Electrochemical determination of activation energies for methanol oxidation on polycrystalline platinum in acidic and alkaline electrolytes
Jamie L.
Cohen
,
David J.
Volpe
and
Héctor D.
Abruña
*
Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853, USA. E-mail: hda1@cornell.edu; Fax: +1-607-255-9864; Tel: +1-607-255-4720
First published on 24th November 2006
Abstract
The oxidation pathways of methanol (MeOH) have been the subject of intense research due to its possible application as a liquid fuel in polyelectrolyte membrane (PEM) fuel cells. The design of improved catalysts for MeOH oxidation requires a deep understanding of these complex oxidation pathways. This paper will provide a discussion of the literature concerning the extensive research carried out in acidic and alkaline electrolytes. It will highlight techniques that have proven useful in the determination of product ratios, analysis of surface poisoning, anion adsorption, and oxide formation processes, in addition to the effects of temperature on the MeOH oxidation pathways at bulk polycrystalline platinum (Ptpoly) electrodes. This discussion will provide a framework with which to begin the analysis of activation energy (Ea) values. This kinetic parameter may prove useful in characterizing the rate-limiting step of the MeOH oxidation at an electrode surface. This paper will present a procedure for the determination of Ea values for MeOH oxidation at a Ptpoly electrode in acidic and alkaline media. Values from 24–76 kJ mol−1 in acidic media and from 36–86 kJ mol−1 in alkaline media were calculated and found to be a function of applied potential and direction of the potential sweep in a voltammetric experiment. Factors that influence the magnitude of the calculated Ea include surface poisoning from MeOH oxidation intermediates, anion adsorption from the electrolyte, pH effects, and oxide formation processes. These factors are all potential, and temperature, dependent and must clearly be addressed when citing Ea values in the literature. Comparison of Ea values must be between systems of comparable electrochemical environment and at the same potential. Ea values obtained on bulk Ptpoly, compared with other catalysts, may give insight into the superiority of other Pt-based catalysts for MeOH oxidation and lead to the development of new catalysts which lower the Ea barrier at a given potential, thus driving MeOH oxidation to completion.
1. Introduction
1.1 General comments on fuel cell research and development
Recently, increasing monetary support and scientific research have focused on alternative energy sources in order to diminish not only dependence on oil and natural gas, but to also decrease waste and pollution coming from combustible fuels and non-rechargeable batteries. Much of the research has focused on the development of clean, energy efficient power sources. One of the most encouraging of these is fuel cells. Fuel cell research has been ongoing for many years but has recently been brought to the forefront as the demand has increased for working devices for use in everything from cell phones and computers, which require 1–10 W of power, to automobiles and backup power stations, which require between 1 kW–10 MW of power.1–4There are many advantages afforded by fuel cell technologies. Some of the most commonly highlighted are that fuel cells convert chemical energy directly into electrical energy with efficiencies not limited by the Carnot cycle. Fuel cell efficiencies can be greater than 80% under certain operating conditions, have few moving parts, and are mechanically simple. Fuel cells also generate relatively innocuous waste products, depending on the fuel and oxidant used.1,5,6 One of the simplest and efficient fuel cell systems is the hydrogen–oxygen (H2–O2) fuel cell, which produces water as a waste product. This characteristic is especially important to reduce air pollution and meet emissions requirements for trucks and automobiles.1,6,7 Finally, depending on the type of fuel cell employed, the energy density could surpass that of some batteries. This aspect is important for smaller fuel cells, which could be used to replace batteries in portable electronics applications.1,4,7–10
1.2 Types of fuel cells—where does methanol fit in?
There are a variety of fuel cells being developed. These fuel cells offer a wide range of power outputs, use a variety of fuels, and operate in vastly different temperature regimes. High temperature solid oxide (SOFC), molten carbonate (MCFC), and phosphoric acid (PAFC) fuel cells operate between 200–1000 °C and are used for high-power applications in the kilowatt to megawatt range.1,3,5–8,11 Fuel cells more suitable for automobile and portable electronics applications are polyelectrolyte membrane fuel cells (PEMFC).1,2,4,10,12–16 PEMFCs are particularly attractive because they typically run at temperatures ranging from 20 to 100 °C and can use gaseous or liquid fuels. The gaseous H2/O2 fuel cell system mentioned previously can be employed in a PEMFC.1,4 The H2 used in these PEMFC systems must be very pure, which makes it difficult to manufacture. Primary fossil fuels via reforming and cracking, biofuels and biological breakdown of fuels, hydrogen storage materials such as metal hydrides, and water electrolysis are all ways in which H2 gas can be obtained.1 The H2 gas obtained usually must be further purified in order to remove sulfur and carbon-containing species, as well as other impurities. This purification is costly and time-consuming. The steam reformation of MeOH has been found to produce H2 under mildly exothermic conditions.1 The use of MeOH in the production of H2 gas has brought it attention as a possible fuel for a PEMFC.Liquid fuels and/or oxidants can be used in a PEMFC. A type of PEMFC, the direct methanol fuel cell (DMFC), uses liquid MeOH as the fuel. Methanol has a relatively high net energy density (5.26 kWh kg−1) which surpasses that of pure H2 that can be made directly from MeOH (2.3 kWh kg−1) and has an energy density comparable to gasoline.1,2,12–14 Methanol as a liquid fuel also has an important advantage in that the infrastructure for supporting a liquid fuel is already in place in terms of fuel pumping stations and fuel transportation, making the refueling of a liquid fuel cell facile. DMFCs do suffer drawbacks.1,7,13–19 One of the most prevalent is the need for better catalysts for the anode, where MeOH fuel oxidation is carried out. The oxidation of MeOH proceeds via a variety of intermediates, depending on the reaction conditions, thereby lowering the efficiency of the fuel cell and, in some instances, poisoning the catalyst surface with tightly bound carbon-containing intermediate poisons, such as carbon monoxide (CO). While there are numerous advantages and disadvantages of DMFCs, as well as a great number of components that require further optimization, the DMFC appears most promising for multiple applications. Much of the current literature cited above, and in general, has focused on elucidating the complete pathway of MeOH oxidation on current catalysts in order to better select catalyst components to enhance the power output and fuel efficiency of the DMFC.
The following section will provide an overview of the vast literature concerning the elucidation of the MeOH oxidation pathways, including the numerous electrochemical techniques that have been employed. While it is nearly impossible, and not the intent of this review, to be all-inclusive, this work will highlight individual techniques that have been benchmarks for the understanding of the oxidation pathways. Additionally, it will provide relevant articles, including reviews, which provide useful references pertaining to the research being carried out concerning the MeOH oxidation pathway. This paper will focus exclusively on smooth bulk Ptpoly electrodes, although much of the literature cited contains information about other Pt-containing catalysts. A detailed pathway for MeOH oxidation in both acidic and alkaline electrolyte will be introduced based on the literature as well as on our own research. Complications with the determination of this pathway, as well as details yet to be resolved, will be discussed throughout the text. In response to some of these complications, and unresolved details, the often overlooked topic of activation energy (Ea) values will be discussed as a means of further inquiry into the individual steps in MeOH oxidation, as well as a means of comparison between different electrocatalysts being developed for improved oxidation of MeOH. An in-depth Ea study dealing with MeOH oxidation on Ptpoly has been carried out by our lab, which is the first in-depth study to date in both acidic and alkaline media using a smooth bulk Ptpoly electrode. These results will be discussed in terms of the detailed pathway of MeOH oxidation presented in order to determine the possible implications of these values. A strategy for analyzing these Ea values will be presented in terms of the processes that must be taken into account that can affect the magnitude of the Ea, including surface adsorption and poisoning effects, in addition to paying attention to the potential at which the Ea was calculated. By scrutinizing all of the information compiled for the MeOH oxidation process, outlining the factors that control product distributions, and using a new approach of determining Ea values of MeOH oxidation on a Ptpoly surface, optimal catalysts for MeOH oxidation may be found. Even more importantly, a more complete understanding of how new catalysts alter the pathway of MeOH oxidation may emerge. This understanding will aid in choosing new catalyst components that target specific steps in the oxidation pathway, leading to enhanced efficiencies of MeOH oxidation and will further improve the power output of the DMFC, making it a power source that can be reasonably considered for numerous power applications.
2. Methanol oxidation pathway
The pathway of methanol oxidation has proven to be very complex on a smooth Ptpoly surface. While other catalysts (alloys, intermetallics, etc.) that contain Pt as one of the components have been developed, it is important to fully understand the complete oxidation pathway of MeOH at a clean bulk Ptpoly surface before more complex surfaces can be understood. The composition and behavior of other catalysts will therefore not be specifically addressed here. The complete oxidation of MeOH in acid or alkaline media is a 6e− oxidation forming CO2 as shown in eqn (2.1) for MeOH oxidation in acidic media and eqn (2.2) for alkaline media: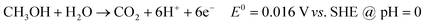 | (2.1) |
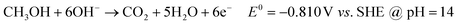 | (2.2) |
2.1 Detailed methanol oxidation pathway
While the complete oxidation of MeOH yields CO2 as the final product, at a bulk Ptpoly surface, the complete 6e− oxidation seldom takes place unless a large overpotential is applied. There are numerous intermediate products, including adsorbed poisons, formed during MeOH oxidation. The MeOH pathway presented in Fig. 1 represents the oxidation pathway in acidic2,12,14–45 and alkaline11,12,20,29,31,34–36,45–59 media and has been compiled from results found in the literature, as well as from our group. A pathway for MeOH dissociation, observed in and derived from UHV experiments, is also included for completeness.21,22,24,60–64 The following discussion will review the techniques used to determine this pathway, and will talk about selected steps of the pathway in detail, noting the intermediates and products formed, Pt sites needed to carry out the reaction taking place, and the nature of these Pt sites including the need for hydroxide/oxide adsorption in order to facilitate MeOH oxidation. In addition, the detriment of CO and anion adsorption onto the Pt surface during the oxidation process will be reviewed. This detailed discussion will aid in the analysis of the Ea energy values discussed in section 3 and those calculated in the studies carried out in section 6. A complete understanding of the complexity of MeOH oxidation will set the framework for determining the factors that are most important to take into account when carrying out experiments to determine Ea values.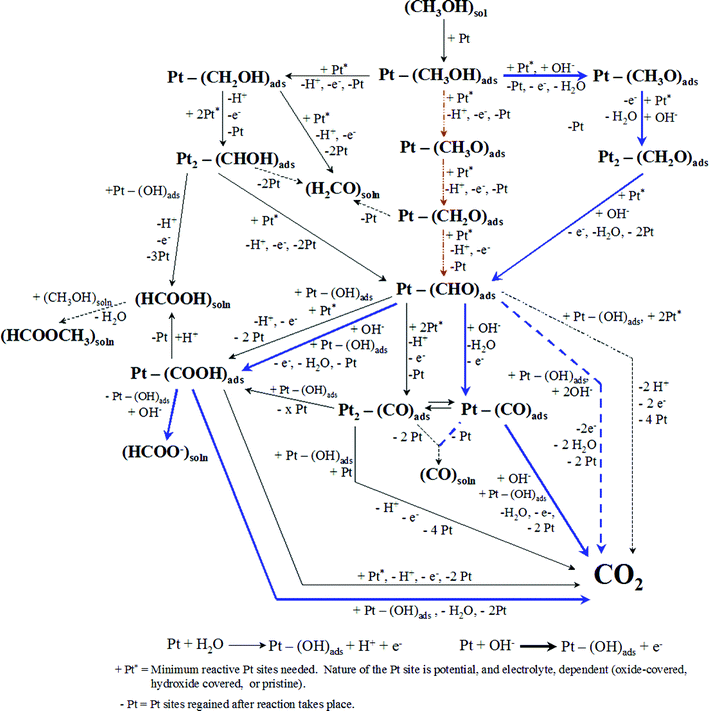 | ||
| Fig. 1 Detailed pathway for the oxidation of MeOH in acid (—) and alkaline (blue solid line) electrolyte. Dashed lines represent pathways that have been suggested in the literature for acidic and alkaline electrolytes respectively, but are unlikely to occur under typical experimental conditions. The pathway (red line) corresponds to the initial dehydrogenation pathway as determined by UHV experiments. | ||
Fig. 1 shows that common reactive intermediates and products that have been determined to contribute to the charge passed during MeOH oxidation include formic acid (HCOOH), formaldehyde (HCHO), formate (HCOO−), and CO, in addition to the final oxidation product, carbon dioxide (CO2). Other solution products formed via chemical reaction between the products above and bulk MeOH can also be detected during bulk electrolysis experiments and under other experimental conditions. For example, methyl formate (HCOOCH3) can be used to detect the formation of HCOOH when running mass spectrometry experiments; see section 2.1.2 for more details. The formation of these intermediates (reactive or poisonous) and products (soluble intermediates including CO2) are strongly dependent on the potential applied, the nature of the Pt surface, and also the electrolyte in which the oxidation is being carried out. In particular, the contributions from Ptpoly surface oxides and anion adsorption from the electrolyte have been subjects of intense research, examining how the nature of the electrode surface and nature of the electrolyte govern the MeOH oxidation pathway. There has also been much research concerning the number of Pt sites needed to carry out each of the steps due to the numerous surface processes that occur during potential excursions in different electrolytes.15,16,18,20,22,35,50,65–69 The minimum number of Pt sites needed to carry out each of the MeOH oxidation steps is designated in Fig. 1. These data have been complied from the literature cited throughout this section, as well as from research in our own labs. When determining the number of electrons transferred in a particular reaction step, one must take into account that every time a Pt–(OH)ads oxide species takes part in the reaction, one electron was produced from the formation of that oxide species, emphasizing the importance of the surface oxide in the reaction mechanisms being considered. The literature has explored the nature of all the intermediates in great detail in acidic and alkaline supporting electrolytes, focusing on H2SO4 and HClO4 at concentrations ranging from 0.1 to 1.0 M, although other extreme cases have been analyzed as well. While there has been some research carried out in alkaline media (NaOH or KOH), that literature is much less extensive compared to the acidic electrolyte literature and much of it has focused on the use of Pt single crystal surfaces.
The works cited concerning the MeOH oxidation pathway will be discussed in detail below in order to highlight many of the techniques used to interrogate the complete MeOH oxidation pathway, not only in aqueous media, but also in UHV-type systems. Much of this literature frames the data obtained about the methanol oxidation process in terms of its importance to DMFC development. While Ptpoly will be the focus in this study, many of the references contain information concerning other Pt-containing catalysts for MeOH oxidation. As the methanol oxidation pathway is discussed below, complications with its analysis will become clear. These complications further underline the difficulties faced in the analysis of Ea values. The contributions from Ptpoly surface processes, such as oxide formation and anion adsorption, often make the electrochemical data difficult to analyze. The poisonous intermediate, CO, brings about difficulties in obtaining oxidation turnover rates at specific potentials and makes obtaining steady state currents at low potentials impossible. Changes in fuel concentration, electrolyte concentration, and pH also lead to varying product distributions throughout the literature due to the numerous products that can be formed during MeOH oxidation. All of these experimental parameters and electrochemical processes must be taken into account when analyzing the Ea values obtained because the data used to calculate these values is a convolution of these factors. These complications, to name a few, make drawing broad conclusions from across the literature difficult. Although the above complications can cause problems during data analysis, recognizing their occurrence, understanding how these processes vary with potential and temperature, how they affect the products formed, and using this knowledge during data analysis will lead to a better understanding of the MeOH oxidation pathway. It will also clarify the factors that govern the individual steps, as well as aid in using Ea values obtained as useful points of comparison for catalyst behavior.
Tafel behavior has been analyzed under a variety of conditions for MeOH oxidation in order to gain insight concerning the electrochemical rate determining steps at a number of different potentials. Electrochemical kinetics information has relied heavily on the use of basic voltammetric techniques and has yielded a wealth of information concerning the electron transfers steps in the fuel oxidation process. Of course, the rate determining electron transfer step seems to vary with reaction conditions, nature of the electrode surface, etc. Section 3 will discuss the kinetic information that can be obtained from Tafel plots using traditional electrochemical methods.
Surface coverages of Pt oxide and Pt–(OH)ads,24,31,34,35,45,54,70,75,77,82–91 CO and other intermediates,14,16,18,20,24,25,29,30,34,35,38,51,60,61,65,70,73,76,81,82,92–101 and of adsorbed anions from the electrolyte13,14,16,22,31,34,36,50,54,70,102–108 have also been studied extensively using voltammetric techniques. These will be discussed in detail throughout the text due to their immense importance in the pathway of oxidation of MeOH, both kinetically, and thermodynamically. The literature pertaining to oxide formation and coverage, as well as anion adsorption, is vast and will only be highlighted in this report. Fig. 1 has many steps that require a Pt site with adsorbed OH− in order to provide an additional oxygen atom to the carbon atom of MeOH. These steps normally also require either a clean Pt site for binding or another Pt site with adsorbed OH− for deprotonation. The electrode potential governs the coverage of Pt–(OH)ads as well as the adsorbed anion coverage. These coverages, in turn, will determine the extent of oxide coverage, as well as Pt sites free to bind MeOH and other intermediates, at a given potential. Cyclic voltammetry has been invaluable in the determination of oxide coverages as a function of the potential of the Ptpoly electrode. The oxide coverage depends on the time the electrode is held at a given potential, the switching potential of the electrode when a potential sweep is being carried out, and pH of the electrolyte. Because the MeOH oxidation pathway in alkaline media requires Pt–(OH)ads as a reactant (Fig. 1), the nature of the oxide on the surface is especially important. Oxide coverages will be discussed in section 7.2.4 when analyzing the Ea values obtained for MeOH oxidation at a number of different potentials. There is a wealth of literature on the subject of oxide formation on a Ptpoly surface in numerous electrochemical environments. The main concern of this paper, with respect to oxide coverages, will be examining the change in oxide coverage with temperature when carrying out Ea experiments.
