mol–1, because during the stretching of the C–H bond the charge transfer strongly increases and stabilizes the transition state . When the C–H bond lengthens, its orbital energy increases strongly, so in the transition state the charge transfer is much stronger than in the reactants complex. We followed the charge transfer during the reaction, as shown in Fig. 8. The reaction coordinate ξ was defined as the relative position of the hydrogen between the carbon and the oxygen atoms (eqn (1)).
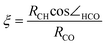 | (1) |
![Charge transfer (Mulliken gross population in 3σ* LUMO) during H-abstraction from CH4 by [FeO(H2O)5]2+, plotted up to the transition state. For the definition of the reaction coordinate, see text. The orbital energy of the CH4HOMO is also displayed.](/image/article/2007/CP/b613182d/b613182d-f8.gif) | ||
| Fig. 8 Charge transfer (Mulliken gross population in 3σ* LUMO ) during H-abstraction from CH4 by [FeO(H2O)5]2+, plotted up to the transition state . For the definition of the reaction coordinate, see text. The orbital energy of the CH4HOMO is also displayed. | ||
4. Results: the effect of solvation on charge transfer and transition state barriers
We have noticed on previous occasions36,37 that in water solution the barrier for H-abstraction is much higher than in the gas phase. This effect was found in Car–Parrinello MD calculations, where the solvent water molecules were represented explicitly and quantum mechanically. An increase of the barrier in solution compared to gas phase, which often occurs with charged reactants, is usually caused by less solvation stabilization in the transition state compared to the reactants complex. In our previous work,36 we have considered the possibility that this differential solvation was causing the barrier increase. However, this could not be established unambiguously as the source of the solvent effect in the present case. We will now consider how the insight we have gained in the electronic structure aspects of the reaction, helps us understand the significant solvent effect for the H-abstraction reaction.In water solution the net charge of the [FeO(H2O)5]2+–molecule complex will induce a reaction field in the solvent, which leads to a destabilization of the orbitals relative to the gas phase. So all orbitals, both of the oxoiron complex itself and of the coordinated substrate molecule, shift upwards in energy. However, one may expect this effect to be largest close to the charged centre. In that case, the orbitals of [FeO(H2O)5]2+, including its 3σ* LUMO , are more destabilized than the orbitals of the substrate molecule. Of course, such a change in the relative energies of the orbitals of the two moieties would imply a change in the charge transfer interaction, resulting in a weakening of the interaction strength and of the stabilization of the transition state . In this model, the water solvent directly affects the electronic interactions and hence the reactivity of [FeO(H2O)5]2+. The effect is not caused by specific interactions of water molecules with the reactants complex and the transition state , but is purely caused by long-range dielectric screening effects of the bulk solvent on the relative level positions of the reactants. Therefore, it should already occur when the solvent is represented by a polarizable continuum model.
To investigate this model, we have repeated the calculations of the reactants complexes and the transition states with a modelled dielectric around the reactants, using a conductor-like screening model (COSMO). The dielectric constant has been chosen as that of water, ε = 78.4. In these calculations we used the same penta-aqua oxo complexes and transition states as before, only now surrounded by a dielectric and reoptimized in this environment.
Fig. 9 contains the geometrical parameters of the reactants complexes in the COSMO environment. The distance between the oxo group and the H of the substrate is much larger than without dielectric (compare to Fig. 2). This is in agreement with the much smaller interaction energies, as given in Table 4. This table also shows that the transition state barriers have strongly increased. The H-abstraction barrier increases in the solvent by 69, 33, and 51 kJ mol–1 for methane, methanol CH-bonded, and methanol OH-bonded, respectively.
![Distances and angles in the [FeO(H2O)5]2+ complexes with methane, methanol, and water, respectively, optimized in the presence of the COSMO (ε = 78.4) environment.](/image/article/2007/CP/b613182d/b613182d-f9.gif) | ||
| Fig. 9 Distances and angles in the [FeO(H2O)5]2+ complexes with methane, methanol, and water, respectively, optimized in the presence of the COSMO (ε = 78.4) environment. | ||
When the solvation is taken into account, simulated with the COSMO model, the complexation of methanol (at the OH group) and water appears to change qualitatively, occurring via the O–H bonds rather than the oxygen lone pairs. However, a hydrogen bond does not form; the interaction is a charge transfer in both cases (from the substrate molecule into the 3σ* orbital of FeO2+).
