Homo- and heterometallic coordination polymers from the f elements
Christopher L. Cahill*ab, Daniel T. de Lilla and Mark Frischa
aDepartment of Chemistry, The George Washington University, 725 21st Street, NW, Washington, DC 20052, USA
bGeophysical Laboratory, Carnegie Institution of Washington, Washington, DC 20015, USA
First published on 4th December 2006
Abstract
Lanthanide and actinide based coordination polymers and metal–organic framework materials present a number of interesting opportunities with respect to their syntheses and properties. Ln elements typically display large coordination numbers and roughly spherical bonding environments whereas An elements (specifically U(VI)) tend to form triatomic species with terminal oxo groups. As such, these features may be perceived by some as obstacles to the development of designed architectures of f-metal containing hybrid materials. We argue that these features, when coupled to the luminescent behavior of these elements actually give rise to a diverse family of compounds with a range of interesting topologies. Presented herein is an overview of the structural features and luminescent behavior of these materials, as well as some synthetic strategies toward producing both homo- and heterometallic compounds.
 Christopher L. Cahill | Christopher Cahill earned a BS in Chemistry and Geochemistry from the State University of New York (SUNY) College at Fredonia (1993) and his Ph.D. in Inorganic Chemistry from SUNY-Stony Brook (1999). After a post-doctoral position at the University of Notre Dame, he joined the faculty at The George Washington University in 2000. His research interests include hydrothermal systems, mineral structures, in situ synchrotron studies and hybrid materials. He is a recipient of a Bender Teaching Award (GW) and active in the American Crystallographic Association and the United States National Committee for Crystallography. |
 Daniel T. de Lill | Daniel de Lill is currently a fourth year graduate student at The George Washington University studying Inorganic Chemistry. He earned his BA in French (1999) and BA in Chemistry (2000) from Lock Haven University of Pennsylvania. His research focuses on the influence of structure-directing agents in the assembly of metal–organic framework materials and lanthanide luminescence. He has been an ARCS Scholar (Achievement Rewards for College Scientists) for the past two years. |
 Mark Frisch | Mark Frisch earned a BS in Chemistry from The Pennsylvania State University (2002) and is currently a fifth year graduate student at The George Washington University pursuing a Ph.D. in Inorganic Chemistry. He joined the Cahill research group in the spring of 2003. His research focuses on synthesizing heterometallic uranium-based coordination polymers along with studying their subsequent fluorescent properties. He is a member of the American Chemical Society, Division of Inorganic Chemistry. |
1. Introduction
The field of coordination polymers and metal–organic frameworks has seen a tremendous expansion in research efforts over the past several years. Briefly, these materials consist of inorganic metal centers (which may or may not be polynuclear) assembled by means of multifunctional organic linker species. These ‘hybrid’ materials display a range of topologies resulting from these pairings, including 1- and 2-dimensional polymeric structures as well as ‘true’ metal–organic frameworks (or MOF) wherein 3-dimensional structures may give rise to accessible void space.1 Structural features such as inherent binding sites, flexibility and void space can be coupled to recognition, electronic and magnetic properties in order to make these materials attractive for a range of potential applications such as gas storage, sensing, catalysis and separations.It is arguable that one may divide this family of materials into two categories: 1) those based on d-element chemistry and 2) those based on f-element compositions. Interestingly, the former group, i.e. transition metal compositions, can be said to be more mature in terms of development. It is perhaps convenient to think of the evolution of MOF and coordination polymer (CP) research in terms of structure, followed by function and ultimately application. With this model, one may observe a progression in the literature from heavy synthetic and characterization efforts, followed by a more refined approach where specific functionality is built into materials of interest. Lastly, an application is paired with a material. For the transition metals, scientists are entering the latter stage. The f-element researchers however, are approximately at the second stage where we are refining our synthetic efforts based on what has been discovered during previous, more exploratory endeavors.