One can also study product distributions by carrying out bulk electrolysis and isotope exchange experiments.33,34,63,95,109–111 The reaction products HCOOH, HCHO, and CO2 have been detected using post-analysis techniques like UV-Vis and fluorometric techniques,14,19,39,40,112 and liquid/gas chromatography after bulk electrolysis was carried out.14,15,22 The drawback to these experiments is while a number of different intermediates may be forming and diffusing away from the electrode surface, they can make their way back to the electrode to undergo further oxidation. Formic acid and formaldehyde can be further oxidized to form CO on the electrode surface or CO2. This effect produces product ratios that are not representative of the actual processes occurring at the electrode surface at a given potential. Also, products from bulk electrolysis can react with species in solution, again giving false product ratios and complicating the final product analysis. The electrode potential during the bulk electrolysis is clearly an important factor, which will determine not only the extent of poisoning of the electrode by CO formation, but also the extent of complete oxidation of MeOH to CO2. The use of flow cells has improved the results of bulk electrolysis experiments, but other techniques may be more suitable for quantitative product distributions as will be described below.95,113
Finally, linear sweep and cyclic voltammetry, in addition to chronoamperometry have been used to carry out experiments at elevated temperatures in order to determine the effects of increased temperature on the kinetics and thermodynamics of MeOH oxidation in terms of product formation, onset potential, current enhancements, and oxide and CO coverage and anion adsorption changes.34,45,54,56,72,73,94,114–122 Many of the temperature-dependence experiments have been carried out on not only bulk Ptpoly but also at single crystal electrode surfaces. While a large volume of literature exists concerning MeOH oxidation on single crystals, it will not be discussed here, although some of the results of these experiments can be carried over and applied to the behavior of bulk Ptpoly as well. All of these processes, surface processes in particular, are extremely important when analyzing Ea values obtained at a Ptpoly surface. Changes in these processes with temperature will change the magnitude of the current measured at a given potential. These current changes are, in turn, used to calculate Ea values. The magnitude of the Ea value would then not be representative of the MeOH oxidation pathway being carried out at that potential, but a convolution of a number of different processes affecting the faradaic current observed.
The wealth of information obtained using these traditional electrochemical methods will be applied when carrying out the Ea experiments presented in section 6. Linear sweep voltammetry at a number of different temperatures will be employed in this work to obtain the data needed to calculate Ea values. The exact nature of the linear sweep voltammetry method will be explained in section 4. Information needed to analyze the Ea values is obtained when comparing linear sweep voltammetry and chronoamperometry experiments in terms of the role that oxide reduction plays in the MeOH oxidation pathway over the potential range of the linear sweep. Chronoamperometry data will also provide valuable information about the CO poisoning occurring on the timescale of the linear sweep. The linear sweep data, when combined with cyclic voltammetry of Ptpoly at a number of different potentials in pure electrolyte, in addition to systems that contain MeOH, give much of the information needed to analyze the contribution of anion adsorption, pH, and oxide formation to the magnitude of the Ea values obtained.
Not only were the volatile intermediates and products detected using DEMS, but the kinetics of CO oxidation have also been studied, and isotope experiments have been carried out to determine the exact nature of the formation of these intermediates.33,111,123–125 It was determined using DEMS and a Pt/C catalyst that CO coverages greater than 1/3 of a monolayer preclude the adsorption of MeOH onto the catalyst surface.33 DEMS has also been able to provide insight into the importance of Pt oxide in the MeOH oxidation and that the reduction of a preformed monolayer of PtO by methanol at open-circuit points to the fact that Pt oxide species are indeed active for methanol oxidation.33 As the surface coverage of PtO begins to be electrochemically reduced, MeOH can adsorb. The subsequent dehydrogenation of MeOH facilitates the reduction of PtO at adjacent Pt sites.59,128 The PtO can be reduced to Pt–(OH)ads species, which in turn aids the oxidation of intermediates, such as CO, to CO2. Steps governing the MeOH oxidation rate, including the oxidation of CO or the adsorption of MeOH, were also investigated at a Pt/C electrode and it was found that the removal of the COads poison is the rate determining step during methanol oxidation, not the dissociation of the C–H bond during the initial MeOH adsorption.33 The potential at which COads begins to be oxidized to CO2 was also determined using DEMS and COads stripping experiments. It was found that its oxidation begins at 0.15 V vs. RHE on a Pt/C catalyst surface.124
It is quite clear that DEMS has been able to provide the most concrete evidence for the importance of catalyst surface structure, fuel and electrolyte composition and concentration, electrode potential, and mass transport when analyzing the intermediate and product concentrations and distributions formed during an experiment. A comparison of data from DEMS and from other techniques, such as bulk electrolysis, provides useful information, but attention must be paid to significant variations in product distributions that can arise from differing experimental conditions. The factors influencing the product distribution must be taken into consideration when analyzing any of the data on MeOH oxidation obtained using DEMS or other traditional electrochemical interrogation methods. Not only recognizing that the product distributions vary, but also what causes this variation, leads to a more clear understanding of the methanol oxidation process at a given potential. This understanding must be applied to the analysis of the Ea results presented in sections 3 and 6 of this Invited Article. DEMS demonstrates that the assignment of Ea values to a specific step in the MeOH oxidation pathway will prove to be nearly impossible. The changes in product distribution with surface structure, potential, and poisoning mean that the pathway for MeOH oxidation is always changing. The Ea at a specific potential will correspond to the chemical rate-determining step at that potential, which varies as the potential is changed. In order to assign Ea values to a specific step in the MeOH oxidation pathway, a detailed map of the processes occurring at the electrode surface, including oxide formation, anion adsorption, and surface poisoning process, is needed at the potential at which the Ea value was calculated. DEMS must be used to carry out such experiments, but the same conditions must be employed in the DEMS experiments as in the electrochemical experiments used to determine the Ea values. An added complication arises from the possibility of changes in the MeOH pathway as the temperature is increased. The determination of Ea values is carried out by increasing the temperature of the electrolyte and measuring the current as a function of temperature at a given potential. DEMS experiments need to be carried out at a variety of temperatures in order to observe changes in the product distribution as a function of temperature and potential. Only with these detailed data will it be possible to correlate Ea values with specific steps in the oxidation pathway of MeOH.
The formation and oxidation of COads has been studied extensively and the differentiation between bridge-bonded and linearly bonded CO has been made using variations on IR spectroscopy at a number of different catalyst surfaces, often comparing pure Pt to Pt–Ru alloy compounds.17,23,65,79,92,112,129,131,134,135 The adsorption coverages of CO have been studied in CO-saturated solutions of various electrolytes and the CO stripping/oxidation potentials have been investigated in many different electrochemical environments.24,54,92,96,99,112,130,141,142 This intermediate is one of the most important and troublesome intermediates due to its strong adsorption to Pt surfaces up to saturation coverages of θ = 0.5–0.68.81,124,130,131,134,141 The CO formed from MeOH oxidation poisons the electrode surface, inhibiting additional MeOH adsorption, which diminishes the current density at a given potential, and shifts the onset of oxidation to more positive potentials. The adsorbed CO can only be removed at high potentials (depending on the surface roughness, composition, etc), and it requires a Pt–(OH)ads species following the Langmuir–Hinshelwood mechanism.17,54,81,92 The different bonding conformations (geometries) of CO include linear, bridged, and at threefold hollow sites. The relative coverages of CO in the linear and bridge-bonded forms can be detected using the above mentioned surface sensitive techniques, and interconversion between linear and bridge-bound CO has been found to occur, depending on the surface coverages of CO and of other adsorbed species, which also makes this interconversion a function of potential. Coverage as a function of potential has been studied rigorously using FT-IR, more specifically single potential alteration IR spectroscopy (SPAIRS).134 The electrolyte strongly influences the extent of CO adsorption following the trend that CO adsorbs more strongly to the Pt surface in electrolytes HClO4 > H2SO4 > H3PO4.99 Alloys and intermetallics containing Pt with other non-noble metals have been studied to facilitate oxide formation at more negative potentials in order to promote the oxidation of CO at lower potentials to mitigate poisoning.92 These materials may also be able to bypass the CO formation pathway completely and proceed mainly via the formic acid pathway presented in Fig. 1. It is virtually impossible to review all of the literature about CO, its adsorption, and oxidation due to the vast amounts of research reported and being carried out. A good review of research up to 1990 is found in Beden et al.,112 a recent review of CO surface electrochemistry is found in Mayrhofer et al.,137 and, along with the references provided throughout the text above, a broad view of the research to date can be obtained.
The coverage and oxidation of CO has been monitored as a function of temperature and potential in FT-IR and cyclic voltammetry experiments by a number of research groups although very few contain results for bulk Ptpoly. It was determined by Kardash et al. that the coverage of CO during the oxidation of 0.3 M MeOH in 0.1 M HClO4 did not change appreciably as the temperature was increased from 25 to 50 °C, although the CO formed at higher temperatures was more resistant to oxidation.130 This fact was attributed to the probability of other surface poisons forming at higher coverages at these temperatures and precluding the activation of water to form an oxide on the Ptpoly surface. In the absence of these other surface poisons, the CO coverages were observed to diminish slightly as the temperature was increased due to the thermal activation of Pt–(OH)ads formation. As has been mentioned in sections 2.1.1 and 2.1.2, the calculation of Ea values requires measurement of the current of MeOH oxidation as a function of temperature at a given potential. It was also mentioned that surface processes, such as CO poisoning and anion adsorption, were measured as a function of temperature using traditional electrochemical techniques. Surface-sensitive techniques used in conjunction with these traditional electrochemical techniques lend powerful insight into how surface processes change as the potential and temperature are modulated. The analysis of the Ea values will require the understanding of how CO poisoning is influenced by increasing the temperature of the MeOH solution. In situ IR is one of the most powerful tools that can be used in order to understand how anion adsorption influences CO adsorption at a given potential and also how CO adsorption and oxidation processes behave as a function of temperature. Again, much like in the case of DEMS, in situ IR studies that monitor CO adsorption and oxidation not only as a function of potential, but also as a function of temperature at a given potential will aid in the determination of the relevance of Ea values calculated at a given potential under comparable experimental conditions (pH, potential, etc.).
3. Temperature dependence and activation energy studies
Section 2 mentioned a number of temperature-dependent MeOH oxidation studies that have been carried out on a variety of surfaces and surface compositions.56,59,72–74,94,114–119,121,130,132,143–146 The focus of many of these studies included examining the change in surface coverage of CO and other poisonous intermediates, changes in anion adsorption and oxide coverages, current density, and turnover rate enhancements, in addition to onset of oxidation potential shifts afforded by increased solution temperatures. The rate determining steps of the oxidation process have also been studied as the temperature is modulated, and recently high pressure electrochemical cells have been used in order to prevent MeOH evaporation so that the system behavior could be evaluated at temperatures above 65 °C.73,94 Commonly, Tafel plots are constructed at given potentials and can be constructed over a variety of temperatures and over a range of pHs.45–48,57,71,81,94,110,114,117,143,147 A discussion of the effect of temperature on Tafel slopes is presented in a paper by Damjanovic.148 Changes in the overall slope brought about by changes in temperature yield insight into the electron transfer rate determining step of the MeOH oxidation pathway at that potential and its dependence on temperature.115,148 These results depend on the electrochemical environment and parameters under which the experiment is carried out, leading to a variety of conclusions throughout the literature concerning the electrochemical rate determining steps in MeOH oxidation. In general, changes in the slope within a given Tafel plot point to mechanistic differences within a given potential range at a given temperature.57,115 It has been mentioned in the literature that the usual fundamental meaning of the Tafel slopes for MeOH oxidation does not apply due to the lack of linearity often observed when E vs. log current density is plotted.71,72 Nonetheless, the literature provides instances where a linear portion of the Tafel plot has been analyzed. For example, using Pt(111) and Pt(110) electrodes, the Tafel slope was measured to be 120 mV per decade over the potential range of 0.0 to 0.25 V vs. Ag/AgCl, indicating that the rate-determining step was the C–H bond scission associated with an electron transfer process; the initial dehydrogenation of MeOH as it adsorbs to the electrode surface in acidic media. On the other hand, the Tafel slope for a Pt(100) electrode was 60 mV per decade over the same potential range, indicating a possible change in the rate-determining step.24,70 This lower value of the Tafel slope is generally related to the oxidation of an adsorbed intermediate (usually CO) rather than an adsorption process. Values of the Tafel slope for Pt–Ru alloy catalysts range from 100–115 mV per decade at room temperature up to 40 °C to 60–195 mV per decade between room temperature and 60 °C, depending on the support used.71,117 Tafel plots in alkaline media were obtained by Volkovich et al.56,57 These plots had four different regions of linearity depending on the concentration of MeOH, making the analysis of these plots very confusing. All of the values for the Tafel slope tend to be obtained at potentials below 0.5 V vs. SHE as this is the most linear region. As the potential increases, the methanol adsorption step tends to become rate-determining.71 Obviously, there is a large range of data and the values of the slope can depend on temperature, fuel concentration, and catalyst surface. The interpretation of these results is difficult to generalize throughout the literature.Chronoamperometry is a valuable tool used to look at changes in current as a function of time, which can be used to further study the kinetics of certain oxidation reactions and poisoning events. Chronoamperometry has been used to determine if the extent of poisoning varies with temperature.72–74,81,94,149 Monitoring current levels with time, as well as the magnitudes of steady-state currents at elevated temperatures compared to room temperature, gives rise to a better understanding of the behavior of surface processes with temperature. Chronoamperometry has also been used to study the extent of CO poisoning on Ptpoly as a function of time by calculating a time-dependent parameter that is a function of the MeOH decomposition charge found using chronoamperometry divided by the CO oxidation charge found from fast cyclic voltammetry.32,73 The result was the determination that other products are being formed besides those that arise via a CO intermediate, which is evidence of a dual pathway mechanism. A study analogous to the one presented above would be beneficial at multiple fuel temperatures in order to look closely at the temperature dependence of CO poisoning over time.
Other kinetic parameters, including transfer coefficients (α ∼ 0.5 under a number of reaction conditions), adsorption rates, and reaction orders of different species contributing to the oxidation MeOH, have also been extracted for systems that use MeOH as the fuel and Pt and Pt–Ru as the catalyst species.25,29,44,45,57,60,70,74,94,110,115,117,118,143,150–152 These parameters all vary with the reaction conditions, electrochemical environment, and the catalyst surface being employed. As an example of the information that can be extracted, reaction orders can be calculated from plots of the log current vs. log concentration at a specific potential. It was determined in a study using a Pt–Ru catalyst for MeOH that values less than 1 corresponded to a rate determining step that involved the adsorption of organic species.115 Reaction order values were also found to depend on potential, with lower reaction orders obtained at low potentials. At high potentials, the reaction order approached 1. In between the extremes, the MeOH oxidation reaction at a Pt–Ru surface is without order, which means that one reaction does not dominate as the rate-determining step.117 Another example of the evaluation of kinetic information was the determination of the standard rate constant and anodic transfer coefficient for the oxidation of MeOH to formic acid, an important step during MeOH oxidation. The analysis was carried out using normal pulse voltammetry. The rate constant was found to be of the order of 10−9 cm s−1, a reasonable value for a slow completely irreversible kinetic process. The transfer coefficient was found to be independent of concentration of MeOH and was ∼0.35, further proof of a slow kinetic process occurring at the anode.150
In many of these temperature-dependent MeOH oxidation studies, an apparent Ea has been calculated. An apparent Ea value determined using electrochemical techniques gives information concerning the chemical rate determining step. These values are simple to calculate by plotting the change in current at a specific potential as a function of the change in temperature using the Arrhenius eqn (3.1). The slope of a linear fit gives the Ea of the process occurring
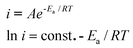 | (3.1) |
Numerous experimental Ea values for MeOH oxidation have been found throughout the literature and have quite a wide range of values. Values found on a bulk Ptpoly surface are not as common as those for Pt–Ru and for nanoscale Pt, and experiments done in acidic media far surpass those done in alkaline electrolytes. In 1969, Volfkovich et al. performed a study of the electrooxidation of MeOH at a smooth Pt electrode in alkaline media.56 The values obtained in 0.05 M MeOH and 1.0 M KOH ranged from 62.8 kJ mol−1 at +0.2 V vs. SHE to 54.4 kJ mol−1 at +0.5 V vs. SHE. These were compared to values found in acidic media on platinized platinum determined by Stenin and Podlovchenko.157 The values in 5.0 M MeOH and 0.1 M H2SO4 were found to range from 93.7–63.6 kJ mol−1 as the potential was varied from +0.35 to +0.5 V vs. SHE. It was determined that Ea values in alkaline media are lower than those in acidic media, but this conclusion is difficult to justify due to the differences in electrolyte and fuel concentration, the nature of the different electrode surfaces, as well as the low potentials at which these experiments were carried out. Poisoning may have played an important role in the determination of these Ea values. In 1981 Raicheva et al. (and references therein) performed one of the few experiments to determine the Ea for MeOH oxidation at given potentials on smooth bulk Pt electrodes in acidic media.119 They recognized that Ea values are potential dependent and differ depending on whether the data were obtained on the anodic or cathodic sweep. The Ea values ranged from 41 kJ mol−1 between +0.86 to +0.97 V vs. SHE, when sweeping the potential to more positive values, to 56 kJ mol−1 between +0.65 V to +0.75 V vs. SHE, when sweeping the potential in the negative direction. The sweep rate was 100 mV s−1, which may be too fast to ignore current transients and would enhance the contribution from surface processes. Wakabayashi and co-workers determined the Eas for MeOH oxidation on thin film Pt using cyclic voltammetry.116 Their values were lower than those obtained by Raicheva et al. and ranged from 15 to 24 kJ mol−1 over the potential range of +0.60 to +0.70 V vs. RHE. These values were obtained in acidic media, and it was observed that Ea increased with increasing overpotential as the potential was swept anodically, but further investigation into this trend was not carried out. An even higher Ea value was reported for Pt/C–Nafion catalysts and was around 70 kJ mol−1 at a rather low potential of +0.4 V vs. SHE.94 These values were compared to others cited previously for Pt/C of 60–70 kJ mol−1, but the potential at which these values were determined was not mentioned. Other references have reported on the variation in Ea values ranging from 22 to 95 kJ mol−1 for a number of Pt, and Pt–alloy surfaces in MeOH.72,74,92,115,117,118,144 These values have been correlated to rate determining steps that range from activation–adsorption controlled, surface diffusion processes, to strictly adsorption controlled, depending on the potential.