To verify that the solvent indeed acts by way of differential level shifts of the ironoxo complex and the substrate molecules, Table 4 gives the orbital energies of the relevant frontier orbitals , and Fig. 10 shows orbital interaction diagrams for gas phase and solvated systems in the case of methanol interacting with [FeO(H2O)5]2+. Table 4 shows that the orbital energies of the isolated neutral substrate molecules in the COSMO calculation differ very little from the gas phase values in Table 3. The energy levels of these orbitals shifted by the field of the [FeO(H2O)5]2+ species are also given, again using the lower lying non-bonding orbitals to obtain an estimate for this shift. Remarkably, the downward shift is now almost negligible, so the screening effect of the solvent on the 2+ charge of [FeO(H2O)5]2+ is apparently very effective. This means that the orbitals of the substrate molecules are lying considerably higher (5–7 eV) in the COSMO-surrounded complexes compared to the complexes in the gas phase, cf. Table 4. However, the 3σ*α of charged [FeO(H2O)5]2+ shifts upwards even more, from –13.6 eV in the gas phase to –5.4 eV in the COSMO environment. So there is a considerable differential shift of the substrate levels and the [FeO(H2O)5]2+ levels upon COSMO solvation: the latter have undergone a relative destabilization with respect to the substrate orbitals. As a result, the 3σ*α of [FeO(H2O)5]2+ is no longer an effective acceptor orbital for the HOMO of methanol. The effect is quite large, the admixture of the 3σ*α into the methanolHOMO in the reactants complex, which was quite strong in the gas phase (45%), is strongly reduced to only 2% in water solution, with a concomitant reduction in complexation energy from –70 to only –9 kJ mol–1.
![Schematic orbital interaction diagrams showing the effect of water solution on the interactions of [FeO(H2O)5]2+. The net charge is shielded by the dielectric effect of the solvent, causing the orbitals to shift upwards in energy. Because this shift is largest for the charged fragment, [FeO(H2O)5]2+, the methanol orbitals are relatively stabilized.](/image/article/2007/CP/b613182d/b613182d-f10.gif) | ||
| Fig. 10 Schematic orbital interaction diagrams showing the effect of water solution on the interactions of [FeO(H2O)5]2+. The net charge is shielded by the dielectric effect of the solvent, causing the orbitals to shift upwards in energy. Because this shift is largest for the charged fragment, [FeO(H2O)5]2+, the methanol orbitals are relatively stabilized. | ||
Such a small complexation energy in the solvent environment does not mean that the transition state cannot be lowered considerably by charge transfer interaction with [FeO(H2O)5]2+. As previously observed, during the reaction the C–H bond is elongated and the corresponding orbital shifts upward. It will then start to interact more strongly with the 3σ*α LUMO of [FeO(H2O)5]2+, so there will be a much larger interaction energy of methanol with [FeO(H2O)5]2+ in the transition state , leading to the still modest barrier of 35 kJ mol–1.
The weakened complexation interactions in the calculations with the COSMO dielectric agree with observations in the Car–Parrinello simulations36,37 with explicit water molecules: Although the interaction energy could not be measured in the simulations, it was observed that none of the substrates formed a stable complex with FeO2+, and each substrate was replaced by a water molecule. This is in agreement with interactions of 6–9 kJ mol–1 for methane and methanol and 19 kJ mol–1 for water (Table 4).
The reaction barriers found in the COSMO calculations compare very well, both for methane and for methanol, with the free energy barriers found in the Car–Parrinello simulations. This indicates that entropy effects are fairly small and that the solvation effect can indeed be explained from the effect of the dielectric screening on the electronic structure mechanism of the reaction.
