Electron-induced reactions in condensed films of acetonitrile and ethane
Imre
Ipolyi
,
Wilfried
Michaelis†
and
Petra
Swiderek
*
Universität Bremen, Institute of Applied and Physical Chemistry, Fachbereich 2 (Chemie/Biologie), Leobener Straße/NW 2, Postfach 330440, Fachbereich 2 (Chemie/Biologie), 28334, Bremen, Germany
First published on 21st November 2006
Abstract
Reactions in pure and mixed films of C2H6 and CD3CN deposited on a Au surface at 35 K have been induced by low-energy electrons and investigated by Thermal Desorption Spectrometry (TDS). The incident electron energy (E0) was varied between 5 and 16 eV and a number of different products were identified. Beside the main products, CD4, CD3H, and C2D6, molecules resulting from atom scrambling during radical chain reactions (C2H5D) and recombination products (CD3CD2CN and C2H5CD3) were identified while others were characteristically absent. The quantity of the different products varied with E0. The observed electron-driven processes are in accord with previous findings from gas phase experiments on dissociative electron attachment and electron impact ionization. On this basis, reaction mechanisms leading to the formation of the observed products are suggested for different ranges of E0.
1. Introduction
Functionalized surfaces are important because they allow to tailor the properties of a material or provide chemical groups to which more complex molecular architectures can be attached. This calls for the development of processes to modify the chemical composition of surfaces in a controlled way. It has been shown that exposure to low-energy electrons can be used to functionalize semiconductor surfaces.1,2 This approach is motivated by the finding that specific chemical reactions can be initiated by proper tuning of the energy of electrons incident onto a molecule or surface. A very fine example is provided by recent in depth investigations of electron-induced reactions in gaseous thymine.3 Therefore, it should be possible to achieve a better control of surface modifications than in the case of high-energy radiation such as X-rays or keV-electrons, which abundantly produce secondary electrons with energy below 20 eV but with a wide energy distribution.4Processes for surface modification are not only of interest for inorganic but also for organic materials. While simple hydrocarbon materials like polymers and alkanethiols or alkanesilanes for the formation of self-assembled monolayers are readily available, more complex molecules, which carry the desired functional groups already before being deposited or self-assembled on a surface, usually require a sophisticated synthetic strategy and are therefore costly. Thus, it is desirable to establish processes that allow to couple functional groups to hydrocarbon materials after their deposition on a surface.
This work pursues the idea to functionalize a hydrocarbon by producing, through an electron-induced reaction, species that would both activate the hydrocarbon and bind to the resulting radical site. While examples of intermolecular reactions have been studied both in gaseous clusters5 and condensed samples,6,7 attachment of specific molecular groups to a hydrocarbon could not be demonstrated so far. As a first effort to demonstrate functionalization of hydrocarbons, an investigation of the reactions induced by low-energy electrons in pure and mixed films of ethane (C2H6) and deuterated acetonitrile (CD3CN) and monitored by use of Thermal Desorption Spectrometry (TDS) is presented here. This system served as a model for materials consisting of a hydrocarbon surface (represented by C2H6) and a reactive agent (CD3CN) that is expected to functionalize the hydrocarbon material upon electron exposure. CD3CN was chosen as example because electron-induced processes in the non-deuterated molecule have been well characterized in the gas phase.8–10 The deuteration was not expected to fundamentally modify these processes but helped to unravel the reaction mechanisms. Multilayer films of CD3CN and C2H6 condensed onto a Au surface cooled to 35 K in ultra-high vacuum have been exposed to low-energy electrons. Desorbing products were detected by mass spectrometry in the TDS experiments. The results of these experiments show that intermolecular reactions are, in fact, initiated. The underlying mechanism are investigated by variation of the incident electron energy.
2. Experiment
The experiments were performed in an UltraHigh Vacuum (UHV) chamber pumped by a turbomolecular pump to a pressure of about 10−10 Torr, with the residual gas consisting mainly of hydrogen. If necessary, additional pumping can be provided by a titanium sublimator. Low-energy electron-induced reactions in both mixed (with a ratio of 1 : 1) and pure films of C2H6 and CD3CN condensed onto a polycrystalline Au substrate were monitored by TDS using a Quadrupole Mass Spectrometer (QMS) residual gas analyser (Stanford, 200 amu). The substrate is held at 35 K by means of a closed-cycle He cryostat. The temperature was measured using a thermocouple type E. A commercial fload gun delivering currents of the order of a few μA cm−2 as measured at the sample was used for electron exposure. The energy (E0) of the non-monochromatized electron beam with an estimated resolution of the order of 0.5–1 eV was varied between 5 and 16 eV. Typical exposure times yielding 5000 μC ranged from 5 min at 15 eV to 1 h at 5 eV. At the relatively high incident current densities used here, charging of the irradiated molecular films is likely. While this modifies the exact value of E0, we have at present no established procedure to determine exactly the amount of this effect. This is due to the divergence of the non-focussed electron beam that increases upon decrease of E0. This prohibits the measurements of the current onset that could be used to calibrate E0. Therefore, all stated E0 are merely nominal energies as deduced from the difference in potential between the filament of the electron gun and the substrate.For each experiment, thin films of C2H6 (Messer-Griesheim, purity 99.95%) and/or CD3CN (Acros Organics, NMR-grade, nominal purity 99 atom % D) were deposited onto the Au substrate by introducing gases or vapours via a gas-handling manifold. The manifold consists of precision leak valves and a small calibrated volume where the absolute pressure is measured with a capacitance manometer. For each film deposition, a calibrated amount of gas or vapour, measured as a pressure drop, is leaked via a stainless steel capillary whose end is located just in front of the substrate. Prior to each deposition, the substrate is cleaned by resistive heating to 270 K. The thickness of the films was estimated from TDS measurements performed on pure and non-exposed CH3CN (Riedel-de Haen, 99.9%) for increasing amounts of deposited vapour as described in Section 3.1.