Activation energies for some surface processes, such as MeOH dissociation and CO oxidation, have been determined. For example, values for CO oxidation have been determined experimentally on single crystal Pt surfaces and are very high, ranging from ∼111 to 140 kJ mol−1 depending on the surface and the electrolyte.141 The Ea for surface diffusion of CO has been measured to be between 20–30 kJ mol−1. Dissociative adsorption has been approximated at ∼60 kJ mol−1. As was mentioned in section 2, correlation between these values and experimental Ea for MeOH oxidation cannot be carried out until other processes that affect the determination are addressed.
The difficulty in accurate Ea calculations arises from the fact that some of these numbers are given without stating the potential at which they were determined. As was mentioned numerous times in section 2, Ea is potential dependent because a number of different surface processes and adsorption processes that affect the magnitude of the current are potential dependent and also time dependent. The potential at which the Ea value is determined is critical to ascertaining the mechanistic significance of this value. While many of the references that quote values for Ea point out that these values are influenced by surface coverages and other parameters, they do not follow up on how they might influence the Ea calculation. Because over time, and over the potential range of the data collection, the surface becomes covered with adsorbed anions, poisonous intermediates from MeOH oxidation, and oxide, the sweep rate and direction of the potential sweep are parameters that must also be noted when presenting Ea results. Finally, in order to get the most information from the Ea values calculated for nanoscale Pt and Pt-based alloys, a rigorous determination is needed of the Ea of MeOH oxidation on bulk Ptpoly in acidic and alkaline media. In this way, one can make relevant comparisons of how Ptpoly and Pt-based alloys alter the MeOH oxidation pathways at different potentials and under different electrochemical conditions. The following sections will present a complete set of Ea values obtained in acid and alkaline media for the oxidation of MeOH at a bulk Ptpoly surface. A discussion of the factors that influence these Ea values will follow. These factors include anion adsorption, CO poisoning, and oxide formation and reduction. These parameters are all potential dependent, which justifies the trend that Ea values vary as the potential is varied. Because Ea values do vary with potential, but are also calculated by measuring the changes in current as a function of temperature, the potential dependence of these variables will be discussed first, and then a discussion of their temperature dependence will follow. A final analysis of the calculated Ea values will be given taking into account the potential and temperature dependencies of each of the above factors. General guidelines for the analysis of Ea obtained at a given potential will also be suggested in order to facilitate the comparison of Ea values obtained throughout the literature for different catalyst surfaces.
4. Materials and methods
4.1 Electrode and solution preparation
A smooth bulk polycrystalline Pt electrode was used for all experiments. The Pt slug (3 mm diameter) was press fitted into a Teflon mount and electrical contact was made using a machined stainless steel connector as described elsewhere for use in rotating disk voltammetry, as well as steady state, experiments.158 Prior to each electrochemical experiment, the electrode was polished using a polishing wheel and 800 grit and 1200 grit sandpaper (Buehler). The electrode was subsequently polished using a slurry of 1 μm diamond polishing compound and polishing extender (MetaDi-Buehler) and washed with ultra pure water (Millipore Milli-Q, 18 MΩ cm) in order to achieve a mirror finish. The Pt electrode was then immersed in an electrolyte of 0.1 M H2SO4 (99.999% ultra pure, Sigma-Aldrich) or perchloric acid (70%, redistilled to 99.999%, Sigma-Aldrich), depending on the experiment being carried out, and cycled from −0.2 to +1.2 V (all potentials referenced to a Ag/AgCl, sat. NaCl reference electrode fabricated in-house without regard for the liquid junction potential) for 10 min at 100 mV s−1 in order to form a pristine Pt surface. The Pt electrode was cycled from −0.95 to +0.6 V prior to experiments carried out in potassium hydroxide electrolyte (pellets, Mallinckrodt Chemicals). All current densities are calculated using the geometric electrode area. A Pt mesh (30 mm × 5 mm, polished in a propane flame prior to use) spot welded to a 10 cm long Pt wire was employed as the counter electrode for all experiments. All analytical solutions were prepared using ultra pure water. All solutions were deaerated with nitrogen (high purity, Airgas, Inc.) for at least 10 min prior to carrying out an experiment. The N2 was flowed through a hydrocarbon trap (Labclear, No. RGF-250-200) and an oxygen (Messer-Griesheim) trap prior to use. During the experiments, N2 gas was flowed over the solution to minimize the dissolved O2 in solution.4.2 Electrochemistry
All (CV), chronoamperometry, and linear sweep voltammetry (LSV) were carried out using a CV-27 potentiostat (Bioanalytical Systems). The data were acquired using a computer with a National Instruments IEEE-488 general purpose interface card (GPIB) and software written in-house using a Lab Windows platform. Rotating disk experiments were run using a modulated speed rotator and MSRX speed controller (Pine Instrument Company). The shaft and steel connector were both covered with Teflon™ tape for all elevated temperature experiments to prevent contamination. The oxidation of MeOH (high purity, HPLC grade, Burdick and Jackson) was studied using LSV starting at +1.2 V and sweeping negative to −0.2 V at 10 mV s−1. The potential was held at +1.2 V for 5 s prior to commencing the LSV experiment. CV was also run starting from +1.2 V and cycling between +1.2 and −0.2 V at 10 mV s−1. Chronoamperometry was carried out by holding the Pt working electrode at +1.2 V for 5 s, then sweeping to the desired potential at 10 mV s−1, where the potential was held for the remainder of the experiment. The current at that final potential was monitored as a function of time.Temperature-controlled LSV experiments were carried out in order to calculate the Ea of MeOH oxidation at a variety of potentials. The electrode was rotated in these experiments at 3000 rpm, unless otherwise stated, in order to prevent accumulation of CO2 bubbles at the electrode surface. These experiments (as well as CVs and chronoamperometry) were carried out in a double-jacketed electrochemical cell as shown in Fig. 2. The reference electrode was placed in a cell that was jacketed and into which water at a constant temperature of 25 °C was flowed (blue compartment in Fig. 2). In this way, correction of the reference potential due to changes in temperature of the solution is not required. The working and counter electrodes were placed in separate compartments of a second beaker, that was also jacketed (red compartment in Fig. 2), into which temperature-controlled water was circulated using a thermostatic bath (Fisher-Scientific, ±0.01 °C). The reference and counter electrode compartments were separated from the working electrode by medium porosity glass frits. The desired temperature for the MeOH–electrolyte solution was entered into the thermostatic bath controller, and the water was then left to equilibrate to that temperature. The temperature of the MeOH solution was monitored using a digital thermometer until it reached the temperature of the water circulating in the jacketed cell. The cell was equipped with an inlet on the front used for insertion of N2 in order to continually keep the system under a N2 atmosphere. Prior to carrying out the linear sweep voltammetry, the electrode was immersed into the solution and the potential was held at the positive-most potential for 5 s to ensure reproducible oxide coverages before commencing each potential sweep. The desired linear sweep voltammetry measurement was subsequently carried out. Between each temperature increase, the Pt electrode was removed from the MeOH solution, washed with degassed electrolyte and then placed in fresh electrolyte solution until the next experiment was run. This process ensured that the surface did not undergo any changes during the time it took to raise the temperature of the solution. The temperature range studied was from 20 to 60 °C.
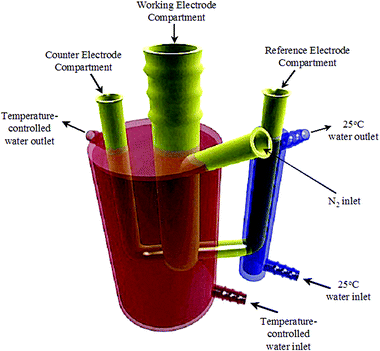 | ||
| Fig. 2 Double-jacketed electrochemical cell used for LSV, CV, and chronoamperometry experiments. The red compartment housed the working and counter electrodes and the temperature of the MeOH solution was adjusted by circulating temperature-controlled water. The blue compartment housed the reference electrode and the solution was kept at constant temperature at all times by circulating room temperature water. | ||
The electrochemical data were plotted and analyzed, and the Ea data were plotted using the Arrhenius eqn (3.1) and were fit using Origin 6.1 graphing software. Error bars represent standard errors, which were calculated within the 95% confidence interval for each data set.
5. Cyclic voltammetry
5.1 Cyclic voltammetry of bulk Ptpoly in various electrolytes at different temperatures
Prior to carrying out the LSV to evaluate the Ea for MeOH oxidation at a Ptpoly surface, CV was carried out at different temperatures in sulfuric, and perchloric acid and potassium hydroxide. These electrolytes varied in pH (from pH = 0.9 in 0.1 M H2SO4 to pH = 13 in 0.1 M KOH) and anion adsorption characteristics (HSO4 > ClO4−). They were used as supporting electrolytes when evaluating Eas for MeOH oxidation. Fig. 3(A–C) presents cyclic voltammograms of clean Ptpoly in each of the three electrolytes at T = 20 °C and 50–55 °C. This temperature range was chosen for subsequent MeOH oxidation experiments due to the low boiling point of MeOH (64.6 °C). As has been observed previously, and evident in Fig. 3, the shapes of the cyclic voltammograms do not change as the temperature of the electrolyte is increased.119,159,160 Likewise, the potentials for hydrogen adsorption/desorption and for oxide formation do not shift significantly with temperature. There is an increase in the amount of oxide formed as the temperature increases. This increase in coverage depends on the nature of the electrolyte. For example, a 16% increase in the oxide reduction charge was measured from Fig. 4A when the temperature was increased from 20 to 70 °C. Fig. 4B shows the integrated charge density vs. temperature for the oxide reduction peak in the voltammetry in Fig. 4A. The charge density increase proved to be linear with temperature within the temperature range of these experiments. The slope of the regression line is 3 μC cm−2 °C−1. It has been found that the adsorption of anions may preclude the formation of platinum surface oxide.75 In the case of perchloric acid, the ClO4− anion is much less adsorbing than bi(sulfate), which should allow for the increase in oxide coverage with increasing temperature to surpass that of an adsorbing electrolyte, such as sulfuric acid. This was the case as observed in Fig. 3C, as the cyclic voltammetry carried out in 0.1 M HClO4, considered a non-adsorbing electrolyte, showed a large increase in oxide reduction charge within a smaller range of temperatures when compared to sulfuric acid. For HClO4, a 19% increase in oxide reduction charge (6 μC cm−2 °C−1) was measured when the temperature was increased from 20 to 50 °C. Because there is no anion adsorption in alkaline media, and free OH− groups adsorb to the Ptpoly surface facilitating oxide formation, the oxide formation in 0.1 M KOH was greatly enhanced with temperature as well. A 28% increase (7 μC cm−2 °C−1) in oxide formation from 20 to 55 °C was observed when using 0.1 M KOH as the electrolyte. Table 1 gives a summary of these results for each electrolyte within the given temperature range of each experiment. This increase in oxide charge with temperature suggests that the adsorption of OH− increases as the temperature is increased, likely representing a delicate interplay of anion adsorption and oxide formation. This factor is important to consider when carrying out temperature dependent experiments where the OH− may be a reactant in the overall oxidation of the fuel of interest. Where other anion adsorption events may be deleterious to fuel oxidation currents, the adsorption of OH− may be advantageous to not only fully oxidize the fuel to the final products, but also to aid in the oxidation of any poisonous intermediates (such as CO) that may form as well. Finally, it can also be observed that the oxide reduction peak becomes sharper as the temperature is increased. This increase in sharpness can be attributed to the enhancement in attractive surface interactions between the adsorbed oxide species, in addition to increased kinetics as the temperature is increased.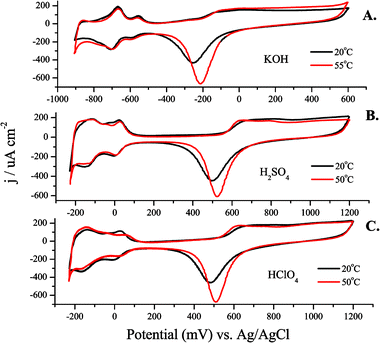 | ||
| Fig. 3 Cyclic voltammogram for a Ptpoly electrode in (A) 0.1 M KOH, (B) 0.1 M H2SO4, and (C) 0.1 M HClO4 electrolyte at different electrolyte temperatures. The sweep rate for each experiment was 100 mV s−1. The sweep was started at 0.0 V and the potential was swept anodically. The Ptpoly electrode was 3 mm in diameter for each experiment. | ||
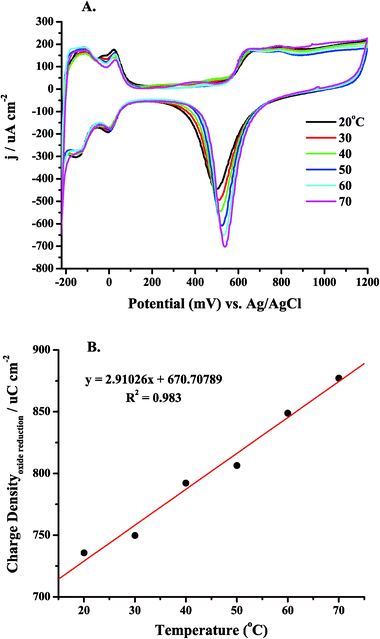 | ||
| Fig. 4 (A) Cyclic voltammetry of a Ptpoly electrode in 0.1 M H2SO4 within the temperature range of 20–70 °C. The sweep rate was 100 mV s−1. The potential sweep was initiated at 0.0 V and was swept anodically. (B) The Pt electrode oxide reduction charge (μC) versus electrolyte temperature (°C) for a Ptpoly electrode in 0.1 M H2SO4. The Ptpoly electrode was 3 mm in diameter for each experiment. | ||
It can also be recognized from Fig. 4A that the Pt oxide reduction peak potential on the cathodic sweep shifts to more positive potentials as the temperature is increased. This shift in oxide reduction potential has clear consequences when looking at the full cyclic voltammogram for MeOH oxidation at the Ptpoly surface. Because the onset of MeOH oxidation observed in the cathodic sweep coincides with the reduction of the surface oxide, a concomitant shift in the onset of MeOH oxidation will also be observed. Fig. 5 plots the Epeak (mV) of the Ptpoly oxide reduction versus temperature (°C) in 0.1 M H2SO4 electrolyte. The slope of this plot is linear over this particular temperature range and predicts a potential shift of +0.7 mV °C−1 change in temperature for the reduction of the Pt surface oxide in 0.1 M H2SO4, further supporting the fact that the oxide formation (and thus reduction) is activation controlled at a bulk Ptpoly surface. The results in 0.1 M HClO4 are comparable to those obtained in sulfuric acid, with a shift in potential of ∼+0.9 mV °C−1. The alkaline electrolyte, on the other hand, shows an even greater shift with temperature. This value is ∼+1.1 mV °C−1. Table 1 presents a summary of these results for each electrolyte. The increase in oxide charge, as well as the potential shift to more positive potentials during oxide reduction, plays a role in the cyclic voltammetric behavior of MeOH oxidation at different temperatures. This will be discussed briefly in section 5.2 and in more detail in section 6.