One possible reason for this gap in the evolution of these materials may be explained by the coordination chemistry of the lanthanide and actinide elements themselves (Fig. 1). The Ln elements tend to have larger coordination numbers (typically 8–12), are primarily ionic in their bonding and display no crystal field effects, thus resulting in spherical coordination geometries. This is in stark contrast to the d-metals where at least somewhat predictable coordination geometries may be anticipated.2 An elements (specifically, U(VI) for this Highlight) may also present a challenge (or opportunity!) as these often occur as linear triatomic cations with terminal axial bonds and accordingly promote coordination in two dimensions only. Further, hydrolysis to form polynuclear species is often observed.
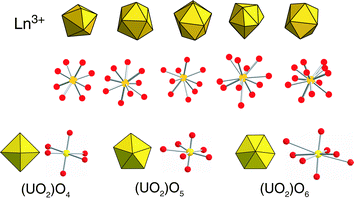 | ||
| Fig. 1 Coordination geometries for the lanthanide elements (top) and U(VI) (bottom). U(VI) commonly forms as the uranyl cation, UO22+, with terminal axial oxygen atoms to give rise to bipyramidal geometries. | ||
The purpose of this Highlight is therefore to give a snapshot of some of our efforts to capitalize on the coordination geometry of these materials. Our group, as well as several others, has made some important observations with respect to controlling the geometries of f-element based materials, including appropriate choice of linker molecules, inclusion of guest and/or templating species to direct structure formation and the pairing with d-elements to tune both structure and properties.
Several other reviews of metal–organic framework and coordination polymer materials (focused primarily on those containing transition-metals) have appeared recently and we encourage readers to explore those, especially for a more fundamental discussion of design, structural features and general vocabulary.3–21 Moreover, a Ln-specific review of coordination frameworks by Champness et al. has been published, many of the sentiments of which are echoed in this contribution.22 Herein, however, we aim to highlight both homo- and heterometallic coordination polymers constructed from either Ln/An elements or d-/f- metal pairings. In addition to the structural features already mentioned (and to be discussed below), these elements have particular relevance to sensor or display technologies, as well as gas storage materials. We will opt not to discuss theses applications in any great detail and instead point readers to some recent publications that address some of this potential. For example, several coordination polymers containing Eu(III), Tb(III) or other, less easily sensitized lanthanide elements, have been explored due to their attractive emissive properties.23–32 Additionally, certain crystal structures of lanthanide-containing CPs have the potential to produce second-harmonic generation, which could lead to applications in non-linear optics.33,34 Since the lanthanide ions are predominately in the 3+ oxidation state, examinations into the magnetic properties of these materials have also been conducted.35–40 Aside from the specific properties induced through the lanthanide metal center, structurally derived characteristics such as porosity,41–43 sorption,27,44,45 and thermal stability24,41,46 have been explored, as have efforts to produce helical and columnar structures.47,48
Uranyl containing coordination polymers are being explored for their luminescent properties as well. Emission from the uranyl center may be sensitized in a fashion similar to the Ln elements using the “antenna effect” (below).49–52 Photoexcited uranyl cations have long been known to oxidize organic compounds53–55 and U(VI) containing coordination polymers have most recently been applied to DNA and RNA cleavage as well as to the degradation of pollutants.56 Additionally, and more generally speaking however, the hydrothermal synthesis of U(VI) containing hybrid materials advances our understanding of this element under environmentally relevant conditions and as such may contribute to the nuclear fuel cycle as well as long-term storage and stewardship of spent nuclear fuel.57–59
2. Examples
As mentioned above, the general philosophy behind MOF and CP construction is to assemble metal centers, or more generally speaking, inorganic building units into extended architectures through the use of multifunctional (or ‘multitopic’) organic linkers (Scheme 1).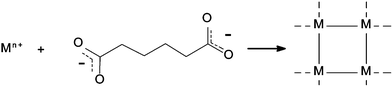 | ||
| Scheme 1 A schematic of coordination polymer or metal–organic framework assembly. A multifunctional organic linker (in this case a dicarboxylic acid) is used to assemble metal centers into an extended topology. | ||
A fundamental requirement of the organic species is that the functional groups must be sufficiently spaced from one another to avoid chelation. In addition, the metal center and functional group must have a coordination preference for one another. At present, most of our work has been focused on carboxylate functionalized linkers owing to the affinity of Ln(III) and U(VI) cations for these anions. Further, the diversity of structure types observed within these pairings, both from our group as well as others, has in some respects kept considerable attention on O-donor species. As such, we offer the following sections as representative examples of compounds synthesized with this philosophy in mind. Sections are organized according to homo- vs. heterometallic compounds and demonstrate an evolution in synthetic strategies to produce novel structure types. When appropriate, properties are discussed in the context of structure–property relationships.