After an irradiation time corresponding to a specific electron exposure, stated as an accumulated charge, thermal desorption was induced by increasing the substrate temperature from 35 K up to 250 K at a rate of 1 K s−1 (exposure experiment). This was achieved by resistive heating of two thin Ta ribbons spotwelded to the thicker Au substrate. The temperature was controlled using a standard power supply, which is computer-controlled by software developed in our lab. In each experiment, TDS curves were recorded for four different molecular masses, among which in all cases the masses of the parent positive ions of the investigated molecules formed after 70 eV electron impact at the entrance of the QMS. The other chosen signals were characteristic of specific product molecules and also correspond to the parent positive ions. As reference, a TDS measurement was performed using a non-exposed film of the same composition and thickness prior to each exposure experiment (denoted as 0 μC). TDS measurements at 44 amu (parent ion of CD3CN, see section 3.2) and 43 amu (parent ion of CD2HCN, not shown) recorded on pure and non-exposed films of CD3CN both revealed a single desorption peak at 140 K with very similar shape and an intensity ratio of 100 : 5. Assuming CD3CN and CD2HCN to have the same ionization efficiency, this suggests that some isotope exchange has occurred prior to the experiments. This needs to be taken into account in the discussion of the results.
3. Results and discussion
3.1. Thickness calibration
TDS measurements of films of CH3CN produced from increasing amounts of gas were performed to calibrate the monolayer coverage. The obtained asymmetric desorption peaks were fitted using two Gaussians with maxima at 135 K and 148 K. An example is shown in Fig. 1a. The integral signal corresponding to the monolayer should increase linearly with the amount of leaked vapour, up to a point where the monolayer is complete, and remain constant at higher doses. This behaviour is observed for the higher temperature desorption signal (Fig. 1c). In contrast, the lower temperature peak only starts to grow upon completion of the monolayer and increases linearly with the dose thereafter as a characteristic of the formation of a multilayer. A dose of 0.14 ± 0.03 mTorr is required for monolayer formation as deduced from linear least square fits, depicted as a dashed and a compact line in Fig. 1b and c, to TDS curves from measurements for amounts of leaked vapor ranging from 0.02 to 1.2 mTorr. From this calibration, the thickness of the CD3CN films as used in most of the electron exposure experiments was estimated to be of the order of 15 monolayers (MLs). Some experiments aiming at larger product molecules were performed at a thickness of 30 ML to achieve a larger signal. At these thicknesses, effects from the Au surface during electron exposure can be largely excluded. The metal may nonetheless contribute to the thermal desorption signal, as will be discussed later. The thickness of the mixed layers is estimated to be of similar magnitude due to the roughly comparable size of CD3CN and C2H6. | ||
| Fig. 1 (a) TDS measurement at 41 amu for a film of CH3CN produced by depositing an amount of vapor corresponding to a pressure drop of 0.4 mTorr in the manifold. The desorption signal is fitted by two Gaussian with maxima at 148 K and 135 K, ascribed to the monolayer and the multilayer signal. Integrated multilayer (b) and monolayer (c) signal as a function of the amount of vapor leaked into the chamber. | ||
3.2. Reactions in pure CD3CN films
Each specific molecule can be identified in a TDS curve corresponding to a characteristic molecular mass by the temperature at which desorption from the surface occurs. Under the conditions of the present experiments, the partial pressure of CD3CN typically starts to increase at 120 K and peaks at or near 140 K. Assuming that overlap with signals due to other molecules with fragments of identical mass does not occur, the intensity of the desorption peak is proportional to the amount of its neutral precursor molecule on the surface. The magnitude of this signal thus reveals the variation of the amount of CD3CN on the substrate under electron exposure.
Fig. 2a shows representative TDS curves at 44 amu (parent ion of CD3CN) without exposure and after exposure at three different E0. Fig. 2b summarizes the peak intensities of the 44 amu desorption signal after electron exposure of 5000 μC, normalized to the same signal in the corresponding reference experiment, i.e., prior to exposure. The results indicate that the rate of loss of CD3CN increases with increasing E0, to become noticeable around 10 eV and to reach an order of 50–60% after exposure to 5000 μC at and above 14 eV. Assuming first order kinetics, a cross section σ for the reaction can be calculated from
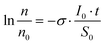
 | ||
| Fig. 2 (a) TDS measurements at 44 amu for 15-layer films of CD3CN before and after electron exposure of 5000 μC at the stated incident energies. The magnification is the same for all curves. (b) Peak intensities of the desorption signal after exposure, each normalized to the same intensity for a non-exposed film recorded immediately before. (c) Depletion cross sections for CD3CN under electron exposure estimated from the data in (b). | ||
The mass spectra of CD3CN also show a small signal at 28 amu corresponding to a fragment ion DCN+. The intensity of this signal amounts to about 6% of the 44 amu peak. Fig. 3a shows representative TDS measurements for 28 amu, while the intensity of this signal relative to the 44 amu data from the same experiment (Fig. 2) is plotted in Fig. 3b. Above a threshold of roughly 10 eV, the relative intensity of the 28 amu signal is found to increase. At the same time the desorption peak broadens and, at 15 eV, the maximum shifts to 130 K. This gives evidence of the production of another species with a signal at 28 amu that desorbs at a similar temperature as CD3CN. As a first guess, the literature was searched for data on DCN. CD3CN has a reported vaporization enthalpy in the 30–35 kJ mol−1 range, while 36–38 kJ mol−1 are reported for DCN.11 In a pure film, DCN should thus desorb at a higher temperature than CD3CN. If DCN is only present as a small amount in CD3CN, hydrogen bonding must be less pronounced. Thus, we suggest that the 28 amu signal actually gives evidence of the production of a certain amount of DCN under electron exposure starting around E0 = 10 eV.