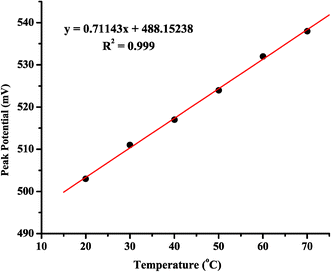 | ||
| Fig. 5 Peak potential (Epeak) for the Ptpoly electrode oxide reduction versus electrolyte temperature (°C) in 0.1 M H2SO4. Data were obtained from the cyclic voltammograms presented in Fig. 4A. The Ptpoly electrode was 3 mm in diameter for each experiment. | ||
5.2 Cyclic voltammetry of methanol oxidation at different temperatures
Initially, CV was carried out for 0.5 M MeOH in three different electrolytes by sweeping the potential anodically. The reproducibility of the onset of oxidation potential of MeOH, as well as the magnitude of the current, was low for the acidic electrolyte systems, especially in the case of 0.1 M H2SO4. This irreproducibility was attributed to the fact that poisonous intermediates, such as CO, would adsorb to the electrode surface at low potentials, poisoning the electrode and altering the nature of the surface. This poisoning causes a diminution in current as well as a positive potential shift (typical of poisoning) in the onset of MeOH oxidation. In the case of 0.1 M H2SO4, anion adsorption would also cause changes in free surface sites for MeOH binding and subsequent oxidation. Instead of initially sweeping anodically, CV was carried out by sweeping cathodically as shown in Fig. 6. The potential sweep for the CV carried out in 0.5 M MeOH in the three different electrolytes was initiated at +1.2 V (0.6 V in alkaline media, Fig. 6C). These results were much more reproducible in terms of the magnitude of the currents, as well as the potential of the onset of MeOH oxidation. As the temperature of the fuel and electrolyte were increased, the currents increased reproducibly, and the onset of oxidation was consistently observed at more negative potentials. This behavior was difficult to obtain when the CV was carried out by initially sweeping in the anodic direction. Because of this reproducibility, the Ea studies were carried out by doing LSV starting at positive potentials and sweeping cathodically. By sweeping cathodically, the Pt surface was covered with an oxide layer, precluding significant anion adsorption and preventing MeOH dissociation from forming CO. If the LSV were carried out by sweeping anodically, the electrode would be partially poisoned by CO at low potentials, the coverage of which would be a function of how long the electrode had been immersed in the solution prior to commencing the potential sweep. Although less critical at low potentials, adsorption of anions should be taken into account depending on the potential at which the electrode is held and for how long it is poised prior to carrying out the experiment. These effects would drastically alter the Ea data obtained, because the current change brought about by the change in temperature would be inextricably convoluted by these un-quantifiable surface adsorption processes.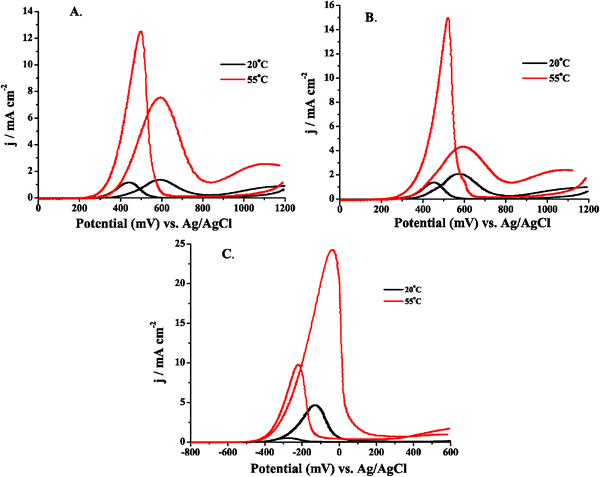 | ||
| Fig. 6 Cyclic voltammograms of 0.5 M MeOH in (A) 0.1 M H2SO4, (B) 0.1 M HClO4, and (C) 0.1 M KOH electrolyte. All sweep rates were 10 mV s−1 and the potential sweep was initiated at +1.2 V (0.6 V in alkaline media) and the potential was swept cathodically. The Ptpoly electrode was 3 mm in diameter for each experiment. | ||
The cyclic voltammograms presented in Fig. 6 exhibit the hysteresis commonly found during MeOH oxidation at a Ptpoly surface. This hysteresis has been attributed to the difference in surface oxide coverages on the anodic and cathodic sweeps.68,161 When the potential was initially swept cathodically, a sharp MeOH oxidation peak was observed coinciding with the reduction of Ptpoly surface oxides as observed in Fig. 4A. The maximum peak current in the acidic electrolytes occurred on the cathodic sweep. It can also be observed in Fig. 6A and B that the onset of oxidation observed on the anodic sweep ranged from +0.325 V to +0.4 V in acidic electrolyte and, in Fig. 6C, from −0.5 V to −0.4 V in alkaline media depending on the temperature of the fuel. As the temperature of the solution was increased, the onset of oxidation potential was shifted negative in all cases. The onset of oxidation in alkaline media was ∼100 mV more negative than the onset of oxidation in acidic media when the difference in pH is taken into account, as has been observed previously.35,36,56 After the onset of oxidation, the current on the anodic sweep increased coinciding with the initiation of Pt oxide formation, which aids in MeOH oxidation. The current then decreased sharply as the Pt oxide became a poison and covered the surface, precluding the adsorption and subsequent oxidation of MeOH. It is interesting to note that the maximum peak current obtained during the cathodic sweep was greater than that observed for the anodic sweep for both acidic electrolytes as observed in Fig. 6A and B. However, in the case of the alkaline media in Fig. 6C, the reverse was observed so that the anodic sweep exhibited a higher current than the cathodic sweep. This also makes sweeping cathodically during the LSV experiments even more interesting because differences in the Eas may be observed that correlate with the different behavior between electrolytes of high and low pH. It was also observed that the current (and thus current density as the same electrode was used for all experiments) for MeOH oxidation was higher on the anodic sweep in alkaline media than in either acidic system. This was attributed to the lack of poisoning from anion adsorption and to the adsorption of OH−, which may help to diminish CO poisoning and also facilitate the oxidation of MeOH to HCOO− and to CO2. It has also been observed that due to OH− adsorption at low potentials in alkaline media, the adsorption of MeOH is lower than in acidic media, but the rate of oxidation of MeOH is enhanced when compared to that in acidic electrolyte, leading to the current enhancements observed.57 The lower current on the cathodic scan is often ascribed to (bi)carbonate formation and its surface adsorption, which then causes the decrease in current on the cathodic sweep in alkaline media.1,31,34,46–50,53,55,149
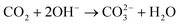 | (5.1) |
Eqn (5.1) shows how carbonation can occur in alkaline media. Even though this surface poisoning may occur, the currents observed on the cathodic sweep in alkaline media are comparable to those observed in acidic media, as shown in Fig. 6, further supporting the observation that MeOH oxidation kinetics are enhanced in alkaline media. The current was also larger for MeOH oxidation in 0.1 M HClO4 when compared to MeOH oxidation in 0.1 M H2SO4, ostensibly because of (bi)sulfate’s stronger anion adsorption.
6. Activation energies for methanol oxidation
It was observed in Fig. 6A and B that the maximum peak current obtained for MeOH oxidation in acidic media occurs on the cathodic sweep. This maximum current is obtained at potentials more negative than the maximum current obtained on the anodic sweep. The ability to maximize the current density of a fuel at a minimum overpotential is especially desirable for fuel cells. Information that can be gained about the surface processes, as well as the MeOH oxidation pathway being followed at that potential of maximum current, will be useful in the design of new catalysts. These new catalysts can be designed to enhance and/or enable those surface processes which are key in obtaining high current values, and can also be designed to preclude certain detrimental processes. Ea values obtained in this region of maximum activity may help to point to specific processes that are rate-determining in acidic media. Studying the Ea values obtained in alkaline media, as in Fig. 6C, will allow comparisons to be made concerning the MeOH oxidation mechanism in acidic and alkaline media. It is desirable to be able to obtain in acidic media the enhanced performance observed in alkaline media. The determination, and especially the comparison, of Ea values can give insight into the performance differences between the electrolytes in terms of specific steps in the MeOH oxidation pathway that are being carried out in each media. The Ea information, as well as an in-depth understanding of the surface processes that govern Ea, may allow catalysts to be designed that will be able to generate the same maximum current observed on the cathodic sweep for MeOH oxidation over a broader potential range, making MeOH a more viable fuel for DMFCs.E a values have been determined using CV and chronoamperometry throughout the literature as cited in section 3. Normally, cyclic voltammograms are obtained by sweeping the potential anodically at a given sweep rate and the data for the Ea determination is taken at the onset potential for MeOH oxidation. This procedure introduces complications due to surface processes like anion adsorption and CO adsorption that can occur at low potentials. Another way to determine Ea would be to perform a series of potential step experiments, at a number of temperatures, at the potentials of interest by initially holding the potential at either a very positive or very negative value and then stepping to the potential of interest. While this procedure may remove the possibility of surface poisons at the time of the initial step, other complications are inherent to this experiment. A potential step experiment would introduce complications because the Ea values are calculated by measuring the current increase as a function of temperature. The magnitude of the current is sensitive to the surface poisons formed during MeOH oxidation, i.e. CO, as well as anion adsorption. If a potential step experiment were carried out, a certain delay between the potential step and the current measurement would be necessary to minimize contributions to the measured current from double layer charging. This delay would also allow anions to adsorb and CO species to form, which would distort the current measured. These adsorption processes would also be affected by the change in temperature, making deconvolution of these distortions impractical. Initial experiments carried out by such potential step techniques did not provide reliable data from which to calculate Ea values.
In order to minimize the poisoning factors, as well as take advantage of studying the potential range over which the maximum MeOH current was observed, LSV was carried out. Ea values were determined from a numerous LSV experiments at 5 °C intervals from 20 to 60 °C. The procedure for carrying out these measurements was described in section 4. The potential was swept cathodically for reasons described in section 5.2. In order to minimize anion adsorption and CO poisoning, as well as to minimize any contributions from double layer charging, the potential was swept cathodically at a relatively slow sweep rate of 10 mV s−1 in order to attenuate the poisoning effects and to obtain currents that were representative of MeOH oxidation.
During the LSV experiments, the electrode was rotated in order to remove any bubbles (ostensibly CO2) that would form at the electrode surface at elevated temperatures, as described in section 4.2. The rotation also mitigated the diffusion of any soluble intermediates, such as HCOOH and HCHO, back to the electrode surface for further oxidation after their initial formation. The rotation rate chosen was 3000 rpm. In order to conclude that the Ea values calculated did not depend on rotation rate, experiments were carried out at a slower rotation rate of 1000 rpm. The Eas obtained fell within the error of the measurements made at 3000 rpm. The only difference between the two experiments was that the magnitude of the current was larger in the case of 3000 rpm, as would be anticipated due to an increase in mass transport. Finally, the experiments were carried out using 0.5 M MeOH at all times. In order to ensure that Ea did not change appreciably with changes in MeOH concentration, an experiment was carried out using 1.0 M MeOH. This experiment yielded Ea values that fell within the error of the measurements obtained in 0.5 M MeOH and 0.1 M H2SO4. A concentration of 0.5 M MeOH was chosen for all experiments in order to be consistent. No such MeOH concentration experiments were carried out in KOH or HClO4 electrolytes. A more exhaustive concentration-dependence study of Ea will be carried out in future work, as it has been shown in section 3 that the concentration of MeOH can affect the oxidation product ratios, and thus the MeOH oxidation pathway followed. The electrolyte concentrations were kept low in order to minimize anion adsorption, but comparisons can still be made between H2SO4 (an adsorbing electrolyte) and HClO4 (non-adsorbing).
The improved kinetics for MeOH oxidation, as well as the lower surface poisoning of the bulk Ptpoly species in alkaline media, made it possible to carry out LSV experiments sweeping both anodically and cathodically. Sweeping anodically is useful because unlike acidic media, the maximum current for MeOH oxidation was observed on the anodic sweep as was pointed out in section 5. Similar experiments in acidic media were attempted, but did not give reliable and reproducible results, as predicted in section 5.2, due to anion adsorption and CO poisoning at low potentials prior to the onset of MeOH oxidation.
6.1 E a results obtained in acidic supporting electrolyte
Fig. 7A and B present the linear sweep voltammograms for 0.5 M MeOH in 0.1 M H2SO4 and 0.1 M HClO4, respectively, over a range of temperatures. The LSV was initiated at +1.2 V and the potential was swept to −0.22 V vs. Ag/AgCl. As was observed in the cyclic voltammogram of clean bulk Ptpoly in pure supporting electrolyte with no MeOH, the peak potential, Epeak, for MeOH oxidant (and concomitant oxide reduction) shifted to more positive potentials as the temperature was increased. This shift in 0.5 M MeOH was measured from the LSV in Fig. 7, as well as from the cyclic voltammograms in Fig. 6. In 0.1 M H2SO4, the shift was 1.6 mV °C−1 and in 0.1 M HClO4 it was 1.9 mV °C−1. These values are quite similar to each other and are both larger than the shifts observed in section 5.1, using just pure electrolyte. This larger potential shift can be attributed to the enhanced turnover rate of adsorbed MeOH, and subsequent enhanced surface oxide consumption, as the temperature of the solution is increased. Oxide reduction is an activated process, as is the adsorption and oxidation of MeOH. The oxidation peak observed in the LSV in Fig. 7A and B was attributed to the concomitant reduction/consumption of surface oxides as MeOH was being oxidized.36,68,161 As the potential was swept cathodically, MeOH was adsorbed and oxidized at an increasingly rapid rate as the temperature was increased. Because MeOH was adsorbed at increasingly more positive potentials, it follows that surface oxides are consumed at more positive potentials in order to form CO2. In other words, as the turnover rate for MeOH oxidation increases, the consumption of surface oxide is increased. The oxide is consumed more quickly at a given potential in a MeOH-containing electrolyte compared with the results in pure electrolyte and will be consumed more quickly as the temperature is increased due to the activation of MeOH oxidation and oxide reduction with temperature. These effects shift the peak potential to even more positive values than was observed for a Ptpoly electrode in pure electrolyte. As can be observed from the magnitude of the currents in Fig. 7, anion adsorption was still a factor that must be considered due to the significantly lower currents consistently observed in 0.1 M H2SO4 electrolyte when compared to 0.1 M HClO4.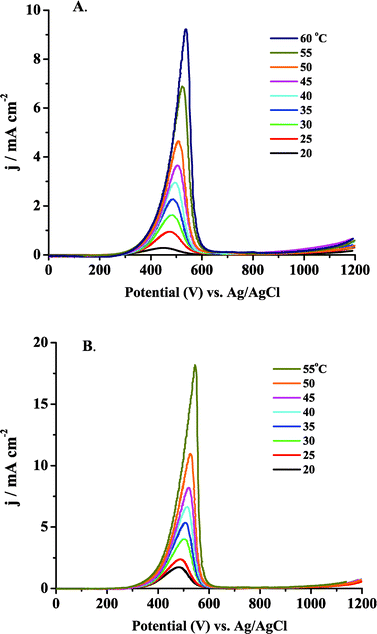 | ||
| Fig. 7 LSV in 0.5 M MeOH in (A) 0.1 M H2SO4 and (B) 0.1 M HClO4 at different temperatures. The sweep rate was 10 mV s−1. The LSV was carried out by sweeping cathodically. The Ptpoly electrode was 3 mm in diameter for each experiment. | ||
Apparent Ea values were obtained from the data in Fig. 7 by plotting ln (current) as a function of 1/T at a given potential. Fig. 8A and B show representative Arrhenius plots for 0.5 M MeOH in 0.1 M H2SO4 and in 0.1 M HClO4, respectively. The slope, intercept, and R values of the linear fits are displayed on each graph. The potential at which the specific data points were obtained is also labeled on the plot. Table 2 provides apparent Ea values at 8 potentials for each electrolyte. A large range of Ea values is observed for each medium. At the most positive potentials, as the current increased abruptly, governed by the commencement of oxide reduction/consumption, the highest apparent Eas are observed. As the potential was swept more negative, Ea decreased. They approached their lowest values as the current sharply dropped to very low values at potentials where there was little driving force for MeOH oxidation. It can be observed from Table 2 (and will be discussed further in section 7) that the apparent Ea values for both acidic media were similar. Fig. 9A and B show the apparent Ea values for 0.5 M MeOH in 0.1 M H2SO4 and 0.1 M HClO4, respectively, as a function of the potential at which these Ea values were obtained. The shapes of the plots in Fig. 9 look similar to the LSV responses observed in Fig. 7. The apparent Ea obtained at the peak potential for 0.5 M MeOH was approximately 69 ± 2 kJ mol−1 in 0.1 M H2SO4 and 66 ± 5 kJ mol−1 in 0.1 M HClO4, respectively. That the apparent Ea was a function of potential is clear, but this important fact has been only mentioned in the literature and not elaborated upon. The importance of the potential at which the Ea is calculated dramatically affects the values obtained and thus limits the comparisons that can be made between different experiments, as well as between different catalyst materials. Section 7 will discuss the possible reasons why the apparent Ea values do indeed depend on potential, as well as the phenomena that need to be taken into account when analyzing the final Ea values obtained. Factors such as anion adsorption, intermediate poisons, and oxide formation, must be analyzed for each individual system prior to comparing Ea values for different materials in different electrochemical environments. It must also be determined which Ea gives the most insight into the oxidation pathway being followed at a specific potential (i.e., one minimally affected by adverse surface processes).