It has been suggested that solvents with smaller dielectric constants, which will exhibit smaller screening effects, may be expected to lower the barrier for H-abstraction considerably compared to water.36 We have verified this conjecture by calculating the barrier for our model substrates with increasingly less polar solvents, represented in the COSMO model with dielectric constants 24.3 (ethanol) and 4.8 (chloroform). For simplicity we neither changed the first coordination sphere ligands (water molecules) nor the COSMO parameter for the radius of the solvent molecules. Table 5 displays the calculated barriers. In agreement with our explanation for the mechanism of the solvent effect, the barriers are considerably lowered, in particular for the low dielectric constant of 4.8 (chloroform). However, the effect for ethanol solvent is small. A more effective method, possibly in combination with tuning the dielectric effect of the solvent, may be to influence the precise energy level of the 3σ* orbital by varying the ligands, in particular the axial ligand.52
5. Conclusion
The formal description of iron(IV)oxo as Fe4+ and O2– suggests that the oxo oxygen would be nucleophilic. However, the FeO bond in FeO2+ has a strong covalent character, and FeO2+ actually turns out to be an extremely electrophilic (acidic) species. Therefore, it is not capable of acting as an electron donor and forming hydrogen bonds with H-bond donors. Its extreme electrophilicity makes FeO2+ an electron acceptor even for poor electron donors such as C–H bond orbitals. Strong charge transfer interactions are established, in which charge is donated out of the HOMO (the C–H bond orbitals) of a substrate into the 3σ*α LUMO of FeO2+. As a result of the considerable charge transfer (up to 50%) out of the C–H bond, the C–H bond is strongly activated for the rebound mechanism in hydroxylation of C–H bonds.The actual strength of the interaction and the effectiveness of the activation depend on the precise level of the HOMO of the incoming molecule: When all occupied orbitals of the substrate molecule are significantly lower lying in energy than the FeO2+LUMO , the charge transfer is only moderate, and so is the activation. However, when the substrate HOMO lies at equal energy or even higher than the FeO2+LUMO , a huge charge transfer occurs. Typically, the transition state is stabilized even more, because the C–H bond orbital shifts upwards as a result of the C–H bond lengthening that occurs in the transition state , leading to surprisingly low reaction barriers. For gas phase [FeO(H2O)5]2+ the H-abstraction barriers range from a stunning 2 kJ mol–1 for methanol to 23 kJ mol–1 for methane.
We have analyzed the electronic structure origin of the remarkably effective electron acceptor property of FeO2+. Of course, the effect of the 2+ charge is important, though reduced by screening effects of the solvent (see below). However, the LUMO of FeO2+ is particularly low because the antibonding character of the σ* of FeO2+ is less strong than the antibonding character in, for instance, a 2pz–2pz antibonding orbital. The σ* is therefore not driven up to such high energies as for instance the 2pz–2pz antibonding orbital of O2.
As a result, the chemistry of FeO2+ is completely driven by this low-lying σ* orbital. The related FeO+ ion is believed to react via a two-state-reactivity (TSR) mechanism and is commonly compared with the O2 molecule (being a π biradical ),23–25 and for FeO2+ complexed with a heme group, for instance in cytochrome P450, the same mechanism has been suggested.2 However, for high-spin FeO2+ the extremely low-lying empty σ* orbital is much more important than the π biradical character. In this respect FeO2+ is more analogous to the F2 molecule.
We elaborate further on the differences between low-spin or high-spin and heme or non-heme FeO2+ elsewhere.52
The gas phase barriers for the H-abstraction by [FeO(H2O)5]2+ are much lower than those in water solvent.36,37 This difference is not caused by specific (H-bond) interactions of outer sphere water molecules, but by long range screening effects of the dielectric medium, which are rather strong for a 2+ charged species like [FeO(H2O)5]2+. The polarization of the dielectric affects the relative orbital energies. The 3σ* LUMO of [FeO(H2O)5]2+, which was so effective in the C–H bond activation in the gas phase, is shifted almost out of reach for the substrate HOMOs by the solvation. This finding highlights the sensitivity of the activation of C–H bonds to a truly low-lying acceptor orbital, which in the form of the FeO2+ 3σ*α is present in the gas phase, but is much less available in solution. Of course, when the C–H bond is lengthened and the C–H bonding orbital rises in energy, the interaction with 3σ* does set in, and the H-abstraction barrier, although much higher than in the gas phase, is still relatively modest, in particular for methanol in the CH-bonded geometry.