 | ||
| Fig. 3 (a) TDS measurements at 28 amu for 15-layer films of CD3CN before and after electron exposure of 5000 μC at the stated incident energies. The magnification is the same for all curves. (b) Peak intensity of the desorption signal at 28 amu before and after exposure, each normalized to the intensity at 44 amu in the same experiment, as function of incident electron energy. | ||
Production of CD4 in CD3CN exposed to electrons is deduced from a desorption peak around 55–60 K in the TDS scan for 20 amu, as shown in Fig. 4 for representative E0. This signal remains small through the 6–9 eV range, with a somewhat higher intensity at 6 and 7 eV, and starts to increase around 10 eV to reach a new constant level at 12 eV. The observation of CD4 suggests that cleavage of the C–C bond in CD3CN occurs and the resulting CD3 species reacts with hydrogen from its environment. In order to trace the origin of this hydrogen, the signal at 19 amu corresponding to CD3H was also recorded. The fact that this signal was very small or even absent at all E0 shows that CD3CN, and not H2 from the residual gas of the vacuum chamber, is the source of hydrogen. It must be noted that both the 19 amu and the 20 amu signal yield a flat baseline in most experiments when the TDS measurement is performed without electron exposure. A signal above 150 K, appearing upon electron exposure and becoming stronger at higher E0, may stem from gas accumulated on other parts of the sample holder than the substrate or from fragments that attach to the metal surface and are released in a reactive process at higher temperature. The position and shape of this signal changed somewhat between the single measurements. The same applies to the signal in the 130–135 K range seen at higher E0. While this signal is absent from all measurements without electron exposure, its position shifted between different measurements. The fact that the Au surface itself was not freshly prepared prior to each measurement may explain these changes if one assumes that the higher temperature signal stems from molecules or fragments in immediate contact with the metal. Chemisorption on polycrystalline or stepped Au surfaces has been reported previously for CO.12 In the absence of sputter cleaning, such chemisorption processes may change the state of the metal surface from one experiment to the next, thus accounting for the variations in the TDS curves. The detailed investigation of this effect was beyond the scope of the present work and therefore not pursued.
 | ||
| Fig. 4 TDS measurements at 20 amu for 15-layer films of CD3CN before and after electron exposure of 5000 μC at the stated incident energies. For comparison, measurements at 19 amu for both a fresh film and a film after the same exposure are included. The magnification is the same for all curves. | ||
As a third product from CD3CN under electron exposure, this time performed at 6 eV, 10 eV, and 15 eV, C2D6 could be observed as concluded from a 36 amu desorption signal in the 75–80 K range (Fig. 5). All reference measurements, i.e., experiments without electron exposure, show a flat baseline throughout the investigated temperature range. Similar to the results for CD4, the intensity of the 36 amu signal is higher at 15 eV than at lower E0 but, in contrast, it displays a minimal intensity at 10 eV. The energy dependence of C2D6 formation will be investigated more closely in the case of mixed films, as described in the next section.
 | ||
| Fig. 5 TDS measurements at 36 amu for 15-layer films of CD3CN before and after electron exposure of 5000 μC at the stated incident energies. The magnification is the same for all curves. | ||
In addition, as will be discussed in detail in Section 4, it was anticipated that larger products like CD3CD2CN and NCCD2CD2CN might be formed. This possibility was investigated at E0 = 7 eV. As mass spectra from the NIST database11 usually resemble those acquired on the present apparatus, these data were used as reference for the identification of the two compounds. For CD3CD2CN, the parent cation (60 amu) and fragments 58 amu and 56 amu are thus expected with relative intensities of approximately 15 : 85 : 9, while the strongest signal for NCCD2CD2CN should be 56 amu. Fig. 6a shows the TDS curves for 44 amu (CD3CN) as well as 56 amu, 58 amu, and 60 amu after an exposure of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μC onto a 30 ML film of pure CD3CN. While measurements for the latter three masses exhibit a flat baseline prior to exposure (not shown), desorption signals are observed at 140 K and around 180 K for 58 amu. A very weak increase of the signal is observed around 180 K for 60 amu. The relative intensities of the signals at 180 K corresponds roughly to that expected for CD3CD2CN, according to the database.11 We thus ascribe the observed desorption signals to this compound. Assuming similar ionization efficiencies for CD3CD2CN and CD3CN, we estimate, from a comparison of the peak intensities for 44 amu and 58 amu, that of the order of 0.1% of CD3CD2CN was formed. The very close agreement of the lower temperature peak with the desorption signal of CD3CN can be rationalized by the similar vaporization enthalpies reported for the two compounds.11 Also, it is possible that some CD3CD2CN could be driven to the vacuum by evaporation of the matrix material, CD3CN. The higher temperature desorption signal may tentatively be assigned to recombinative desorption of fragments that are initially trapped at the metal surface. The lack of signal for 56 amu indicates that formation of NCCD2CD2CN cannot be observed within the sensitivity of the present experiment. It must be noted that at the larger film thickness and exposure time, while the intensity of 44 amu (CD3CN) has roughly doubled, the intensities of CD4 and C2D6 have now increased by factors of 4 and 8 (Fig. 6b). This can be traced back not only to the larger amount of material and longer exposure but also to the somewhat higher E0, as compared to the experiments described earlier (Fig. 4 and 5). Fig. 6b also demonstrates that while the amount of CD3CD2CN appears to be smaller than that of CD4 and C2D6, as far as can be judged from the mere comparison of the mass spectral signals, it is nonetheless not negligible.