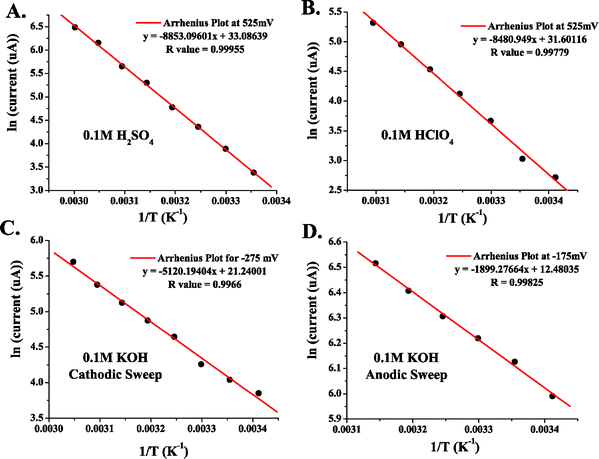 | ||
| Fig. 8 Arrhenius plots for 0.5 M MeOH in (A) 0.1 M H2SO4, (B) 0.1 M HClO4, and (C) 0.1 M KOH for LSV carried out by sweeping cathodically. Plot (D) is in 0.1 M KOH and was obtained from LSV data taken when sweeping anodically. | ||
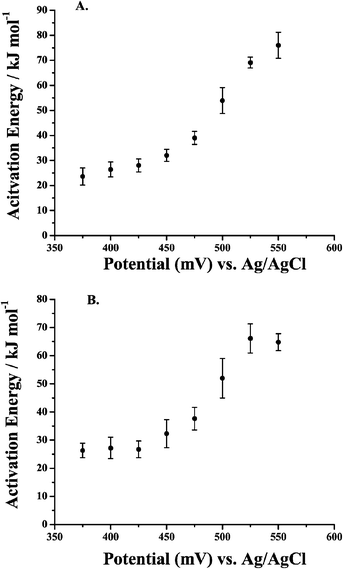 | ||
| Fig. 9 Activation energies as a function of electrode potential for 0.5 M MeOH in (A) 0.1 M H2SO4 and (B) 0.1 M HClO4. | ||
| E at which Ea values were calculated in 0.1 M acid electrolyte/mV | E a values obtained during the cathodic potential sweep from +1.2 V to −0.22 V in 0.1 M H2SO4 electrolyte/kJ mol−1 | E a values obtained during the cathodic potential sweep from +1.2 V to −0.22 V in 0.1 M HClO4 electrolyte/kJ mol−1 |
|---|---|---|
| 375 | 24 ± 3 | 26 ± 3 |
| 400 | 26 ± 3 | 27 ± 4 |
| 425 | 28 ± 3 | 27 ± 3 |
| 450 | 32 ± 2 | 32 ± 5 |
| 475 | 39 ± 3 | 38 ± 4 |
| 500 | 54 ± 5 | 52 ± 7 |
| 525 | 69 ± 2 | 66 ± 5 |
| 550 | 76 ± 5 | 65 ± 3 |
6.2 E a results obtained in alkaline supporting electrolyte
Fig. 10 show LSV data for 0.5 M MeOH in 0.1 M KOH. In Fig. 10A, the potential was swept from an initial potential of −0.9 to +0.6 V. In Fig. 10B, the potential sweep was initiated at +0.6 V and swept to −0.9 V. As was noted in section 5, the magnitude of the current on the anodic sweep was larger than on the cathodic sweep. This is the reverse of the behavior observed in acidic media. The data obtained on the anodic sweep was more reproducible and an Arrhenius-type plot yielded highly linear results. This behavior was due to the lack of competitive anion adsorption accompanying oxide formation in the KOH electrolyte, as well as that OH− adsorption to form Pt oxide occurs at much lower potentials (starting in the Hads region) than in acidic media. This facilitates CO oxidation, and/or lowers the probability of following the pathway to CO formation, instead forming the formate intermediate. In much of the literature on MeOH oxidation in alkaline media, an explanation for the difference in the magnitude of the current on the anodic and cathodic sweep has rarely been discussed. The lower current observed on the cathodic potential sweep has often been attributed, in part, to the formation of (bi)carbonate at high potentials (eqn (5.1)).1,31,34,46–50,53,55,149 The (bi)carbonate formed can prevent a complete surface oxide layer from forming. As the potential is swept cathodically, the oxide is reduced, but the (bi)carbonate remains adsorbed onto the Ptpoly surface. With a lower number of surface sites available for MeOH adsorption as compared to the anodic sweep, a decrease in current is observed. As the potential is swept to more negative values, the adsorbed anions are removed. Significant surface coverages of (bi)carbonate formed in KOH electrolyte have not been cited. It is not clear whether rotation of the electrode can preclude some of the carbonation effects, and only small amounts have been detected using IR techniques.53,55 For now, it can only be speculated that under these experimental conditions, (bi)carbonate adsorption can be considered one of the factors that causes a diminution in the MeOH oxidation current during a cathodic sweep in KOH electrolyte.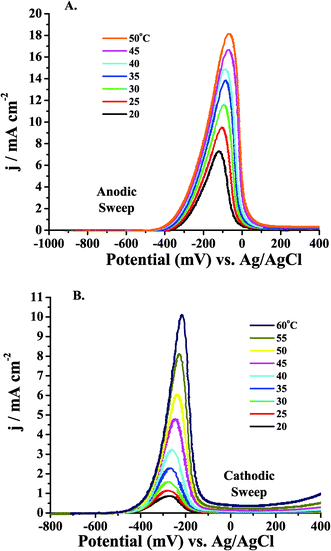 | ||
| Fig. 10 LSV in 0.5 M MeOH in 0.1 M KOH sweeping at different potentials (A) anodically and (B) cathodically. The sweep rate was 10 mV s−1. The Ptpoly electrode was 3 mm in diameter for each experiment. | ||
Using the data in Fig. 10, apparent Ea values could be calculated at specific potentials. Those results are presented in Table 3. Linear Arrhenius plots were obtained for a wide range of potentials on the forward and reverse potential sweeps. An example of these plots is given in Fig. 8C and D. The potentials in Table 3 are presented both as they are observed in alkaline electrolyte vs. Ag/AgCl and also with a correction factor in order to shift the values to make them comparable to the acidic electrolyte media results. The linear sweep voltammograms were taken over a range of temperatures. Because the reference electrode potential is dependent on temperature, the working electrode potential will shift depending on the temperature employed. In order to compare the voltammetry within each data set obtained for a single electrolyte, between alkaline and acidic electrolytes, and also to be able to calculate Ea at specific potentials, the voltammetry must be referenced to one specific temperature, with this potential shift taken into account. Because the reference electrode in these experiments was kept isolated at a constant temperature, no correction factors must be applied to the LSV data in order for comparisons to be made concerning the potentials. All LSV data obtained was reference to a Ag/AgCl reference electrode held at a constant 298.0 ± 1 K. The correction factor for the alkaline system takes into account the pH difference between the alkaline and acidic electrolytes. This correction factor was calculated using the Nernst equation and the potential of the alkaline results were shifted 710 mV (0.059 V for each pH unit at 298 K) more positive in order to make direct comparisons with the acidic electrolyte results given in Table 2.162
| E at which Ea values were calculated in 0.1 M KOH electrolyte/mV | E for 0.1 M KOH data +710 mV to compare with acidic electrolyte/mV | E a values obtained during the anodic potential sweep/kJ mol−1 | E a values obtained during the cathodic potential sweep/kJ mol−1 |
|---|---|---|---|
| −375 | 335 | — | 36 ± 4 |
| −350 | 360 | 39 ± 4 | 33 ± 5 |
| −325 | 385 | 38 ± 5 | 34 ± 3 |
| −300 | 410 | 33 ± 7 | 37 ± 3 |
| −275 | 435 | 27 ± 2 | 45 ± 4 |
| −250 | 460 | 22 ± 2 | 50 ± 5 |
| −225 | 485 | 22 ± 4 | 60 ± 6 |
| −200 | 510 | 20 ± 3 | 74 ± 4 |
| −175 | 535 | 18 ± 4 | 71 ± 1 |
| −150 | 560 | 18 ± 4 | 86 ± 10 |
| −125 | 585 | 16 ± 1 | — |
| −100 | 610 | 19 ± 1 | — |
| −75 | 635 | 23 ± 2 | — |
The Ea values shown in Table 3 are plotted in Fig. 11 as a function of the potential at which they were obtained. It can be observed that the Ea values calculated for the anodic sweep differ greatly from those calculated on the cathodic sweep. As the potential was swept anodically, the Ea values decreased with potential and then began to increase again as shown in Fig. 11A. The maximum value for the Ea on this sweep was 39 ± 4 kJ mol−1 obtained at the most negative potential of −350 mV, corresponding to the onset of MeOH oxidation. The minimum Ea obtained on the anodic sweep was 16 ± 1 kJ mol−1 at −125 mV, which corresponds to the potential at which the current reached 95% of its maximum value on the anodic sweep. As the current reached its maximum value on the anodic sweep, the apparent Ea values begin to increase again, concomitant with oxide formation replacing the process of OH− adsorption. On the cathodic sweep, Ea results similar to those obtained in acidic electrolyte were observed. As the potential is swept cathodically, the Ea values decrease, reaching a minimum value of 33 ± 5 kJ mol−1 at potentials where the driving force for MeOH oxidation is lower. The largest value for the Ea obtained on this sweep was nearly 90 kJ mol−1. This value is obtained at the onset of MeOH oxidation around −200 mV, following the initiation of oxide reduction on the Ptpoly surface. This maximum value on the cathodic sweep is significantly larger than the maximum value obtained on the anodic sweep by about 50 kJ mol−1. At the potential corresponding to the maximum peak current, the Ea obtained was 50 ± 5 kJ mol−1. This is dramatically different from the value of 19 ± 1 kJ mol−1 obtained at the potential corresponding to the maximum peak current on the anodic sweep in alkaline electrolyte. In order to see more clearly the differences in Ea obtained on the anodic and cathodic sweeps in alkaline media, Fig. 12 plots the Ea as a function of the potential at which they were obtained superimposed on a cyclic voltammogram for 0.5 M MeOH in 0.1 M KOH at a sweep rate of 10 mV s−1. The values of the apparent Ea clearly depend on the applied potential, as well as on the direction of the sweep. The Ea values intersect around 35 kJ mol−1, at ca. −325 mV, where the onset of oxidation of MeOH occurred on the anodic sweep and where the current for MeOH oxidation began to decrease on the cathodic sweep. The magnitude of the current also differs at this intersection point. On the cathodic sweep, the current is nearly 1.1 mA cm−2 greater than on the anodic sweep, as has been observed previously.68 The dependence on the directionality of the sweep is clearly an important point, which will be discussed in more detail in section 7. The processes occurring at the Ptpoly surface, as well as the oxidation intermediates formed during the potential sweep, play a role in the value of the apparent Ea and must be taken into account when using the Ea value to compare results from different experiments and different materials.
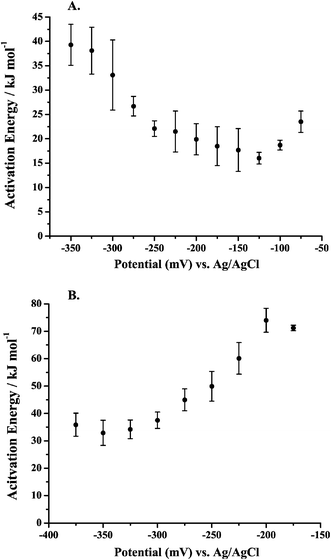 | ||
| Fig. 11 The activation energies, as a function of electrode potential, of 0.5 M MeOH in 0.1 M KOH supporting electrolyte on the (A) anodic sweep, and (B) cathodic sweep. | ||
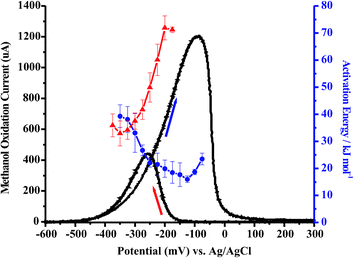 | ||
| Fig. 12 Complete set of Ea data as a function of potential for 0.5 M MeOH in 0.1 M KOH. The blue circles represent Ea values obtained when sweeping the potential anodically. The red triangles represent Ea values obtained when sweeping the potential cathodically. A cyclic voltammogram obtained in 0.5 M MeOH with 0.1 M KOH as supporting electrolyte is superimposed in order to show the shape of the voltammetry as it corresponds to the Ea values obtained at a given potential. The sweep rate was 10 mV s−1. | ||
7. Discussion
7.1 Comparison of apparent activation energy values in different electrolytes
Fig. 13 plots the apparent Ea values obtained in acidic and alkaline media as a function of the potential at which the values were obtained when the potential was swept cathodically. The potentials for the alkaline system have been shifted positive by 710 mV in order to account for the pH difference between 0.1 M KOH and the acidic electrolytes so the data can be directly compared to acidic media. Fig. 13 shows not only the difference in Ea values obtained in alkaline media and acidic media, but also shows that the Ea values obtained in 0.1 M H2SO4 and in 0.1 M HClO4 are nearly equal. The values obtained in alkaline media are between 10–20 kJ mol−1 greater than those obtained in acidic media. Differences in these Ea values can be attributed to the extent of anion adsorption, the behavior of surface oxide processes, and the formation/adsorption of poisonous intermediates during the oxidation of MeOH (i.e. CO). Clearly, these processes are potential dependent, which reflects the fact that in all electrolyte media, the Ea values obtained are a function of the potential at which they are calculated. These potential dependent processes also explain the directionality of the apparent Ea data. It has been observed that the cyclic voltammograms on the anodic and cathodic potential sweeps are different, owing to different processes occurring at the electrode surface and different intermediates being formed during MeOH oxidation depending on the potential. These processes also are time dependent, which in the case of LSV and CV, corresponds to being sweep rate dependent. As the sweep rate decreases, the coverage of adsorbed species increases at low potentials. Thus, when comparing Ea values, not only is the potential at which these values were obtained important, but also how one got to that potential (i.e. sweep rate, sweep direction, etc.) is clearly important and can have a large impact on the magnitude of the Ea values obtained. These processes must be considered when comparing Ea values. These underlying processes that affect the adsorption of MeOH, compounded with the complexity of the MeOH oxidation pathways, make the assignment of Ea values to specific steps in the MeOH oxidation process quite challenging. The use of Ea values for comparison between different catalyst surfaces is a laudable goal, but the parameters indicated above must be taken into account, at least qualitatively, in order to make an effective comparison.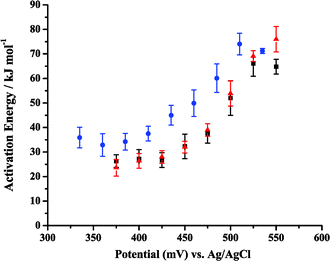 | ||
| Fig. 13 Comparison of apparent Ea values as a function of the potential at which they were obtained in 0.5 M MeOH with different electrolytes. The potential was swept cathodically at a sweep rate of 10 mV s−1. Black squares represent 0.1 M HClO4, red triangles represent 0.1 M H2SO4, and blue circles represent 0.1 M KOH data. | ||
Three of the most important processes pertaining to this system that depend on potential, and possibly on temperature, will be discussed in more detail in the following sections, beginning with the role that anion adsorption plays in the determination of Ea values. Poisonous intermediates, specifically COads, will also be discussed in terms of the LSV experiments carried out here. Finally, the role of oxide coverage will be discussed.
7.2 Factors that influence activation energies
In an electrochemical context, Ea values are determined experimentally by varying the system temperature and measuring the resultant change in current. Implicit in this procedure are the twin assumptions that (1) the only effect of the temperature change is on the kinetics of the process of interest and (2) that any variation in current is exclusively the result of the change in temperature. While these assumptions hold in an ideal case, in relatively complex systems, such as the one under study here, other factors become relevant and can distort the calculated Ea. More specifically, Ea is considered to be an intensive property. The turnover rate (molecules of MeOH oxidized multiplied by Pt site−1 s−1) for a Ptpoly electrode at a given temperature and potential is also an intensive property. Current, and even current density calculated using geometric area, is an extensive property. Changes in the electrode area due to adsorption processes do not change the turnover rate of the system, but can change the magnitude of the current measured. The Ea values in the present case are calculated by varying the temperature and measuring the change in current, not turnover rate. In order to carry out the Ea calculation using turnover rate, the exact electrochemically active area for each linear sweep must be determined, which implies knowledge of the exact surface coverages of each adsorbed intermediate at each temperature and potential of interest in order to calculate the turnover rate. Because the LSV experiments facilitate the extraction of currents, and not turnover rates, surface coverages of adsorbates can introduce distortions into the Ea calculation. These surface coverage changes do not change the turnover rate, but do change the magnitude of the current, which is being used in the Ea calculation. The current measured in the LSV experiments depends upon potential, adsorption of intermediate poisons formed during MeOH oxidation, anion adsorption from the electrolyte, and oxide coverage changes. While the potential of the experiment can be controlled, and the Ea values can be calculated at a specific potential, the effects of surface adsorption processes cannot be deliberately controlled.In order to minimize the distortions introduced into the Ea calculation by using current values, and not turnover rates, it is imperative to determine whether changes in current at a given potential are the result of only the temperature increase. For instance, if increasing the temperature of the system decreases the coverage of COads (see section 7.2.3 for more on the coverage of CO as a function of temperature), then the portion of active surface area of the electrode will increase. In this case, an increase in current with increasing temperature is actually a convolution of a change in kinetics (which is the desired information) and a change in surface area. While both of these will affect the calculated, “apparent” Ea, only the change in kinetics affects the “true” Ea. It is clear that as long as the surface processes occurring during MeOH oxidation are not appreciably affected by temperature, i.e., if adsorption coverages stay constant as the temperature is increased, then the Ea can be calculated without worrying about the effects of adsorption. This would provide an Ea comparable to one calculated using turnover rate.
While it is experimentally difficult to keep all parameters (potential, adsorption of poisons, anion adsorption, and oxide coverage, see sections 7.2.1–7.2.4) under control, a useful check can be applied to determine if a given change in the system affects the “true” Ea, or only the “apparent” Ea. Current is the product of an active site’s “turn-over rate” and the “number of active sites.” Only system changes that affect the “turn-over rate” affect the “true” Ea, while a change to either factor affects the “apparent” Ea. Therefore, in the previous example, where increasing temperature decreased COads coverage, it is clear that the decrease in CO coverage increases the number of active sites, not the turn-over rate, thereby producing an “apparent” Ea higher than the “true” Ea. Such an analysis makes clear not only what variables distort the Ea calculations, but also the direction in which the distortion occurs. In addition, because both potential and temperature are changing throughout the LSV experiments, the contributions of both of these processes to the current enhancements observed must be deconvolved. It is therefore important to determine how these surface adsorption processes depend on the applied potential, timescale and direction of the potential sweep, in addition to how they are affected by temperature.
The next sections will address the behavior of surface processes as a function of potential and the timescales over which these processes are occurring. The temperature dependence will also be addressed for anion adsorption, CO adsorption, and oxide formation processes.
Other surface processes, specifically CO adsorption and anion adsorption, and their behavior as a function of potential, can be studied by carrying out “sweep and hold” chronoamperometry experiments at a given temperature. The cathodic LSV in Fig. 7 and Fig. 10B show that the current decreases as the potential becomes more negative. This decrease could be due to surface adsorption processes, such as CO poisoning, in addition to the decrease in driving force. Chronoamperometric experiments can be carried out to determine the time dependence of the current decrease at a given potential. This current decrease, measured over a certain timescale, can be analyzed to demonstrate that on the timescale of the LSV experiments used to calculate Ea, the current should not decrease appreciably due to surface poisoning (COads or anion adsorption). This finding would eliminate the complication of time-dependent surface coverage changes. If the surface processes are not playing a major role in the current decrease, the interpretation of the Ea results may be easier because only changes in surface coverages of poisons with temperature need to be considered.