We emphasize that instead of the more common solvation effect of different solvation energies for reactants and transition state , here the solvent influences directly the electronic structure mechanism of the reaction. Since it is a long-range screening effect, an implicit solvent model of solvation (like COSMO) suffices to account for this effect almost quantitatively. The solvation effect occurs via a relative shift of the 3σ*α orbital, strongly influencing the charge transfer. A similar effect, in either direction, could be achieved by varying the ligands, especially the axial ligand. Based on our results, the effect of such variations can be predicted from the resulting relative energy level of the 3σ*α orbital of the FeO2+ moiety.52
In this work, we have neglected the possible effect of counter anions. When close to the ironoxo complex (otherwise they are screened by the polar solvent ), they could have a similar effect as the described solvation effect, as the negative charges also shift the orbitals upward in energy. However, the relative shift of the FeO2+ orbitals and the substrate orbitals would strongly depend on the precise position of these counter anions, and could result in an effect in either direction.
Ultimately, insight, derived from the electronic mechanism, into how the solvent affects the reactivity, and how the reactivity can be tuned by ligand effects, may help in the design of useful catalysts based on FeO2+ or other metaloxo species. Understanding the solvation effects is a key element in this process.
References
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS.
- J. C. Schöneboom, S. Cohen, H. Lin, S. Shaik and W. Thiel, J. Am. Chem. Soc., 2004, 126, 4017–4034 CrossRef.
- J. T. Groves and G. A. McClusky, J. Am. Chem. Soc., 1976, 98, 859–861 CrossRef CAS.
- J. T. Groves and M. Van Der Puy, J. Am. Chem. Soc., 1976, 98, 5290–5297 CrossRef CAS.
- L. Que, J. Biol. Inorg. Chem., 2004, 9, 684–690 CAS.
- A. Bassan, M. R. A. Blomberg, T. Borowski and P. E. M. Siegbahn, J. Inorg. Biochem., 2006, 100, 727–743 CrossRef CAS.
- A. Karawajczyk and F. Buda, J. Biol. Inorg. Chem., 2005, 10, 33–40 CrossRef CAS.
- G. I. Panov, A. K. Uriarte, M. A. Rodkin and V. I. Sobolev, Catal. Today, 1998, 41, 365–385 CrossRef CAS.
- H. J. H. Fenton, Chem. News, 1876, 190 Search PubMed.
- P. Wardman and L. P. Candeias, Radiat. Res., 1996, 145, 523–531 CrossRef CAS.
- H. B. Dunford, Coord. Chem. Rev., 2002, 233, 311–318 CrossRef.
- F. Gozzo, J. Mol. Catal. A: Chem., 2001, 171, 1–22 CrossRef CAS.
- J. T. Groves, J. Inorg. Biochem., 2006, 100, 434–447 CrossRef CAS.
- F. Buda, B. Ensing, M. C. M. Gribnau and E. J. Baerends, Chem.–Eur. J., 2001, 7, 2775–2783 CrossRef CAS.
- B. Ensing, F. Buda, P. Blöchl and E. J. Baerends, Angew. Chem., Int. Ed., 2001, 40, 2893–2895 CrossRef CAS.
- B. Ensing, F. Buda, P. E. Blöchl and E. J. Baerends, Phys. Chem. Chem. Phys., 2002, 4, 3619–3627 RSC.
- B. Ensing and E. J. Baerends, J. Phys. Chem. A, 2002, 106, 7902–7910 CrossRef CAS.
- O. Pestovsky, S. Stoian, E. L. Bominaar, X. Shan, E. Munck, L. Que and A. Bakac, Angew. Chem., Int. Ed., 2005, 44, 6871–6874 CrossRef CAS.
- J. Kaizer, E. J. Klinker, N. Y. Oh, J. U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Munck, W. Nam and L. Que, J. Am. Chem. Soc., 2004, 126, 472–473 CrossRef CAS.