000 μC onto a 30 ML film of pure CD3CN. While measurements for the latter three masses exhibit a flat baseline prior to exposure (not shown), desorption signals are observed at 140 K and around 180 K for 58 amu. A very weak increase of the signal is observed around 180 K for 60 amu. The relative intensities of the signals at 180 K corresponds roughly to that expected for CD3CD2CN, according to the database.11 We thus ascribe the observed desorption signals to this compound. Assuming similar ionization efficiencies for CD3CD2CN and CD3CN, we estimate, from a comparison of the peak intensities for 44 amu and 58 amu, that of the order of 0.1% of CD3CD2CN was formed. The very close agreement of the lower temperature peak with the desorption signal of CD3CN can be rationalized by the similar vaporization enthalpies reported for the two compounds.11 Also, it is possible that some CD3CD2CN could be driven to the vacuum by evaporation of the matrix material, CD3CN. The higher temperature desorption signal may tentatively be assigned to recombinative desorption of fragments that are initially trapped at the metal surface. The lack of signal for 56 amu indicates that formation of NCCD2CD2CN cannot be observed within the sensitivity of the present experiment. It must be noted that at the larger film thickness and exposure time, while the intensity of 44 amu (CD3CN) has roughly doubled, the intensities of CD4 and C2D6 have now increased by factors of 4 and 8 (Fig. 6b). This can be traced back not only to the larger amount of material and longer exposure but also to the somewhat higher E0, as compared to the experiments described earlier (Fig. 4 and 5). Fig. 6b also demonstrates that while the amount of CD3CD2CN appears to be smaller than that of CD4 and C2D6, as far as can be judged from the mere comparison of the mass spectral signals, it is nonetheless not negligible.
 | ||
| Fig. 6 (a) TDS measurements at 44 amu as well as 56 amu, 58 amu, and 60 amu for 30-layer films of CD3CN after electron exposure of 10000 μC at E0 = 7 eV. The magnification is the same for the lower three curves. (b) Direct comparison of desorption signals at 20 amu, 36 amu, and 58 amu recorded under the same conditions. The magnification is the same for all curves. | ||
3.3. Reactions in mixed CD3CN/C2H6 (1 : 1) films
Mixed films of CD3CN and C2H6 were produced by leaking into the UHV chamber a mixture of the vapors containing equal amounts of both gases. Again, TDS curves prior to and after electron exposure of 5000 μC at different E0 were recorded. Fig. 7a summarizes the peak intensities of the 44 amu desorption signal after electron exposure of 5000 μC normalized to the same signal without exposure. Again, the rate of loss of CD3CN increases with increasing E0 to become noticeable above 10 eV and reach an order of 60–70% after exposure to 5000 μC at 15 eV. C2H6 exhibits a similar behavior to reach a loss of 50–60% at 15 eV (Fig. 7b). 30 amu was used to quantify the amount of C2H6, despite the fact that the 28 amu signal is stronger for this compound, in order to avoid possible overlap with the DCN+ signal from CD3CN. Again, some formation of DCN under electron exposure is deduced from an increase of the 28 amu signal around 140 K after exposure at 15 eV (Fig. 8). At the same time, the signal near 80 K ascribed to a fragment of C2H6 decreases under exposure. While the 28 amu signal at 140 K might also be due to a higher temperature channel for production of C2D6, the decrease of the 30 amu signal at the same temperature gives evidence against this interpretation. | ||
| Fig. 7 (a) Peak intensities of the desorption signal at 44 amu for 15-layer films of CD3CN/C2H6 mixtures (1 : 1) after exposure, each normalized to the same intensity for a non-exposed film recorded immediately before. (b) Peak intensities of the desorption signal at 30 amu for the same films after exposure, each normalized to the same intensity for a non-exposed film recorded immediately before. | ||
 | ||
| Fig. 8 TDS measurements at 30 amu and 28 amu for 15-layer films of a CD3CN/C2H6 mixture (1 : 1) before and after electron exposure of 5000 μC at E0 = 15 eV. The curves for 0 μC are offset for clarity. | ||
As in pure CD3CN, production of methane in the mixed films under exposure of the sample to 5000 μC is obvious from desorption peaks between 50 and 55 K, this time for both 19 and 20 amu (Fig. 9). This result indicates that both CD4 and CD3H are formed under exposure to the electron beam. At 15 eV, production of CD3H is now even more important than CD4. Again, experiments performed without exposure of the film (upper frame of Fig. 9) do not show any signal and CD4 and CD3H desorption signals at higher temperatures are not discussed further here. It must be noted that the desorption of CD4 and CD3H occurs at a slightly lower temperature than in the pure CD3CN films, most probably due to the weaker polarization forces resulting from the smaller average amount of CD3CN present in the environment of a single product molecule.
 | ||
| Fig. 9 TDS measurements at 19 amu and 20 amu for 15-layer films of CD3CN/C2H6 mixtures (1 : 1) before and after electron exposure of 5000 μC at the stated incident energies. The magnification is the same for all curves. | ||
Formation of C2D6 under exposure to electrons is again observed in the mixed films. In this case, a larger number of E0 was investigated (Fig. 10). This time, the signal reaches its maximum at E0 = 7 eV before dropping to a roughly constant lower level at higher E0. Desorption takes place in the 70–75 K range. This is again, and in agreement with the result for CD4 and CD3H, slightly lower than in the pure CD3CN film.