In this case, chronoamperometry was carried out at 30 °C in 0.5 M MeOH and either 0.1 M H2SO4 or 0.1 M HClO4. The chronoamperometric experiments were carried out much like the LSV experiments in that the electrode was rotated at 3000 rpm and the potential was swept cathodically at 10 mV s−1 after holding the electrode at +1.2 V for 5 s. Once the potential at which the experiment was to be carried out was reached, the potential sweep was stopped and the potential was held for 2000 s. Fig. 14 shows the first 200 s of those experiments at 8 different potentials that span the range observed for MeOH oxidation on the cathodic sweep in each electrolyte. As can be seen in Fig. 14, the current decreases as a function of time, at a given potential. This current decrease can be attributed to adsorption of poisonous intermediates, anion adsorption in the case of the H2SO4 system, and the decrease in production of CO2 as the surface oxide was consumed, leading to the formation of other soluble intermediates that are removed from the electrode surface with the rotation of the electrode. An induction period prior to a decrease in current can be observed when the potential was stopped and held between 500–560 mV. This induction period will be commented upon further in section 7.2.4 and is present due to the way the experiment was carried out. As the potential was swept cathodically, reduction of surface oxides occurred. When the potential was held between 500–560 mV, complete surface oxide reduction did not take place and substantial oxide coverage was still present. As the potential was held, the initial current increase was due to MeOH oxidation being facilitated by the residual oxide at the Ptpoly surface. After this oxide was depleted, the current then began to decrease.
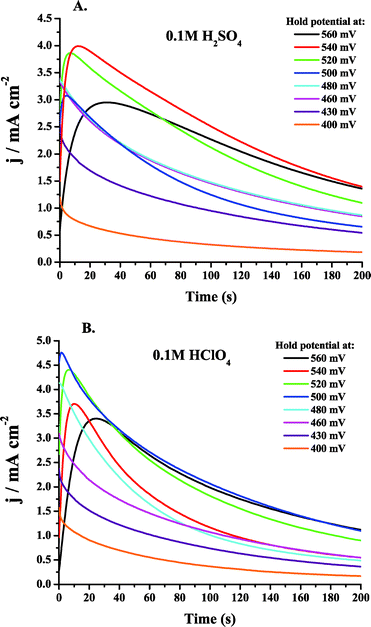 | ||
| Fig. 14 Chronoamperometry for the oxidation of 0.5 M MeOH in (A) 0.1 M H2SO4 and (B) 0.1 M HClO4. The potential was swept negatively from +1.2 V until the holding potential was reached. The potential sweep was then stopped and held at the potentials labeled above. The initial sweep to the holding potential was at 10 mV s−1, and the electrode was rotated at 3000 rpm. The Ptpoly electrode was 3 mm in diameter for each experiment. | ||
The current decrease was more rapid at early times and, as the poisoning progressed, became slower. In the H2SO4 system, the initial current decrease was ∼0.9% s−1, but at longer times near 200 s, the current decrease drops to 0.4% s−1. In the case of the HClO4 system, the initial current decreased 1.2% s−1, and at the latter times it dropped to 0.5% s−1. Considering the timescales of the LSV experiments in section 6, where every 10 s corresponds to a 100 mV change in applied potential, it is clear that the poisoning coverages are going to be minimal. Simple analysis of this current decrease is given in Table 4 for 0.5 M MeOH in 0.1 M H2SO4. Results for 0.5 M MeOH in 0.1 M HClO4 are presented in Table 5. The time at which the potential sweep was stopped and the electrode was held at a specific potential is designated t0 = 0 s. It can be determined that at the lower potentials, where oxide coverages are low, the time it takes for the current to decrease to 75% is nearly 30 s in 0.1 M H2SO4 and 20 s in HClO4. As the potential is decreased in both acidic media, the time it takes the current to reach 75% of the initial value decreases. This is due to the lack of oxide on the Ptpoly surface. The oxidation of adsorbed CO is not facilitated at progressively more negative potentials, causing the surface to become poisoned at a faster rate, in turn causing a decrease in current.
| Potential/mV | Time to reach 75% of initial i/s | Time to reach 50% of initial i/s | Time to reach 25% of initial i/s | Time to reach 10% of initial i/s | ||||
|---|---|---|---|---|---|---|---|---|
| t 0 = 0 s | t 0 = 10 s | t 0 = 0 s | t 0 = 10 s | t 0 = 0 s | t 0 = 10s | t 0 = 0 s | t 0 = 10 s | |
| 560 | — | — | — | — | — | — | — | — |
| 540 | — | 71 | — | 141 | — | 271 | — | 650 |
| 520 | — | 55 | — | 113 | — | 222 | — | 500 |
| 500 | — | 38 | — | 80 | — | 174 | — | 456 |
| 480 | 25 | 38 | 78 | 97 | 202 | 228 | 460 | 536 |
| 460 | 25 | 38 | 78 | 97 | 202 | 228 | 460 | 495 |
| 430 | 21 | 36 | 80 | 94 | 194 | 224 | 430 | 493 |
| 400 | 4 | 30 | 25 | 68 | 111 | 177 | 290 | 380 |
| Potential/mV | Time to reach 75% of initial i/s | Time to reach 50% of initial i/s | Time to reach 25% of initial i/s | Time to reach 10% of initial i/s | ||||
|---|---|---|---|---|---|---|---|---|
| t 0 = 0 s | t 0 = 0 s | t 0 = 0s | t 0 = 0 s | t 0 = 0 s | t 0 = 0 s | t 0 = 0 s | t 0 = 0 s | |
| 560 | — | — | — | — | — | — | — | — |
| 540 | — | 32 | — | 58 | — | 125 | — | 309 |
| 520 | — | 35 | — | 79 | — | 170 | — | 366 |
| 500 | 26 | 37 | 78 | 93 | 85 | 204 | 384 | 408 |
| 480 | 17 | 27 | 45 | 55 | 97 | 112 | 235 | 270 |
| 460 | 15 | 32 | 55 | 79 | 148 | 181 | 320 | 361 |
| 430 | 16 | 31 | 55 | 75 | 140 | 165 | 297 | 330 |
| 400 | 12 | 27 | 41 | 58 | 102 | 129 | 231 | 260 |
These data can also be analyzed by taking t0 = 10 s as the starting time, in order to eliminate any contributions from residual surface oxides that had not yet been reduced (eliminating the time during the induction period between 500–560 mV). The diminution of the current to 75% at t0 = 10 s takes nearly 40 s in 0.1 M H2SO4 and over 30 s in HClO4. The chronoamperometry in Fig. 14 shows that although H2SO4 is an adsorbing electrolyte, the experiments carried out in HClO4 showed a more rapid decrease in current over time, even though the current started out higher in magnitude. This has been observed previously in the literature and can be attributed to the anion adsorption phenomena.70 When (bi)sulfate adsorbs to the Ptpoly surface diminishing the overall current, it also prevents the formation of COads. Therefore, (bi)sulfate inhibits MeOH adsorption, resulting in a lower initial current for the MeOH in H2SO4, but it also inhibits CO adsorption, resulting in a slower current decay. In HClO4, there is little, if any, anion adsorption. Thus, once the oxide has been reduced, the Ptpoly surface is clean, providing more surface area for MeOH adsorption, and hence a higher overall current. Hence, that also allows for increased COads, causing the current to decrease more rapidly. If the chronoamperometry timescale is compared to the timescale of the potential sweep in the LSV, it takes 30 s for the potential to be swept 300 mV. The potential range between the maximum current and the potential at which the current falls to essentially zero is less than 250 mV during the LSV experiment. Clearly, this 100% current decrease is not caused by surface poisoning processes because after 30 s in both acidic media, the current has only decreased by, at most, 25%. Thus, we believe that the time-dependence of surface poisoning processes can be neglected when analyzing the Ea values calculated.
These data are representative of the behavior in acidic media, but the behavior in alkaline media has been shown in the literature to be analogous.46–48,50,149 The current is expected to decrease at a rate that is even slower than observed above because the oxidation of the surface poison CO can occur at even lower potentials in alkaline media than in acidic media. The current decrease will not be influenced by anion adsorption as in the case of H2SO4, but may be affected by the formation of carbonate build-up at the electrode surface (vide supra). This should be minimal though due to the rapid rotation of the electrode during the experiment.
These chronoamperometric experiments do indeed demonstrate that the current should not decrease appreciably due to surface poisoning (COads or anion adsorption) on the timescale of the LSV experiments. They also provide an indication that, in the LSV, when the current begins to decrease, this decrease is not governed by adsorption of poisons as much as it is governed by the decrease in driving force for the electrochemical oxidation of MeOH. This makes the interpretation of the Ea values obtained during the decreasing current portion of the LSV experiment easier to analyze. The changes in COads and adsorbed anion coverages over time complicate their analysis because those changes distort the magnitude of the current used to calculate the Ea. In the LSV experiment above, the sweep started out with an electrode covered by a “protective” oxide layer that precluded anion adsorption, and the sweep started at a potential where COads would be oxidized, if formed. If, on the other hand, the potential sweep was started at more negative potentials, COads could build up, along with anion adsorption, thus influencing the current and onset potential. It will be shown in the next sections that the surface poisoning effects which occur when the potential is swept anodically will become even more problematic when temperature is introduced as a second variable. The next step in the analysis is to determine if these coverages vary with changing temperature and to apply the information to the LSV experiments carried out here, as well as in the literature.
Anion adsorption is an especially important concern when carrying out the Ea studies in the following way. As the temperature is increased, the extent of anion adsorption may change at a Ptpoly surface. This, in turn, changes the surface area of the Pt electrode. The measured current is then not an accurate representation of the variation in current with temperature. Temperature dependent anion adsorption experiments are found in the literature most commonly for single crystal surfaces in sulfuric acid electrolyte. The behavior of these surfaces varies. For Pt(111) and Pt(110), it has been observed that as the temperature is increased, the adsorption of anions remains relatively constant with a maximum coverage (packing density) of 0.2 monolayers.103,106,107,170 On the Pt(100) surface, the anion adsorption behavior is drastically affected by temperature, showing a decrease in anion adsorption with increasing temperature. The adsorption charge decreases by ∼1/3, but this adsorption charge is a convolution of Hupd and anion adsorption.102 An exponential decrease in adsorbed anion coverage with increasing temperature was not observed.
In the case of the cathodically swept LSV experiments at a Ptpoly surface, the linearity of the Arrhenius plots for 0.5 M MeOH in 0.1 M H2SO4 seems to support the idea that the anion adsorption coverage changes with temperature are small or negligible. It must be recognized, though, that the Arrhenius plots would also be linear if the anion adsorption coverages increased exponentially with temperature. More importantly, the similarity in Ea values obtained in 0.1 M HClO4, a non-adsorbing electrolyte, and in 0.1 M H2SO4 support the contention that anion adsorption is negligible for Ea determination under any conditions. The temperature dependent anion adsorption at potentials of interest in these experiments can also be considered negligible due to the directionality of the LSV (cathodic sweep) because the electrode is covered with a substantial oxide layer prior to starting the experiment, precluding significant anion adsorption coverage. Any significant decreases in current observed after the maximum current has been reached are more likely due to a diminution in surface oxide coverage and decreased driving force as was shown in section 7.2.1, over the timescale of these LSV experiments.
On an anodic sweep in acidic media, CO coverages can increase to an appreciable level, significantly lowering the current obtained from MeOH oxidation and significantly precluding MeOH adsorption. The coverages of CO formed during MeOH oxidation on bulk Pt were probed using SPAIRS by Markovic et al., and they found that measurable surface coverages form at potentials negative of the onset of MeOH oxidation in 0.05 M MeOH and 0.1 M HClO4.134 The CO coverage increases to over θ = 0.5 around 300 mV vs. Ag/AgCl. The coverage of CO then gradually decreases as oxide formation on the Pt surface facilitates oxidation, and CO is completely removed by 600 mV vs. Ag/AgCl. On the cathodic sweep, the coverage of CO does not become measurable until after 150 mV vs. Ag/AgCl at a sweep rate of 2 mV s−1, which coincides with nearly complete deactivation for MeOH oxidation due to lack of driving force, as well as the complete reduction of any appreciable coverage of surface oxide. While the SPAIRS experiments were carried out in HClO4, it was noted that there was very little difference in the behavior of the system in 0.1 M H2SO4. The main difference was in the diminution of the overall current, which was also observed in the voltammetry in section 5.2. The initial adsorption of CO may occur at slightly more positive potentials in H2SO4 due to anion adsorption precluding CO formation, but the potential was not expected to be significantly more positive than in HClO4. While there was no data to support the behavior of CO in alkaline media, it is assumed to be quite similar to the non-adsorbing perchloric acid electrolyte data. It was also noted that these surfaces are highly active for MeOH oxidation, which was also observed above in section 5.2 when discussing the CV of MeOH oxidation. These results from the literature also support the chronoamperometry experiments presented in section 7 in that the contribution from adsorbed intermediates will not contribute significantly to the changes in current over the potential sweep.
The LSV experiments carried out in order to determine the Ea values in section 6 take advantage of what was observed in the SPAIRS experiments, mainly that when the potential is swept cathodically, the surface remains essentially free of CO. This statement is true as long as CO adsorption processes are not affected significantly by changes in temperature from room temperature to 55 °C. If CO adsorption changes appreciably with temperature, then this surface coverage change must be taken into account as it would cause the number of Pt sites available for MeOH adsorption to change with temperature. If this number indeed changes, then the current measured at each temperature would not only be a function of the temperature increase, but of the changing surface area available for MeOH oxidation. Various experiments using FT-IR and a Ptpoly electrode surface have been able to provide insight into the temperature dependence of CO coverage. Kardash, et al. reported that for a fixed MeOH concentration, an increase in temperature from 25 to 50 °C did not affect the equilibrium surface coverage of CO in 0.3 M MeOH and 0.1 M HClO4. On the other hand, when the temperature was increased to 75 °C, the detectable CO adsorbed on the surface decreased, pushing the onset of MeOH oxidation to more negative potentials.130,131 This change in CO coverage would indeed complicate the experimental results if the potential was swept anodically during the LSV experiments. This is because at low potentials, an appreciable CO coverage is measured. Indeed, much of the results in the literature have calculated Ea values on the forward sweep of a linear sweep or cyclic voltammogram. In these cases, the values of the Ea may be, and likely are, influenced by this change in CO coverage with temperature. In the LSV experiments carried out here, and over the temperature range investigated, the decrease in CO coverage should be minimal and should not affect the results because on the cathodic sweep, there is not a measurable coverage of CO in the potential region of interest. Difficulties, however, would arise if the CO coverage was found to increase as the temperature was increased, but this was not observed in the FT-IR experiments alluded to earlier. Thus, the Ea values obtained above for the LSV swept cathodically do not depend on the CO adsorption coverage.
For the LSV experiment in alkaline media, where the potential was swept anodically, the change in CO coverage may be a factor. Unfortunately, there has been no literature to aid in this determination. It can be said, however, that OH− adsorption in alkaline media occurs at very low potentials, which may prevent CO from building up any significant coverage. Also, the Arrhenius plots for these data were linear, which would seem to support the claim that changes in CO coverages with changes in temperature do not play a significant role in the determination of these Ea values, but, again, this cannot be supported by the literature on alkaline systems at this time.
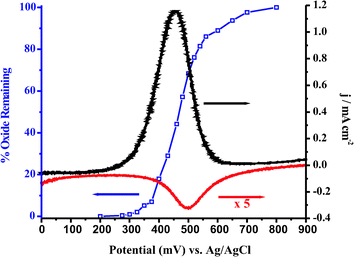 | ||
| Fig. 15 LSV of 0.5 M MeOH in 0.1 M H2SO4 swept at 10 mV s−1 cathodically, superimposed with a cathodic voltammetric sweep of clean Pt in 0.1 M H2SO4 at 10 mV s−1 (current has been multiplied by a factor of 5). These voltammetric profiles are compared to oxide coverages at specific potentials during the cathodic sweep calculated from Pt oxide reduction charge. The Ptpoly electrode was 3 mm in diameter for each experiment. | ||
The complication that changes in oxide coverage with temperature cause in the calculation of Ea obtained in the LSV above can be analyzed most clearly at the very beginning of the potential sweep. As the temperature is increased, the reduction of the surface oxide occurs at increasingly more positive potentials. Thus, at a given positive potential, as the temperature increases, the surface area of the bare Ptpoly increases because the oxide is being reduced at more positive potentials. This increase in surface area translates into an inflated change in current as a function of temperature at these potentials. As was mentioned in the beginning of section 7.2, there is no effect on the turnover rate, but the magnitude of the current is affected. Because current is being used to calculate the Ea, this situation would provide an Ea value that is artificially large. This would occur in both alkaline and acidic electrolytes, but may be more pronounced in alkaline media as discussed in section 5. Again, the reproducible linearity of the Arrhenius plots is evidence that the changes in oxide coverage might be negligible, but they must be at least considered when analyzing these values. A linear Arrhenius plot would also be obtained if the changes in oxide coverage with temperature were exponential. From Fig. 4 and 5, it is noted that the changes are linear within the temperature range applied in these experiments. This allows the assumption to be made that changes in oxide coverage with temperature, while observable, may be negligible in the overall Ea analysis.