- J. U. Rohde and L. Que, Angew. Chem., Int. Ed., 2005, 44, 2255–2258 CrossRef CAS.
- T. A. van den Berg, J. W. de Boer, W. R. Browne, G. Roelfes and B. L. Feringa, Chem. Commun., 2004, 22, 2550–2551 RSC.
- S. Malykhin, I. Zilberberg and G. M. Zhidomirov, Chem. Phys. Lett., 2005, 414, 434–437 CrossRef CAS.
- A. Fiedler, D. Schröder, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 1994, 116, 10734–10741 CrossRef CAS.
- D. Schröder, H. Schwarz and S. Shaik, in Structure & Bonding. Metal-oxo and metal-peroxo species in catalytic oxidations, ed. B. Meunier, Springer Verlag, Berlin, Heidelberg, 2000, vol 97, pp. 91–123 Search PubMed.
- D. Schröder, S. Shaik and H. Schwarz, Acc. Chem. Res., 2000, 33, 139–145 CrossRef CAS.
- Y. Shiota and K. Yoshizawa, J. Am. Chem. Soc., 2000, 122, 12317–12326 CrossRef CAS.
- T. Yumura and K. Yoshizawa, Organometallics, 2001, 20, 1397–1407 CrossRef CAS.
- F. Buda, B. Ensing, M. C. M. Gribnau and E. J. Baerends, Chem.–Eur. J., 2003, 9, 3436–3444 CrossRef CAS.
- A. Decker and E. I. Solomon, Curr. Opin. Chem. Biol., 2005, 9, 152–163 CrossRef CAS.
- A. Decker, J. U. Rohde, L. Que and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 5378–5379 CrossRef CAS.
- D. Kumar, H. Hirao, L. Que and S. Shaik, J. Am. Chem. Soc., 2005, 127, 8026–8027 CrossRef CAS.
- A. Decker and E. I. Solomon, Angew. Chem., Int. Ed., 2005, 44, 2252–2255 CrossRef CAS.
- A. Decker, M. D. Clay and E. I. Solomon, J. Inorg. Biochem., 2006, 100, 697–706 CrossRef CAS.
- A. Ghosh, E. Tangen, H. Ryeng and P. R. Taylor, Eur. J. Inorg. Chem., 2004, 4555–4560 CrossRef CAS.
- F. Neese, J. Inorg. Biochem., 2006, 100, 716–726 CrossRef CAS.
- B. Ensing, F. Buda, M. C. M. Gribnau and E. J. Baerends, J. Am. Chem. Soc., 2004, 126, 4355–4365 CrossRef CAS.
- M. J. Louwerse, P. Vassilev and E. J. Baerends, submitted.
- L. Bernasconi, E. J. Baerends and M. Sprik, J. Phys. Chem. B, 2006, 110, 11444–11453 CrossRef CAS.
- ADF, Amsterdam Density Functional program, Theoretical Chemistry, Vrije Universiteit, Amsterdam, http://www.scm.com Search PubMed.
- E. J. Baerends, D. E. Ellis and P. Ros, Chem. Phys., 1973, 2, 41–51 CrossRef CAS.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 CrossRef.
- G. te Velde, F. M. Bickelhaupt, S. J. A. van Gisbergen, C. Fonseca Guerra, E. J. Baerends, J. G. Snijders and Z. T., J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- A. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- F. Eckert and A. Klamt, AIChE J., 2002, 48, 369–385 CrossRef CAS.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 CrossRef CAS.
- F. M. Bickelhaupt and E. J. Baerends, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. R. Boyd, J. Wiley, New York, 2000, vol. 15, pp. 1–86 Search PubMed.
- K. J. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- L. Bernasconi, M. J. Louwerse and E. J. Baerends, in preparation.
Footnotes |
| † The HTML version of this article has been enhanced with colour images. |
| ‡ Electronic supplementary information (ESI) available: Further detailed results (Fig. S1–S5, Tables S1–S4). See DOI: 10.1039/b613182d |
| This journal is © the Owner Societies 2007 |