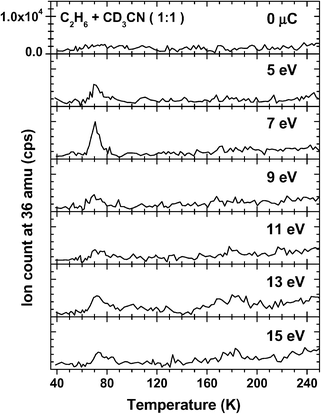 | ||
| Fig. 10 TDS measurements at 36 amu for 15-layer films of CD3CN/C2H6 mixtures (1 : 1) before and after electron exposure of 5000 μC at the stated incident energies. The magnification is the same for all curves. | ||
Assuming that reactions between fragments from CD3CN and C2H6 can occur in mixed films, TDS measurements aiming at a number of different products were performed. First, we searched for some possible adducts between fragments from CD3CN and C2H6. Assuming that CD3 could abstract H from C2H6 and one of the fragments from CD3CN could then react with the resulting radical, we investigated whether C2H5CN or C2H5CD3 are formed for E0 ranging from 6 to 16 eV. Evidence for production of C2H5CD3 was found above E0 = 11 eV (Fig. 11), while at lower E0 a flat baseline was obtained. Evidence for the former could not be found at any of these energies.
 | ||
| Fig. 11 TDS measurements at 47 amu for 15-layer films of CD3CN/C2H6 mixtures (1 : 1) after electron exposure of 5000 μC at the stated incident energies. The magnification is the same for all curves. | ||
Given that the strongest production of C2D6 is observed at E0 = 7 eV (Fig. 10) and formation of C2H5CD3 takes place at higher E0, mixed films were investigated specifically after exposure at 7 eV and 15 eV to find evidence for other products. In analogy to the pure CD3CN film, 30 ML films after exposure to 10000 μC at E0 = 7 eV were searched for traces of CD3CD2CN, NCCD2CD2CN, and, in addition, of n-C4H10. Desorption around 180 K was again observed in the 58 amu curve (not shown) but with smaller intensity, while evidence for NCCD2CD2CN was missing. On the other hand, additional information is now needed to distinguish CD3CD2CN from n-C4H10. From literature data,11 we expect for n-C4H10 a signal at 43 amu that is about an order of magnitude stronger than the parent cation (58 amu). Evidence for such a strong signal at 180 K was not obtained from TDS curves recorded at 43 amu, but a strong desorption peak occurred at 140 K. This signal, on the other hand, can be traced back to an isotopic impurity of about 5% CD2HCN in CD3CN. As recording TDS curves for reference mixtures containing all potential products was beyond the scope of the present work, possible contributions of n-C4H10 to the strong signal at 140 K can not be excluded a priori. On the other hand, a desorption temperature of 110 K has been reported recently for n-C4H10.13 We thus conclude that n-C4H10 was not formed in the present experiment.
As will be discussed in Section 4, scrambling of fragments might lead to products such as CD2HCN or C2H5D. While the former was already present as impurity in the sample of CD3CN, and thus prohibited the observation of a minor product of the same mass, formation of the latter is clearly seen already at E0 = 7 eV (Fig. 12). While the desorption signal for 30 amu decreases slightly upon exposure, the signal for 31 amu increases. Again, this mass was already present as a small isotopic impurity (13C) in C2H6 but the increase of the signal upon exposure must be ascribed to formation of C2H5D. Interestingly, the loss of CD3CN is much more pronounced here than for the pure film at the same thickness and E0, where it amounts to approximately 10%. Formation of C2H5D was also deduced to occur at E0 = 15 eV (not shown). While the desorption signals at 75 K decrease strongly for both 30 amu and 31 amu in a 30 ML pure film of C2H6, and also for 30 amu in a mixed film of CD3CN and C2H6, the same peak in the 31 amu curve of the mixed film remains roughly constant, thus hinting towards formation of C2H5D.
 | ||
| Fig. 12 TDS measurements at 30 amu, 31 amu and 44 amu for 30-layer films of a CD3CN/C2H6 mixture (1 : 1) before and after electron exposure of 10000 μC at E0 = 7 eV. The curves for 0 μC are offset for clarity. | ||
Finally, the films after exposure at E0 = 15 eV were searched for traces of C2H5CD2H as well as CD3CD2CN and CD2HCD2CN. Formation of the former might occur together with C2H5CD3 if more than two fragments are involved in the reaction. Unfortunately, mass 46 (parent cation of C2H5CD2H) is also present as fragment M-H in C2H5CD3. On the other hand, signals at 46 amu and 47 amu should have similar intensity in C2H5CD3 according to the database.11 TDS curves of mixed 30 ML films after 10000 mC exposure at 15 eV did reveal roughly equal intensities for these two masses. Evidence for formation of C2H5CD2H was thus not obtained.
While the formation of CD3CD2CN was already deduced, a reaction mechanism involving more than two fragments could, again, also lead to formation of species like CD2HCD2CN. As the most intense fragment for CD3CD2CN has mass 58 amu, 57 amu should be most intense for CD2HCD2CN. Such a signal was also not detected within the sensitivity of the present experiment.
4. Discussion
Electron-induced chemical modifications in the condensed phase can involve intermolecular reactions of reactive fragments formed during the initial electron–molecule interaction. In order to unravel the reaction sequences in CD3CN or mixed CD3CN/C2H6 films leading to the observed products, we first review the available information on the products of Dissociative Electron Attachment (DEA) and Electron Impact (EI) ionization studies in the gas phase.A recent study on electron attachment cross sections for CH3CN in the energy range from near 0 up to 10 eV has revealed the production of the anionic fragments CH2CN−, CHCN−, CCN−, CN− and CH3− ,8 presumably accompanied by the radicals as follows in reactions (1a–e)
 | (1a) |
 | (1b) |
 | (1c) |
 | (1d) |
 | (1e) |
Electron attachment to C2H6 has only been studied in thin condensed films.15 A characteristic H− signal due to ESD was observed around 10 eV in this case. Other fragments appearing in the same energy range were CH3−, CH2−, and, with a smaller intensity, CH−.