7.3 E a values related to the above anion adsorption and surface process effects
As can be observed from the data given in Tables 1 and 2, a wide range of Ea values can be obtained depending on the potential at which data are collected. This reflects the wide range of values found in the literature on Ea for MeOH oxidation. The values obtained at low potentials in the LSV experiments above, where the potential was swept cathodically, are influenced by the decrease in the driving force for methanol oxidation, as well as the decrease in surface oxide coverage. The electron transfer step in the oxidation of methanol then becomes the rate-determining step. Although the current for MeOH oxidation is low, the overpotential applied is still considerable. At these large overpotentials, it is unlikely that the electron transfer can be activated significantly by an increase in temperature. Thus, it is unlikely that the change in current with temperature will be due to the electron transfer being activated by this temperature increase. Minimal increase in current will be observed due to the rate-limiting electron transfer step over the temperature range applied. The Ea values calculated at these potentials will be distorted to low values because the changes in current are small and will be unrepresentative of any chemical rate-determining step for MeOH oxidation.An unrepresentative Ea for MeOH oxidation can also be observed when Ea values are calculated by sweeping anodically as is often carried out in the literature, and as carried out for the alkaline electrolyte system in section 6.2. Artificially low apparent Ea values can be observed if the data are obtained as the potential is swept anodically due to the adsorption of anions and the build-up of poisonous intermediates, as has been mentioned throughout the above discussions. The sweep rate and starting potential determine the extent of the build-up of these surface species because these surface processes are potential, and time, dependent. These species can influence the onset of oxidation, and the kinetics of the oxidation reaction, which, in turn, influence the current magnitude. These adsorption processes depend on the temperature and change the available surface area and ensemble size available for methanol adsorption and oxidation. If the available surface area decreases as the temperature is increased, then the calculated Ea values will be lower than the true value. This distortion explains the common values around 15–30 kJ mol−1 obtained for nearly all catalysts in a number of different environments at potentials near the onset of MeOH oxidation. Under these conditions, surface processes and the driving force for electron transfer are the main contributors to the Ea values being obtained.
An artificially high Ea can also be obtained if the surface area available for methanol adsorption increases as the temperature increases. Potentials at which the reduction of oxide is initiated are especially affected when sweeping from positive to negative potentials. As the temperature increases, the oxide reduction occurs at more positive potentials. This leads to more surface area being available to carry out methanol oxidation, leading to enhancements in current as the temperature increases due to not only the temperature increase, but also to the increase in electrode surface area. This effect is most obvious at the onset of oxide reduction (over a temperature range) because a clean surface is being revealed that is surrounded by oxide only and not by anions or other methanol molecules (as it would be at more negative potentials further into the potential sweep). Apparent Ea values found in Tables 2 and 3 that may be affected by this are those ranging from 75–90 kJ mol−1 found prior to the potential at which the maximum current value is obtained. Artificially high Ea values can also be found at lower potentials if the potential sweep is carried out anodically, again due to the changing surface area with changing temperature. If CO adsorption is less favorable (or CO desorption/oxidation is more favorable) at elevated temperatures at a given potential, then the available surface area would increase. These phenomena will certainly cause complications when data for Ea calculations are obtained by sweeping anodically. These distortions of the Ea can be eliminated if the turnover rate, instead of current, is used to calculate the Ea. However, difficulties, such as knowing the specific coverage of all surface species at a specific potential and temperature, must be addressed before this can be carried out.
Finally, the Ea found for the peak current potential can be considered the Ea value of greatest interest. The electron transfer is most likely not the rate determining step at this large overpotential. The electrode surface has the optimum oxide coverage to achieve maximum current, but the effect of the change in oxide coverage with temperature is convoluted with the shift in potential at which the maximum peak current is observed during MeOH oxidation. While oxide coverages change as the temperature changes in pure electrolyte, the coverage changes are enhanced when MeOH is introduced into the electrolyte, as was explained in section 6.1. In the case of the LSV carried out cathodically in the above experiments, COads and other poisonous intermediates do not play a significant role in the current level at that potential. The pathway being followed at this specific potential, in addition to what processes are limiting the current at this potential, are of the greatest interest. One would like to be able to drive this pathway over a broad potential range in order to obtain the maximum current over that range. Knowing intimate details of the chemical, as well as electrochemical, rate determining step at that potential would be useful in designing catalysts that can target those steps in order to drive those reactions specifically. The values for MeOH oxidation at the peak current potential determined in this study are Ea = 54 ± 5 kJ mol−1 in 0.1 M H2SO4 and 52 ± 7 kJ mol−1 in 0.1 M HClO4. These are similar to values found by Raicheva et al. at the potential where the maximum peak current was observed on the cathodic sweep.119 In alkaline media, Ea = 60 ± 6 kJ mol−1, determined during the cathodic potential sweep. Fig. 16 sums up the parameters that govern the value of the Ea along the shape of the MeOH oxidation peak on the cathodic sweep.
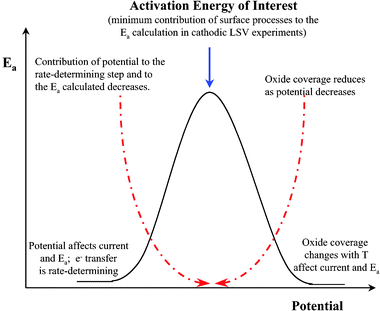 | ||
| Fig. 16 Schematic of the surface processes, and other factors, that must be taken into account when determining the meaning of the Ea value calculated. | ||
The anodic sweep carried out in alkaline media gave very different results when compared to the cathodic sweep (Fig. 12), further amplifying the fact that directionality plays a crucial role. While anion adsorption in alkaline media was not playing a role, the nature of the surface oxide formation, as well as any poisonous intermediates building up at the electrode surface by potentials around −0.075 V played a big role in the Ea values obtained. As the potential was swept more positive of −0.075 V, the Ea began to increase again due to the oxidation of the Pt surface and to the oxidation of COads. Clearly, the surface processes occurring on the anodic and cathodic sweeps are very different, and they lead to Ea values that differ greatly. Further insight into why these values differ so much, as well as what steps the above Ea values may correspond to in the MeOH oxidation pathway, can be gained from further determination of the products formed at the specific potentials of interest. The ratios of product formation on the anodic sweep must be compared to those on the cathodic in order to determine what surface changes are occurring over those potential ranges. The apparent Ea values become an important tool for catalyst comparison only when all of these surface parameters are taken into account, or at least understood and acknowledged, and insight is gained into the specific pathway that MeOH oxidation is following at the potential at which the Ea value was determined.
8. Conclusion
A complete understanding of the MeOH oxidation pathway at a given potential is very important in order to design effective oxidation catalysts and to employ MeOH in DMFCs. The pathways for MeOH oxidation are complicated by the production of numerous soluble intermediates and surface poisons, in addition to the product of complete oxidation, CO2. It has been determined that factors affecting the ratio of product formation include applied potential, temperature, electrode surface structure and composition, nature and concentration of electrolyte, and concentration of MeOH. Comparison of results between various literature sources becomes difficult due to the variety of experimental conditions used and the sensitivity of MeOH oxidation to the above conditions. Careful studies must be carried out using bulk Ptpoly, bulk alloys, and nanoscale catalysts spanning numerous experimental parameters in a controlled fashion in order make useful comparisons.In order to find another way in which to compare the behavior between new catalysts for MeOH oxidation and Ptpoly, the relevance of Ea, a kinetic parameter, was investigated. Ea values used as numerical comparisons between different electrocatalysts throughout the literature, have a wide range of reported values, and have been assigned to many different steps in the MeOH oxidation pathway. The way in which these data were obtained influence the final Ea value, causing some values to be artificially low, or high, depending on the experimental procedure employed. We have carried out a rigorous Ea study using a bulk Ptpoly electrode in acidic and alkaline media in order to address the large range of Ea previously reported, as well as to point out some of the factors that affect the Ea calculation. The Ea values were then analyzed in terms of the surface processes that might influence them.
Apparent Ea values were found to be a function of potential, and depend on the path taken to reach that potential. Therefore the potential at which the Ea is obtained must be included when reporting Ea values. Surface adsorption processes must also be taken into account when analyzing Ea values in both acid and alkaline media. These surface adsorption processes are both potential and temperature dependent. The potential dependence of anion adsorption, COads, and surface oxide oxidation/reduction must be taken into account when determining Ea values. The timescales of the experiments carried out in this work were such that anion adsorption and COads did not play a significant role in any decreases in current observed as the LSV was carried out. Surface oxide coverages are time and potential dependent due to the adsorption of MeOH and subsequent reduction of significant amounts of surface oxides that are not replenished after being consumed. The temperature dependence of anion adsorption, COads, and surface oxide oxidation/reduction must also be analyzed over the specific temperature range of the experiments. In the above experiments, it was determined that these surface adsorption processes do not change significantly as the temperature is increased within the temperature range applied. The Ea values obtained in acidic electrolyte were lower than those obtained in alkaline media on the cathodic sweep by 10–20 kJ mol−1 depending on the potential.
Apparent Ea values determined at low potentials are distorted due to the electron transfer step being rate-determining for MeOH oxidation. The temperature increase will accelerate processes following the electron transfer step, but at low potentials, the barrier for the electron transfer cannot be overcome. Although at these low potentials there is little driving force for MeOH oxidation, a large overpotential is still being applied. The current will not change appreciably as a function of time at those overpotentials because the electron transfer process most likely will not be activated over the temperature range applied. Apparent Ea values obtained at more positive potentials may be influenced by the activation of oxide reduction and consumption as a function of temperature. Changes in surface area as a function of temperature due to changes in oxide coverage may again give Ea values that are not representative of the system. Apparent Ea values obtained at the potential where the maximum peak current is obtained are most interesting because this point is where many of the processes that might affect the Ea are minimized. The value obtained during the cathodic sweep in 0.5 M MeOH was Ea = 54 ± 5 kJ mol−1 in 0.1 M H2SO4 and Ea = 52 ± 7 kJ mol−1 in 0.1 M HClO4. In 0.1 M KOH, Ea = 60 ± 6 kJ mol−1. On the anodic sweep in 0.1 M KOH, Ea = 19 ± 1 kJ mol−1 at the potential at which the maximum anodic peak current was observed. By knowing the nature of the surface at the potential where the maximum current is obtained, as well as doing further research into the product distribution obtained at that potential in the specific electrochemical environment of interest, turnover rates can be calculated, more representative Ea values can be obtained, and these values will be valuable points of comparison among numerous catalysts for the oxidation of MeOH. For now, the Ea values calculated in the above experiments should be used as points of comparison between Ptpoly and other MeOH oxidation catalysts under the same electrochemical conditions.
Acknowledgements
Special thanks to Prof. Juan Feliu and Prof. Enrique Herrero for useful discussions. Jamie L. Cohen would like to thank the NSF Division of Analytical Chemistry and Ford Motor Company (Ford URP) for funding. This work was also funded in part by the Cornell Fuel Cell Institute under a grant from the U.S. Department of Energy (Grant No. DE-FG02-03ER46072), the National Science Foundation (Grant No. CHE 03 46377), and the Cornell Center for Materials Research.References
- J. Larminie and A. Dicks, Fuel Cell Systems Explained, John Wiley & Sons Ltd, Chichester, 2003 Search PubMed.
- W. Vielstich, J. Braz. Chem. Soc., 2003, 14, 503–509 CAS.
- V. Ramani, Electrochem. Soc. Interface, 2006, 41–44 Search PubMed.
- P. Costamanga and S. Srinivasan, J. Power Sources, 2001, 102, 242–252 CrossRef CAS.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS.
- S. Haile, Acta Mater., 2003, 51, 5981–6000 CrossRef CAS.
- A. Heinzel, C. Hebling, M. Muller, M. Zedda and C. Muller, J. Power Sources, 2002, 105, 250–255 CrossRef CAS.
- G. J. K. Acres, J. Power Sources, 2001, 100, 60–66 CrossRef CAS.
- J. S. Wainright, R. F. Savinell, C. C. Liu and M. Litt, Electrochim. Acta, 2003, 48, 2869–2877 CrossRef CAS.
- N.-T. Nguyen and S. H. Chan, J. Micromech. Microeng., 2006, 16, R1–R12 CrossRef CAS.
- E. Gulzow, J. Power Sources, 1996, 61, 99–104 CrossRef.
- G. T. Burstein, C. J. Barnett, A. R. Kucernak and K. R. Williams, Catal. Today, 1997, 38, 425–437 CrossRef CAS.
- C. Lamy, J.-M. Leger and S. Srinivasan, in Modern Aspects of Electrochemistry, Kulwer Academic/Plenum Publishers, New York, 2001, pp. 53–118 Search PubMed.
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J.-M. Leger, J. Power Sources, 2002, 105, 283–296 CrossRef CAS.
- S. Wasmus and A. Kuver, J. Electroanal. Chem., 1999, 461, 14–31 CrossRef CAS.
- A. Hamnett, Catal. Today, 1997, 38, 445–457 CrossRef CAS.
- T. Iwasita, Electrochim. Acta, 2002, 47, 3663–3674 CrossRef CAS.
- J.-M. Leger, J. Appl. Electrochem., 2001, 31, 767–771 CrossRef CAS.
- C. Lamy, E. M. Belgsir and J.-M. Leger, J. Appl. Electrochem., 2001, 31, 799–809 CrossRef CAS.
- D. Cao, G.-Q. Lu, A. Wieckowski, S. A. Wasileski and M. Neurock, J. Phys. Chem. B, 2005, 109, 11622–11633 CrossRef CAS.
- J. Greeley and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 3910–3919 CrossRef CAS.
- E. A. Batista, G. R. P. Malpass, A. J. Motheo and T. Iwasita, J. Electroanal. Chem., 2004, 571, 273–282 CrossRef CAS.
- Y. Zhu, H. Uchida, T. Yajima and M. Watanabe, Langmuir, 2001, 17, 146–154 CrossRef CAS.
- P. Waszczuk, G.-Q. Lu, A. Wieckowski, C. Lu, C. Rice and R. I. Masel, Electrochim. Acta, 2002, 47, 3637–3652 CrossRef CAS.
- T. D. Jarvi, S. Sriramulu and E. M. Stuve, J. Phys. Chem. B, 1997, 101, 3649–3652 CrossRef CAS.
- S. Gojkovic, J. Electroanal. Chem., 2004, 573, 271–276 CrossRef CAS.
- H. Wang, C. Wingender, H. Baltruschat, M. Lopez and M. T. Reetz, J. Electroanal. Chem., 2001, 509, 163–169 CrossRef CAS.
- H. Wang, T. Loffler and H. Baltruschat, J. Appl. Electrochem., 2001, 31, 759–765 CrossRef CAS.
- V. S. Bagotzky, Y. B. Vassiliev and O. A. Khazova, J. Electroanal. Chem., 1977, 81, 229–238 CrossRef.
- K.-I. Ota, Y. Nagakawa and M. Takahashi, J. Electroanal. Chem., 1984, 179, 179–186 CrossRef CAS.
- J. Sobkowski, K. Franaszczuk and K. Dobrowolska, J. Electroanal. Chem., 1992, 330, 529–540 CrossRef.
- G.-Q. Lu, W. Chrzanowski and A. Wieckowski, J. Phys. Chem. B, 2000, 104, 5566–5572 CrossRef CAS.
- Z. Jusys and R. J. Behm, J. Phys. Chem. B, 2001, 105, 10874–10883 CrossRef CAS.
- R. Parsons and T. VanderNoot, J. Electroanal. Chem., 1988, 257, 9–45 CrossRef CAS.
- J. Prabhuram and R. Manoharan, J. Power Sources, 1998, 74, 54–61 CrossRef CAS.
- A. V. Tripkovic, K. D. Popovic, B. N. Grgur, B. Blizanac, P. N. Ross and N. M. Markovic, Electrochim. Acta, 2002, 47, 3707–3714 CrossRef CAS.
- C. Hartnig and E. Spohr, Chem. Phys., 2005, 319, 185–191 CrossRef CAS.
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS.
- C. L. Childers, H. Huang and C. Korzeniewski, Langmuir, 1999, 13, 786–789 CrossRef.
- C. Korzeniewski and C. L. Childers, J. Phys. Chem. B, 1998, 102, 489–492 CrossRef CAS.
- W.-F. Lin, J.-T. Wang and R. F. Savinell, J. Electrochem. Soc., 1997, 144, 1917–1922 CAS.
- P. Gao, S.-C. Chang, Z. Zhou and M. J. Weaver, J. Electroanal. Chem., 1989, 272, 161–178 CrossRef CAS.
- S.-G. Sun, in Electrocatalysis, ed. J. Lipkowski and P. N. Ross, Wiley-VCH, Inc., New York, 1998, pp. 243–290 Search PubMed.
- K. Scott and P. Argyropoulos, J. Power Sources, 2004, 137, 228–238 CrossRef CAS.
- O. A. Petry, B. I. Podlovchenko, A. N. Frumkin and H. Lal, J. Electroanal. Chem., 1965, 10, 253–269 CrossRef CAS.
- A. V. Tripkovic, K. D. Popovic, J. D. Momcilovic and D. M. Drazic, Electrochim. Acta, 1998, 44, 1135–1145 CrossRef CAS.
- A. V. Tripkovic, K. D. Popovic, J. D. Momcilovic and D. M. Drazic, J. Electroanal. Chem., 1998, 448, 173–181 CrossRef CAS.
- A. V. Tripkovic, K. D. Popovic, J. D. Momcilovic and D. M. Drazic, J. Electroanal. Chem., 1996, 418, 9–20 CrossRef CAS.
- A. V. Tripkovic, K. D. Popovic, J. D. Lovic, V. M. Jovanovic and A. Kowal, J. Electroanal. Chem., 2004, 572.
- E. H. Yu, K. Scott and R. W. Reeve, J. Electroanal. Chem., 2003, 547, 17–24 CrossRef.