2.1 Homometallic Ln systems
An early example of a Ln-MOF from our group is GWMOF-1: (C6H8O4)3Nd2(H2O)2 (Fig. 2).60 This compound exemplifies many of the structural features mentioned in the introduction: Each Nd(III) center is coordinated to nine oxygen atoms, eight of which are from adipate anions and the remaining being a bound water molecule. The Nd(III) centers polymerize to form chains that run along [100] which are in turn cross-linked through adipate linkers to form an overall 3-dimensional structure. The polyhedral chain demonstrates the higher coordination number demands of the metal center, in that both edge sharing and decoration via solvent molecules are present to complete the coordination sphere. Such a chain may be considered as a secondary building unit (SBU) as compared to a primary building unit as introduced in Fig. 1 (above).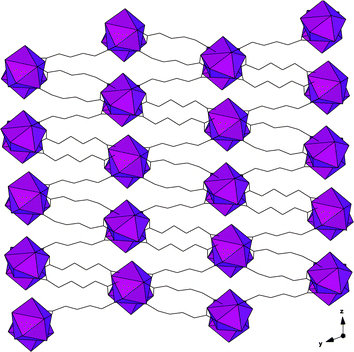 | ||
| Fig. 2 Polyhedral representation of GWMOF-1, (C6H8O4)3Nd2(H2O)2. Purple polyhedra are chains of NdO9 units that are cross-linked through adipate groups (black lines). | ||
Turning toward the analogous Pr–adipate system, we note formation of ([Pr2(C6H8O4)3(H2O)4]·(C6H10O4)(H2O)4) from identical reaction conditions as the previous Nd(III) structure (Fig. 3).61 The slightly larger Pr(III) centers in this compound display the same polymerization to form chains, yet have a total coordination number of 10 when two bound water molecules for each site are counted. The hydrated chains are linked to form an overall 3-dimensional structure (as with the Nd(III) compound). Interestingly, this structure contains neutral, H-bonded adipic acid molecules within the channels defined by the Pr–adipate framework. This was an admittedly serendipitous result, yet one which has served to inspire a new class of materials (below). In comparison to the Nd–adipate, it is clear in this compound that the presence of these extra-framework species has influenced the topology of this material, especially when viewed down the channel directions of both structures.
![Polyhedral representation of ([Pr2(C6H8O4)3(H2O)4]·(C6H10O4)(H2O)4). Chains of PrO10 polyhedra run along [100] to define channels occupied by neutral adipic acid molecules.](/image/article/2007/CE/b615696g/b615696g-f3.gif) | ||
| Fig. 3 Polyhedral representation of ([Pr2(C6H8O4)3(H2O)4]·(C6H10O4)(H2O)4). Chains of PrO10 polyhedra run along [100] to define channels occupied by neutral adipic acid molecules. | ||
In an effort to harness this observation, 4,4′-bipyridine was introduced to reaction mixtures with the intent of having this species serve as a structure directing (or ‘templating’) agent thus resulting in [Pr2(C6H8O4)3(H2O)2]·(C10H8N2) (Fig. 4).61 The 4,4′-bipyridine, as well as related pyridyl species, are very common linker species in d-metal coordination polymers,20 yet was chosen specifically for this system to serve as a guest molecule considering the coordination preference of the Ln(III) ions for harder bases such as carboxylate anions (see Scheme 2). Thus, the 4,4′-bipyridine does not coordinate directly to the metal center and instead remains hydrogen bonded to bound water molecules within the Pr(III) coordination sphere.