Positive ion formation above the ionization threshold11 can also contribute to reactions taking place under electron exposure. The thresholds for gaseous CH3CN, CD3CN, and C2H6 have been reported at 12.2–12.4 eV, 12.2–12.3 eV, and 11.4–11.8 eV. Fragmentation of CH3CN was observed with thresholds around 14 eV (CH2CN+) and 15–16 eV (CHCN+, CH2+). C2H6 produces C2H4+ already near the ionization threshold and C2H5+ near 12 eV, while C2H3+, C2H2+, and CH3+ appear above a threshold between 13.5–15 eV. Dipolar dissociation to CH3+ and CH3− also takes place in gaseous C2H6 starting at 13.6 eV11 but does not noticeably contribute to ESD below E0 = 18 eV.15
Assuming essentially the same behavior for CH3CN and CD3CN, the gas phase electron attachment channel around 7–8 eV producing CN− can explain the formation of CD4 and CD3H at 6–7 eV in the present study. The resonant process which presumably leads to a CD3 radical as a second product (reaction 1d) is likely to be shifted towards lower E0 in the condensed phase due to polarization of the molecular environment. Production of CD4 and CD3H at this energy requires additional abstraction of D or H atoms from adjacent molecules (see also Scheme 1):
 | (2a) |
 | (2b) |
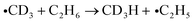 | (2c) |
 | (3a) |
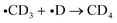 | (3b) |
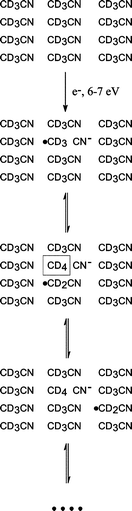 | ||
| Scheme 1 | ||
It is possible that DEA according to process (1d) initiates a radical chain reaction. This might lead formally to migration of a CD2CN radical as depicted in Scheme 1. If this species does not undergo further fragmentation, it could be expected to eventually recombine with CD3 produced by a second DEA event (1d) at another site to form CD3CD2CN or with another migrating CD2CN radical producing NCCD2CD2CN. Among those, the latter should be most probable due to the prediction (Scheme 1) that CD2CN should recur throughout the chain reaction. In contrast, based on the statistical argument derived from Scheme 1, the probability for recombination of two CD3 radicals should be considerably smaller as they are assumed to be consumed upon formation of CD4. On the other hand, C2D6 is, in fact, observed with an intensity comparable to CD4 (Fig. 4 and 5) and CD3CD2CN is formed but not NCCD2CD2CN. This suggests that part of the CD3 radicals can not overcome the activation barrier to D abstraction16 at low temperature, and thus remains immobilized within the film so that recombination with other CD3 or with CD2CN are efficient concurrent reactions to abstraction of a D from an adjacent molecule when the temperature increases. This is reasonable if we assume radical recombination to be barrier-free.17 On the other hand, recombination requires sufficient mobility within the film and CD2CN is probably not mobile enough to recombine with a second CD2CN at a noticable rate. This rationalizes the fact that NCCD2CD2CN is not observed. The results so far suggest that the mobility of fragments is a rate-determining factor in the observed reactions.
In analogy to Scheme 1, Scheme 2 presents a hypothetical reaction sequence induced in the condensed mixed films of CD3CN and C2H6 by DEA process (1d). Here, both of reactions (2b and c) take place as deduced from the simultaneous production of CD4 and CD3H. Again, we start by assuming radical chain reactions following the initial DEA event. In this case, propagation of the chain could produce new products, namely, CD2HCN and C2H5D. Evidence obtained for formation of C2H5D at E0 = 7 eV demonstrates that such chain propagation in fact takes place. In contrast to pure CD3CN, where chain propagation does not lead to new products, the mixed films provide the opportunity to detect such reaction steps.
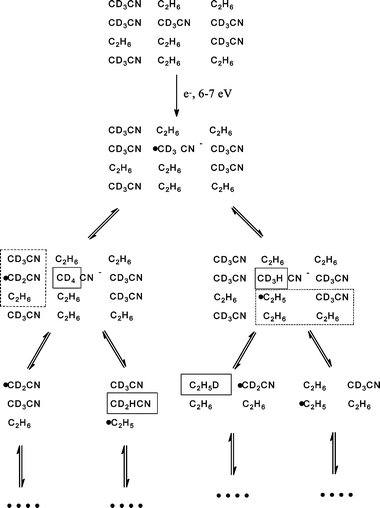 | ||
| Scheme 2 | ||
Based on the finding that chain reactions must take place, a larger variety of recombination processes is expected in the mixed films. Recombination of two CD3 radicals formed as immediate products of DEA explains the observed formation of C2D6, while recombination of the other intermediate radicals would produce CD3CD2CN, CD3C2H5, C2H5CD2CN, NCCD2CD2CN, or n-C4H10. Among those, CD3C2H5 that would be formed according to
 | (4a) |
 | (4b) |
One possible explanation for the fact that CD3CD2CN, but not CD3C2H5, is observed at E0 = 7 eV invokes a different reactivity for CD2CN and C2H5 radicals. While the former can be stabilized by a mesomeric effect, the latter is more reactive and would quickly attack adjacent molecules to abstract a D or H atom. CD2CN can thus survive until recombination with other radicals such as CD3 occurs. This becomes possible upon temperature increase, while C2H5 would react immediately with any neighbor molecule, thus preventing eventual recombination with CD3. On the other hand, if C2H5 is in fact as reactive as assumed here, the same should apply for CD3. This contradicts the interpretation given for the results on pure CD3CN films because such a high reactivity of CD3 should favor the formation of CD4 and CD3H over recombination products, such as CD3CD2CN, which is in contrast to Fig. 6b.
In the light of these contradictions, another explanation needs to be envisaged. The detection of C2H5D in the mixed films clearly demonstrates that chain reactions take place. In the case of pure CD3CN films, we may assume that these reactions lead to accumulation of CD2CN both within the bulk of the film and at the metal surface. After some exposure, newly formed CD3 may encounter not only adjacent CD3CN but also CD2CN frozen in the film. The latter situation could lead to immediate recombination without increase of the temperature. CD3CD2CN formed in this way could freely evaporate upon temperature increase. We attribute the 140 K peak in the 58 amu curve (Fig. 6) to this process. As with CD2CN, CD3 may also get trapped at the metal surface. Trapped CD3 and CD2CN, on the other hand, may recombine through an activated process only at higher temperature, and can thus be held responsible for the 180 K desorption signal.