- T. J. Schmidt, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2001, 105, 12082–12086 CrossRef CAS.
- K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, J. Electrochem. Soc., 2005, 152, A729–A731 CrossRef CAS.
- Z.-Y. Zhou, N. Tian, Y.-J. Chen, S.-P. Chen and S.-G. Sun, J. Electroanal. Chem., 2004, 573, 111–119 CrossRef CAS.
- N. M. Markovic, T. J. Schmidt, B. N. Grgur, H. A. Gasteiger, R. J. Behm and P. N. Ross, J. Phys. Chem. B, 1999, 103, 8568–8577 CrossRef CAS.
- E. Morallon, A. Rodes, J. L. Vazquez and J. M. Perez, J. Electroanal. Chem., 1995, 391, 149–157 CrossRef CAS.
- Y. M. Volfkovich, Y. B. Vassiliev and V. S. Bagotzky, Investiya Akademii Nauk USSR, Seriya Khimicheskaya, 1969, 9, 1898–1905 Search PubMed.
- Y. M. Volfkovich, Y. B. Vassiliev and V. S. Bagotzky, Elektrokhimiya, 1969, 5, 1462–1465 Search PubMed.
- C. Lamy, J.-M. Leger and J. Clavilier, J. Electroanal. Chem., 1982, 135, 321–328 CrossRef CAS.
- D. M. Drazic and V. Drazic, Electrochim. Acta, 1966, 11, 1235–1241 CrossRef CAS.
- M. P. Z. Mallen and L. D. Schmidt, J. Catal., 1996, 161, 230–246 CrossRef CAS.
- R. L. Borup, D. E. Sauer and E. M. Stuve, Surf. Sci., 1997, 374, 142–150 CrossRef CAS.
- C. T. Campbell, G. Ertl, H. Kuipers and J. Segner, J. Phys. Chem., 1980, 73, 5862–5873 CrossRef CAS.
- K. Franaszczuk, E. Herrero, P. Zelenay, A. Wieckowski, J. Wang and R. I. Masel, J. Phys. Chem., 1992, 96, 8509–8516 CrossRef CAS.
- F. Zaera, J. Liu and M. Xu, J. Chem. Phys., 1997, 106, 4204–4215 CrossRef CAS.
- S. Wilhelm, T. Iwasita and W. Vielstich, J. Electroanal. Chem., 1987, 238, 383–391 CrossRef CAS.
- T. H. M. Housmans, A. H. Wonders and M. T. M. Koper, J. Phys. Chem. B, 2006, 110, 10021–10031 CrossRef CAS.
- S. N. Raicheva, E. I. Sokolova and M. V. Christov, J. Electroanal. Chem., 1984, 175, 167–181 CrossRef CAS.
- S. Gilman and M. W. Breiter, J. Electrochem. Soc., 1962, 109, 1099–1104 CAS.
- M. W. Breiter, J. Electrochem. Soc., 1963, 110, 449–452 CAS.
- E. Herrero, K. Franaszczuk and A. Wieckowski, J. Phys. Chem., 1994, 98, 5074–5083 CrossRef CAS.
- S. Gojkovic, T. R. Vidakovic and D. R. Durovic, Electrochim. Acta, 2003, 48, 3607–3614 CrossRef CAS.
- H. A. Gasteiger, N. M. Markovic, P. N. Ross and E. Cairns, J. Electrochem. Soc., 1994, 141, 1795–1803 CAS.
- T. H. Madden and E. M. Stuve, J. Electrochem. Soc., 2003, 150, E571–E577 CrossRef CAS.
- D. Chu and S. Gilman, J. Electrochem. Soc., 1996, 143, 1685–1690 CAS.
- Y. B. Vassiliev, V. S. Bagotzky and V. A. Gromyko, J. Electroanal. Chem., 1984, 178, 247–269.
- C.-F. Mai, C.-H. Shue, L.-Y. O. Yang, Y.-C. Yang, S.-L. Yau and K. Itaya, Langmuir, 2005, 21, 4964–4970 CrossRef CAS.
- T. Biegler, D. A. J. Rand and R. Woods, J. Electroanal. Chem., 1971, 29, 269–277 CrossRef CAS.
- M. C. Santos and L. O. S. Bulhoes, Electrochim. Acta, 2004, 49, 1893–1901 CrossRef CAS.
- A. H. Xia, T. Iwasita, F. Ge and W. Vielstich, Electrochim. Acta, 1996, 41, 711–718 CrossRef CAS.
- J. O. M. Bockris and H. Wroblowa, J. Electroanal. Chem., 1964, 7, 428–451 CrossRef.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. v. Santen, J. Electroanal. Chem., 2002, 524–525, 242–251 CrossRef CAS.
- S. Gilman, J. Phys. Chem., 1963, 67, 1898–1905 CrossRef CAS.
- N.-H. Li, S.-H. Sun and S.-P. Chen, J. Electroanal. Chem., 1997, 430, 57–67 CrossRef CAS.
- J. P. Hoare, Electrochim. Acta, 1972, 17, 593–595 CrossRef CAS.
- R. Thacker and J. P. Hoare, J. Electroanal. Chem., 1971, 30, 1–14 CrossRef CAS.
- L. D. Burke and J. K. Casey, Electrochim. Acta, 1992, 37, 1817–1829 CrossRef CAS.
- G. Jerkiewicz, G. Vatankhah, J. Lessard, M. P. Soriaga and Y.-S. Park, J. Electroanal. Chem., 2004, 49, 1451–1459 CAS.
- T. Biegler and R. Woods, J. Electroanal. Chem., 1969, 20, 73–78 CrossRef CAS.
- K. J. Vetter and J. W. Schultze, J. Electroanal. Chem., 1972, 34, 141–158 CrossRef CAS.
- V. S. Bagotzky and M. R. Tarasevich, J. Electroanal. Chem., 1979, 101, 1–17 CrossRef.
- B. E. Conway, Prog. Surf. Sci., 1995, 49, 331–452 CrossRef CAS.
- F. Maillard, G.-Q. Lu, A. Wieckowski and U. Stimming, J. Phys. Chem. B, 2005, 109, 16230–16243 CrossRef CAS.
- K. Wang, H. A. Gasteiger, N. M. Markovic and P. N. Ross, Electrochim. Acta, 1996, 41, 2587–2593 CrossRef CAS.
- T. H. Madden, N. Arvindan and E. M. Stuve, J. Electrochem. Soc., 2003, 150, E1–E10 CrossRef CAS.
- T. Iwasita, W. Vielstich and E. Santos, J. Electroanal. Chem., 1987, 229, 367–376 CrossRef CAS.
- A. Cuesta, A. Couto, A. Rincon, M. C. Perez, A. Lopez-Cudero and C. Gutierrez, J. Electroanal. Chem., 2006, 586, 184–195 CrossRef CAS.
- N. M. Markovic, B. N. Grgur, C. A. Lucas and P. N. Ross, J. Phys. Chem. B, 1999, 103, 487–495 CrossRef CAS.
- T. Kawaguchi, W. Sugimoto, Y. Murakami and Y. Takasu, Electrochem. Commun., 2004, 6, 480–483 CrossRef CAS.
- E. Herrero, B. Alvarez, J. M. Feliu, S. Blais, Z. Radovic-Hrapovic and G. Jerkiewicz, J. Electroanal. Chem., 2004, 567, 139–149 CrossRef CAS.
- A. B. Anderson and N. M. Neshev, J. Electrochem. Soc., 2002, 149, E383–E388 CrossRef CAS.
- A. B. Anderson, Electrochim. Acta, 2002, 47, 3759–3763 CrossRef CAS.
- A. Zolfaghari and G. Jerliewicz, J. Electroanal. Chem., 1999, 467, 177–185 CrossRef CAS.
- A. Zolfaghari and G. Jerkiewicz, J. Electroanal. Chem., 1997, 422, 1–6 CrossRef CAS.
- A. Zolfaghari and G. Jerkiewicz, J. Electroanal. Chem., 1997, 420, 11–15 CrossRef CAS.
- H. Kita, Y. Gao, T. Nakato and H. Hattori, J. Electroanal. Chem., 1994, 373, 177–183 CrossRef CAS.
- A. Kolics and A. Wieckowski, J. Phys. Chem. B, 2001, 105, 2588–2595 CrossRef CAS.
- E. Herrero, J. Mostany, J. Feliu and J. Lipkowski, J. Electroanal. Chem., 2002, 534, 79–89 CrossRef CAS.
- M. Schell and B. E. K. Swamy, J. Electroanal. Chem., 2005, 584, 157–166 CrossRef CAS.
- C. Coutanceau, F. Hahn, P. Waszczuk, A. Wieckowski, C. Lamy and J.-M. Leger, Fuel Cells, 2003, 2, 153–158.
- A. Wieckowski and J. Sobkowski, J. Electroanal. Chem., 1975, 63, 365–377 CrossRef CAS.
- A. Wieckowski, J. Electroanal. Chem., 1977, 78, 229–241 CrossRef CAS.
- B. Beden, C. Lamy, N. R. de Tacconi and A. J. Arvia, Electrochim. Acta, 1990, 35, 691–704 CrossRef CAS.
- T. Iwasita and W. Vielstich, J. Electroanal. Chem., 1986, 201, 403–408 CrossRef CAS.
- D. B. Hibbert and F. Y. Y. Yon-Hin, J. Electrochem. Soc., 1985, 132, 1387–1389 CrossRef CAS.
- O. A. Khazova, A. A. Mikhailova, A. M. Skundin, E. K. Tuseeva, A. Havranek and K. Wipperman, Fuel Cells, 2002, 2, 99–108 CrossRef CAS.
- N. Wakabayashi, H. Uchida and M. Watanabe, Electrochem. Solid-State Lett., 2002, 5, E62–E65 CAS.
- T. R. Vidakovic, M. Christov and K. Sundmacher, J. Electroanal. Chem., 2005, 580, 105–121 CrossRef CAS.
- P. S. Kauranen, E. Skou and J. Munk, J. Electroanal. Chem., 1996, 404, 1–13 CrossRef CAS.
- S. N. Raicheva, M. V. Christov and E. I. Sokolova, Electrochim. Acta, 1981, 26, 1669–1676 CrossRef CAS.
- Z. A. Rotenberg, J. Electroanal. Chem., 1993, 345, 469–474 CrossRef CAS.
- A. Aramata, T. Kodera and M. Masuda, J. Appl. Electrochem., 1988, 18, 577–582 CAS.
- G. Meli, J.-M. Leger and C. Lamy, J. Appl. Electrochem., 1993, 23, 197–202 CAS.
- Z. Jusys, H. Massong and H. Baltruschat, J. Electrochem. Soc., 1999, 146, 1093–1098 CrossRef CAS.
- Z. Jusys, J. Kaiser and R. J. Behm, Phys. Chem. Chem. Phys., 2001, 3, 4650–4660 RSC.
- S. Wasmus, J.-T. Wang and R. F. Savinell, J. Electrochem. Soc., 1995, 142, 3825–3833 CAS.
- M. Krausa and W. Vielstich, J. Electroanal. Chem., 1994, 379, 307–314 CrossRef CAS.
- Z. Jusys, J. Kaiser and R. J. Behm, Langmuir, 2003, 19, 6759–6769 CrossRef CAS.
- J. E. Oxley, G. K. Johnson and B. T. Buzalski, Electrochim. Acta, 1964, 9, 897–910 CrossRef CAS.
- K. Kunimatsu, J. Electroanal. Chem., 1986, 213, 149–157 CrossRef CAS.
- D. Kardash, J. Huang and C. Korzeniewski, Langmuir, 2000, 16, 2019–2023 CrossRef CAS.
- D. Kardash and C. Korzeniewski, Langmuir, 2000, 16, 8419–8425 CrossRef CAS.
- G. Vijayaraghavan, L. Gao and C. Korzeniewski, Langmuir, 2003, 19, 2333–2337 CrossRef CAS.
- J. G. Love and A. J. McQuillan, J. Electroanal. Chem., 1989, 274, 263–270 CrossRef CAS.
- N. M. Markovic, H. A. Gasteiger, P. N. Ross, X. Jiang, I. Villegas and M. J. Weaver, Electrochim. Acta, 1995, 40, 91–98 CrossRef CAS.
- H. Shiroishi, Y. Ayato, K. Kunimatsu and T. Okada, J. Electroanal. Chem., 2005, 581, 132–138 CrossRef CAS.
- A. Miki, S. Ye and M. Osawa, Chem. Commun., 2002, 1500–1501 RSC.
- K. J. J. Mayrhofer, M. Arenz, B. Blizanac, V. Stamenkovic, P. N. Ross and N. M. Markovic, Electrochim. Acta, 2005, 50, 5144–5154 CrossRef CAS.
- Y. Y. Tong, E. Oldfield and A. Wieckowski, Faraday Discuss., 2002, 121, 323–330 RSC.
- Y. Y. Tong, A. Wieckowski and E. Oldfield, J. Phys. Chem. B, 2002, 106, 2434–2446 CrossRef CAS.
- Y. Tong, H. S. Kim, P. K. Babu, P. Waszczuk, A. Wieckowski and E. Oldfield, J. Am. Chem. Soc., 2002, 124, 460–473.
- E. Herrero, J. M. Feliu, S. Blais, Z. Radovic-Hrapovic and G. Jerkiewicz, Langmuir, 2000, 16, 4779–4783 CrossRef CAS.
- T. E. Shubina and M. T. M. Koper, Electrochim. Acta, 2002, 47, 3621–3628 CrossRef CAS.
- J. Nordlund and G. Lindbergh, J. Electrochem. Soc., 2004, 151, A1357–A1362 CrossRef CAS.
- G. Tremiliosi-Filho, H. Kim, W. Chrzanowski, A. Wieckowski, B. Grzybowska and P. Kulesza, J. Electroanal. Chem., 1999, 467, 143–156 CrossRef CAS.
- A. V. Tripkovic, S. Strbac and K. D. Popovic, Electrochem. Commun., 2003, 5, 484–490 CrossRef CAS.
- A. S. Arico, V. Baglio, A. D. Blasi, E. Modica, G. Monforte and V. Antonucci, J. Electroanal. Chem., 2005, 576, 161–169 CrossRef CAS.
- W. J. Long, R. M. Stroud, K. E. Swider-Lyons and D. R. Rolison, J. Phys. Chem. B, 2000, 104, 9772–9776 CrossRef CAS.
- A. Damjanovic, J. Electroanal. Chem., 1993, 355, 55–77.
- K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, Electrochim. Acta, 2005, 51, 1085–1090 CrossRef CAS.
- W. Xu, T. Lu, C. Liu and W. Xing, J. Phys. Chem. B, 2005, 109, 7872–7877 CrossRef CAS.
- A. Aramata, M. Masuda and T. Kodera, J. Electrochem. Soc., 1989, 136, 3288–3296 CAS.
- S. Sriramulu, T. D. Jarvi and E. M. Stuve, J. Electroanal. Chem., 1999, 467, 132–142 CrossRef CAS.
- A. B. Anderson, Electrochim. Acta, 2003, 48, 3743–3749 CrossRef CAS.
- A. B. Anderson, R. A. Sidik, J. Narayanasamy and P. Shiller, J. Phys. Chem. B, 2003, 107, 4618–4623 CrossRef CAS.
- A. B. Anderson, J. Roques, S. Mukerjee, V. S. Murthi, N. M. Markovic and V. Stamenkovic, J. Phys. Chem. B, 2005, 109, 1198–1203 CrossRef CAS.
- A. B. Anderson and Y. Cai, J. Phys. Chem. B, 2004, 108, 19917–19920 CrossRef CAS.
- V. F. Stenin and B. I. Podlovchenko, Elektrokhimiya, 1967, 3, 481–488 Search PubMed.
- E. Casado-Rivera, Z. Gal, A. C. D. Angelo, C. Lind, F. DiSalvo and H. D. Abruna, ChemPhysChem, 2003, 4, 193–199 CrossRef CAS.
- M. Alsabet, M. Grden and G. Jerkiewicz, J. Electroanal. Chem., 2006, 589, 120–127 CrossRef CAS.
- B. E. Conway, H. Angerstein-Kozlowska and W. B. A. Sharp, J. Chem. Soc., Faraday Trans. 1, 1978, 56, 915–924 Search PubMed.
- M. W. Breiter and S. Gilman, J. Electrochem. Soc., 1962, 109, 622–627 CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Hoboken, NJ, 2001 Search PubMed.
- B. V. Tilak, B. E. Conway and H. Angerstein-Kozlowska, J. Electroanal. Chem., 1973, 48, 1–23 CrossRef.
- H. Angerstein-Kozlowska, B. E. Conway and W. B. A. Sharp, J. Electroanal. Chem., 1973, 43, 9–36 CrossRef CAS.
- D. M. Drazic, A. V. Tripkovic, K. D. Popovic and J. D. Lovic, J. Electroanal. Chem., 1999, 466, 155–164 CrossRef CAS.
- T. Iwasita, A. H. Xia, H.-D. Liess and W. Vielstich, J. Phys. Chem. B, 1997, 101, 7542–7547 CrossRef CAS.
- N. Garcia-Araez, V. Climent, E. Herrero, J. Feliu and J. Lipkowski, J. Electroanal. Chem., 2005, 576, 33–41 CrossRef CAS.
- A. Lopez-Cudero, A. Cuesta and C. Gutierrez, J. Electroanal. Chem., 2003, 548, 109–119 CrossRef CAS.
- G. Horanyi and G. Vertes, J. Electroanal. Chem., 1975, 64, 252–254 CrossRef CAS.
- Z. Radovic-Hrapovic and G. Jerkiewicz, J. Electroanal. Chem., 2001, 499, 61–66 CrossRef CAS.
| This journal is © the Owner Societies 2007 |