![[Pr2(C6H8O4)3(H2O)2]·(C10H8N2) shown along [010]. Neutral 4,4′-dipyridyl molecules occupy channels defined by chains of PrO10 polyhedra.](/image/article/2007/CE/b615696g/b615696g-f4.gif) | ||
| Fig. 4 [Pr2(C6H8O4)3(H2O)2]·(C10H8N2) shown along [010]. Neutral 4,4′-dipyridyl molecules occupy channels defined by chains of PrO10 polyhedra. | ||
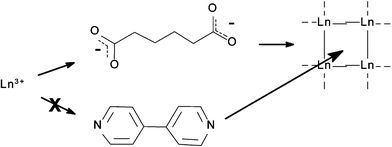 | ||
| Scheme 2 Coordination preferences based on the hard–soft/acid–base distinction.62 Ln(III) ions prefer harder anions such as the carboxylates and will bind to them preferentially over softer bases such at the pyridines. Uncoordinated bipyridine molecules are then free to ‘template’ structure formation. | ||
With respect to properties, the introduction of the aromatic guest molecule can also be seen to behave as a chromophore and ultimately a sensitizing species for the Ln(III) center. As we have already mentioned, much of the promise of f-metal MOFs and CPs stems from their luminescent behavior. Ln elements are notoriously inefficient emitters and the use of sensitizer or ‘antenna’ ligands wherein conjugated species that absorb in the UV transfer their energy to excited Ln states and in turn stimulate emission for the f-metal is a mature route with which to circumvent this issue. This behavior is well known in solution based systems and applications, yet is less studied in the solid state and relatively uncommon (thus far) in the f-metal coordination polymer literature. Fig. 5 shows the emission spectrum of [Eu2(C6H8O4)3(H2O)2]·(C10H8N2) when excited at 236 nm, the absorption maximum of the 4,4′-dipyridyl guest species. It should also be noted that the energy transfer is taking place without direct coordination of the aromatic to the Eu(III) center as the interaction between these species is strictly through H-bonding.
![Emission spectrum of [Eu2(C6H8O4)3(H2O)2]·(C10H8N2). Excitation is at 236 nm, the absorption maximum of the 4,4′-bipyridine guest molecules.](/image/article/2007/CE/b615696g/b615696g-f5.gif) | ||
| Fig. 5 Emission spectrum of [Eu2(C6H8O4)3(H2O)2]·(C10H8N2). Excitation is at 236 nm, the absorption maximum of the 4,4′-bipyridine guest molecules. | ||
Additional examples of this templating phenomenon can be found in GWMOF-9: [Nd2(C6H8O4)3(H2O)2]·(C13H14N2) and GWMOF-10: [Pr2(C8H12O4)3(H2O)2]·(C10H8N2) (Fig. 6). The linker–ligand pairings are adipic acid–1,2-bis(4-pyridyl)propane and suberic acid–4,4′-bipyridine respectively. Whereas the syntheses of these materials remain to be optimized (see Experimental section) and the luminescent properties are not yet explored, we present these compounds here as further examples of this philosophy. Common structural features include neutral frameworks and consequently neutral guests, hydrogen bonding between N atoms on the template and bound framework water molecules and 1-dimensional secondary building units within an overall 3-dimensional structure.