Trapping, i.e. chemisorption of molecules, at Au surfaces is not only known for CO.12 Dissociative adsorption on Au(111) at low temperature has been reported for SiH4.18 Also, electron-induced processes can induce chemisorption as reported for O2 at temperatures below 50 K.19 The scenario suggested above for CD3CN is consistent with such a process, even if the present polycrystalline Au surface is not as well defined as a single crystal surface.
In the mixed films, the situation is more complex. Here, the chain reactions lead to atom scrambling, as witnessed by the formation of C2H5D. This would lead to a larger variety of products. It is thus possible that the signal for each single product may drop below the level of sensitivity of the present experiment. The only remaining signal is the 180 K desorption of CD3CD2CN that was suggested above to involve radicals trapped at the metal surface. This signal is smaller than in pure films of CD3CN, which can be ascribed to both a lower amount of CD3CN but also to scrambling via the chain reactions that consume some of this compound. The fact that the 140 K signal is completely suppressed would thus reflect the lower probability that a CD3 radical is produced in the vicinity of an already present CD2CN.
A further question concerns the absence of a signal due to CD3C2H5 at 7 eV. According to Scheme 2 and the detected amounts of CD4 and CD3H, both CD2CN and C2H5 should be equally present within the mixed films. On the other hand, in the situation where trapping of the radical at the metal surface is important, a different reactivity might again be invoked. CD2CN, due to the possible mesomeric structures, might offer a binding site to the metal even if it is immobilized, and the D abstraction occurs on the far side with respect to the metal. CD3 is equally small and mobile enough to rearrange near the metal so that it may become trapped. C2H5, on the other hand, may not easily rotate to bring the radical site near the metal. This may decrease its lifetime to the point that recombination reactions become less likely than for CD2CN. We must note that CD3CD2CN has been detected here in 30 ML films, while the search for CD3C2H5 has only been performed with 15 ML films. Also, so far, we are not aware of experimental results supporting the suggested different binding efficiency for CD2CN and C2H5 radicals. A final decision on the reason for the absence of detectable amounts of CD3C2H5 can thus not be given here.
It must be noted that production of DCN according to
 | (5a) |
 | (5b) |
 | (5c) |
The rate of production of CD4, CD3H, C2D6 in pure CD3CN, and C2H5CD3 clearly increases above 11 eV. This must be ascribed to the onset of ionisation, which opens cationic reaction pathways. Interestingly, all four products could be most easily explained if CD3 fragments were formed in the initial reaction step. To the best of our knowledge, formation of CD3 from CD3CN or the analogous CH3 from CH3CN does not appear to be important between 11 and 16 eV.11 Formation of CN− would imply also that CD3 is produced, but in the gas phase this process is much less intense at higher E0 than at 8 eV9 and can thus not explain the strong increase in CD3-containing products at higher E0. An intense production of CD3 in this energy range, on the other hand, could stem from reaction (1d) if induced by low energy secondary electrons released upon ionisation according to
 | (6a) |
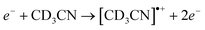 | (6b) |
The production of C2D6 can again be explained by recombination of CD3 radicals upon increase of the sample temperature. On the other hand, the fact that less C2D6 is produced in the mixed film than in pure CD3CN suggests the presence of concurrent recombination channels, for example yielding C2H5CD3 (Fig. 11).
As summarized above, ionization processes also lead to a number of positively charged molecular fragments. Under exposure to the electron beam, these are likely to be neutralized by electrons that are thermalized through inelastic collisions within the film, thus again yielding reactive radicals. C2H5+ formed upon ionization can recombine with CD3 radicals produced by secondary electrons to form C2H5CD3. If this is the dominant reaction pathway, other isotopic isomers are not expected. This product could also be formed by recombination of three fragments, namely CD2+ and C2H5+ neutralized under the electron beam and a free D radical. In this case, not only C2H5CD3 but also C2H5CD2H is expected to form. The missing evidence for the latter suggests that, in fact, deceleration of incident electrons and the release of slow secondary electrons dominates the reaction mechanism.
Recombination processes like the one just described could lead to production of a number of other species. For example, CD2CN+ and CD3, preferably in pure CD3CN, could form CD3CD2CN, while C2H5+ and D would again produce C2H5D that was already predicted to form via the radical chain reactions of Scheme 2 for the mixed films at low E0. Recombination of three species—CD2CN+, CD2+, and H—could possibly form CD2HCD2CN. Again, missing evidence for the latter suggests that such ternary reactions are of minor importance.
While the present results give qualitative information on the reaction mechanism under electron exposure and suggest that secondary electrons are involved at E0 above the ionization threshold, a more quantitative study of this latter effect would require a comparison with experiments performed at lower E0. This way, the assumption that reaction (1a) is also the dominant one in the condensed phase could be verified. Some support that this reaction indeed plays a role has been obtained in a previous study that aimed at the functionalization of hydrogen-terminated diamond.2 At present, we hope to improve our TDS setup in order to achieve electron exposure at lower E0 within acceptable time scales.
Finally, we want to emphasize that there is no clear enhancement of any production rate around E0 = 10 eV. Some production of CD4, CD3H, C2D6, and C2H5CD3 is observed within the range of E0 between 9 and 11 eV, which coincides with the resonant channel producing H− in C2H6,15 taking into account not only the inherent energy width of this process but also the low energy resolution of the present experiments. On the other hand, the production rates of these products increase above 11 eV thus ruling out a dominant role of the H− producing resonance in C2H6 in the formation of the products observed so far. For the same reason, we conclude that DEA to CD3CN, producing D− as observed by ESD above 7 eV with maxima at 9 and 11.5 eV,14 is less efficient than the higher energy reaction channels described before. Also, the fact that a product C2H5CN is not observed at any of the investigated E0 demonstrates that CN− produced by DEA becomes stabilized within the film and is not available for reactions with hydrocarbon radicals.
5. Conclusions
Thermal desorption experiments following exposure to low-energy electrons on pure films of CD3CN and mixed films of CD3CN and C2H6 were performed in order to obtain a detailed picture of the reactions that are initiated by electron impact. It was found that, while the initial events can be explained in line with previous gas phase results on dissociative electron attachment and electron impact ionization, complex intermolecular reactions take place.The electron-driven dissociative process initiates radical chain reactions that lead to atomic scrambling. Products are formed via both abstraction of hydrogen by radicals such as CD3 and also by recombination of radicals. In these processes, the mobility of the participating species is relevant, as witnessed by the prevalence of products resulting from recombination of CD3 with other radicals. Evidence also points towards an influence of intermediate trapping of radicals at the metal. This process increases the lifetime of reactive fragments that are released upon increase of the temperature during the TDS scan.
At energies above the ionization threshold, the observed products point towards a dominant contribution of secondary electrons to the reactions. Again, these low-energy electrons initiate fragmentations that lead to release of CD3, and possibly also D, that can again initiate radical chain reactions but this time with a higher efficiency due to the increased number of electrons.
The results show that intermolecular reactions and formation of larger molecules take place in mixed films of CD3CN and C2H6. The insight obtained from this study will guide future work aimed at the development of new processes for the functionalization of hydrocarbon materials.
Acknowledgements
I. I. acknowlegdes a travel grant from ESF program EIPAM.References
- W. Di, P. Rowntree and L. Sanche, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 16618 CrossRef CAS.
- A. Lafosse, M. Bertin, D. Caceres, C. Jäggle, P. Swiderek, D. Pliszka and R. Azria, Eur. Phys. J. D, 2005, 35, 363 CrossRef CAS.
- (a) H. Abdoul-Carime, S. Gohlke and E. Illenberger, Phys. Rev. Lett., 2004, 92, 168103 CrossRef; (b) S. Ptasińska, S. Denifl, V. Grill, T. D. Märk, P. Scheier, S. Gohlke, M. A. Huels and E. Illenberger, Angew. Chem., Int. Ed., 2005, 44, 1647 CrossRef CAS; (c) S. Ptasińska, S. Denifl, V. Grill, T. D. Märk, E. Illenberger and P. Scheier, Phys. Rev. Lett., 2005, 95, 093201 CrossRef; (d) S. Ptasińska, S. Denifl, P. Scheier, E. Illenberger and T. D. Märk, Angew. Chem., Int. Ed., 2005, 44, 6941 CrossRef CAS.
- V. Cobut, Y. Frongillo, J. P. Patau, T. Goulet, M.-J. Fraser and J.-P. Jay-Gerin, Radiat. Phys. Chem., 1998, 51, 229 CrossRef CAS.
- I. Martin, T. Skalicky, J. Langer, H. Abdoul-Carime, G. Karwasz, E. Illenberger, M. Stano and S. Matejcik, Phys. Chem. Chem. Phys., 2005, 7, 2212 RSC.
- M. Imhoff, L. Parenteau, L. Sanche and M. A. Huels, Phys. Chem. Chem. Phys., 2005, 7, 3359 RSC.
- P. Swiderek, M. C. Deschamps, M. Michaud and L. Sanche, J. Phys. Chem. B, 2003, 107, 563 CrossRef CAS.
- W. Sailer, A. Pelc, P. Limão-Vieira, N. J. Mason, J. Limtrakul, P. Scheier, M. Probst and T. D. Märk, Chem. Phys. Lett., 2003, 381, 216–222 CrossRef CAS.
- M. Heni and E. Illenberger, Int. J. Mass Spectrom. Ion Processes, 1986, 73, 127 CrossRef CAS.
- J. A. Stockdale, F. J. Davis, R. N. Compton and C. E. Klots, J. Chem. Phys., 1974, 60, 4279 CrossRef CAS.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, eds. P. J. Linstrom and W. G. Mallard, June 2005, National Institute of Standards and Technology, Gaithersburg MD, 20899 (http://webbook.nist.gov).
- J. M. Gottfried, K. J. Schmidt, S. L. M. Schroeder and K. Christmann, Surf. Sci., 2003, 536, 206 CrossRef CAS.
- S. Funk, B. Hokkanen, J. Wang, U. Burghaus, G. Bozzolo and J. E. Garcés, Surf. Sci., 2006, 600, 583 CrossRef CAS.
- M. Lachgar, Y. Le Coat, R. Azria, M. Tronc and E. Illenberger, Chem. Phys. Lett., 1999, 305, 408 CrossRef CAS . We note that according to a personal communication by R. Azria, Fig. 3 of this reference indeed shows results for pure CD3CN in contrast to what was erroneously stated in the text of the same reference.
- P. Rowntree, L. Parenteau and L. Sanche, J. Phys. Chem., 1991, 95, 4902 CrossRef CAS.
- B. Kerkeni and D. C. Clary, J. Chem. Phys., 2005, 123, 064305 CrossRef.
- H. Tamura and M. S. Gordon, Chem. Phys. Lett., 2005, 406, 197 CrossRef CAS.
- M. J. S. Spencer and G. L. Nyberg, Surf. Sci., 2004, 573, 151 CrossRef CAS.
- J. M. Gottfried, K. J. Schmidt, S. L. M. Schroeder and K. Christmann, Surf. Sci., 2002, 511, 65 CrossRef CAS.
Footnote |
| † Present adress: Airbus Deutschland GmbH, Hünefeldstr. 1–5, 28119 Bremen, Germany. |
| This journal is © the Owner Societies 2007 |