![GWMOF-9: [Nd2(C6H8O4)3(H2O)2]·(C13H14N2)
(left) and GWMOF-10: [Pr2(C8H12O4)3(H2O)2]·(C10H8N2)
(right). Occluded guest molecules, 1,2-bis(4-pyridyl)propane and 4,4′-bipyridine (respectively) remain neutral and H-bonded to the framework.](/image/article/2007/CE/b615696g/b615696g-f6.gif) | ||
| Fig. 6 GWMOF-9: [Nd2(C6H8O4)3(H2O)2]·(C13H14N2) (left) and GWMOF-10: [Pr2(C8H12O4)3(H2O)2]·(C10H8N2) (right). Occluded guest molecules, 1,2-bis(4-pyridyl)propane and 4,4′-bipyridine (respectively) remain neutral and H-bonded to the framework. | ||
Although not f-element examples, this approach may be extended to Group II metals such as Ca2+ given the similarities in ionic radii.63 [Ca(C6H8O4)(H2O)2]·(C10H8N2) and [Ca(C6H8O4)(H2O)2]·(C12H12N2) make use of 4,4′-bipyridine and 1,2-bis(4-pyridyl)ethane respectively and demonstrate similar structural features introduced for the Ln materials. We have explored the use of non-functionalized template molecules (e.g. benzene, naphthalene), although admittedly not as extensively and it appears that N-functionalized template molecules (e.g. bipyridines) work best for this assembly route.
Templating or structure direction of host topologies via judicious choice of guest molecule is a well known and mature route to new materials in oxide, sulfide, sulfate and related systems. Indeed, this is the route of choice for the synthesis of zeolites and aluminophosphate materials. We hope this approach evolves within the MOF and CP communities to be a robust and generally applicable method to produce new materials. Recognition of guest molecules influencing topologies is increasingly common in the literature, especially for d-metal systems. Many examples are clearly serendipitous, yet have potentially spawned more concerted efforts to incorporate such species explicitly.64–78
2.2 Homometallic U(VI) systems
A representative example of a uranyl containing metal–organic framework material is the uranyl–adipate: UO2(C6H8O4),79 shown in Fig. 7. In this material, a 3-dimensional structure is realized via assembly of [(UO2)2O8] dimers through flexible adipate linker species. As mentioned in the Introduction, the uranyl cation is somewhat pre-disposed toward forming 2-dimensional materials in the solid state owing to the terminal nature of the oxo groups. Rotation around C–C bonds within the adipate backbone however, allows for this 3-dimensional arrangement. It is interesting to note that this dimeric SBU had not been observed previously in U(VI) materials, molecular or otherwise despite the well known and extensive coordination chemistry of this element. A survey conducted in our laboratory of other uranyl–aliphatic dicarboxylates such as glutaric, pimelic, suberic, azelaic and sebacic acids all result in 2-dimensional layered topologies consisting of either these same dimers, or 1-dimensional chains of edge shared hexagonal bipyramids.80,81 Thus there may be competing mechanisms with respect to crystal packing and building unit speciation resulting from the nature of the linker species, further discussion of which has been presented elsewhere.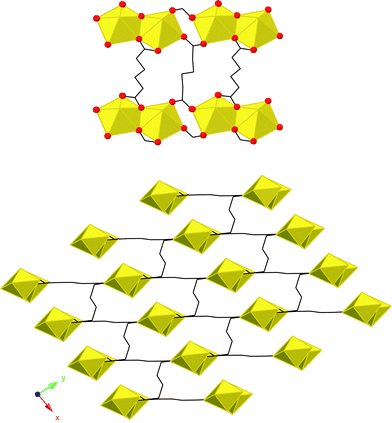 | ||
| Fig. 7 Top: A portion of the UO2(C6H8O4) structure. Yellow polyhedra are UO22+ centered pentagonal bipyramids edge sharing of which produces dimers. Bottom: 3-dimensional view of UO2(C6H8O4). | ||
Efforts to produce templated U(VI) MOFs using the philosophy described above for the Ln system have not been successful. Instead, direct coordination to the uranyl centers by both aliphatic carboxylate anions and bipyridine species is the norm.80Fig. 8 depicts UO2(C6H8O4)(C10H8N2), a structure consisting of three-way interpenetrated sheets constructed from chains of UO22+ cations polymerized through the adipate groups along [001]. These chains are then stitched together through 4,4′-bipyridine groups to form sheets. In light of this observation, we have turned our attention to the construction of U(VI) materials using multiple linker species. As a result, we have produced a diverse family of compounds with a range of topologies. We have recently reported a systematic investigation wherein aromatic N donor species were paired with aliphatic dicarboxylates. Whereas hard–soft/acid–base distinctions are perhaps more clear-cut in the Ln system, the uranyl cation with its potential for formation of polynuclear building units and presence of terminal oxo ligands, may provide for alternative interactions with varying functional groups. In this system, it appears the alipathic carboxylates are always linker species whereas the aromatic pyridyl groups behave as either linkers (when sterically advantageous) or as uncoordinated charge balancing guests.
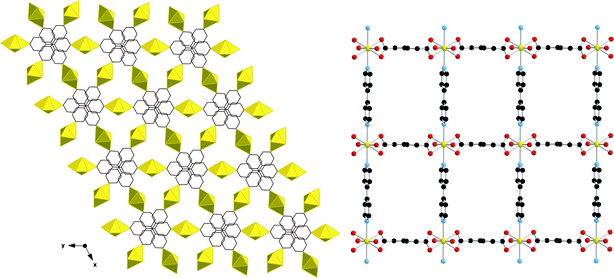 | ||
| Fig. 8 Left: A single sheet of UO2(C6H8O4)(C10H8N2). Right: Overall structure of UO2(C6H8O4)(C10H8N2) showing three-way interpenetration. | ||
3. Heterometallic d-/f- systems
Heterometallic materials, in particular those with d- and f-metal compositions may be prepared by moving toward heterofunctional linker molecules. Ideally, one can tune in a coordination geometry preference within a given linker molecule such as shown in Scheme 3. We have applied this approach to both Ln and An systems. The rationale for heterometallic systems is two-fold: from a structural point of view, the use of a second metal center (in these cases, a transition metal) may provide an additional building unit from which to construct extended topologies. As proposed already, this second metal center may have a more predictable coordination geometry than its Ln (or An) counterpart, and thus make a contribution to the overall structure. Second, the introduction of a d-metal may influence the physical properties of a material, as will be specifically noted with the luminescence features of the following examples.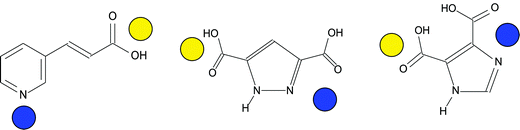 | ||
| Scheme 3 Distinction of metal centers between various functional groups based on coordination preference. Harder cations (e.g. Ln(III) or UO22+, yellow spheres) tend to bind to harder functional groups such as carboxylates. Softer metals (e.g. Cu(II), blue spheres) tend to bind to softer, N-containing groups. Left to right: trans-3-(3-pyridyl) acrylic acid (3-HYPA), 3,5-pyrazoledicarboxylic acid (H3PDC), 4,5-imidazoledicarboxylic acid (4,5-IDCA). | ||
3.1 Heterometallic Ln–d systems
The potential for use of 3-HYPA as a heterofunctional linker was introduced in Scheme 3. [Cu3(C8H6NO2)6Nd2(NO3)6] (Fig. 9) contains NdO9 and CuN2O2 building units that demonstrate HSAB distinctions.82 In a departure from what is observed in many of the Ln centered coordination polymers mentioned thus far however, polymerization of the primary Ln coordination sphere to form chains of polyhedra is not observed. This may be due to a number of factors, including sterics around the Ln center and ultimately not related to the presence of the d-metal. Where the Cu(II) centers may in fact manifest themselves is in the luminescent behavior of these materials. We have prepared homometallic ‘end member’ compounds [Ln(C8H6NO2)3H2O] (Ln = Eu3+, Nd3+), the Eu(III) analogue of which is highly luminescent. Conversely, [Cu3(C8H6NO2)6Eu2(NO3)6] displays no luminescent behavior and it is therefore arguable that the structure provides an alternate pathway for energy dissipation presumably related to the Cu(II) site. Finally, the Ln-only endmembers are 1-dimensional chain structures whereas the heterometallic compounds are 3-dimensional.![3-Dimensional structure of [Cu3(C8H6NO2)6Nd2(NO3)6]. Purple polyhedra are discrete NdO9 units whereas blue squares are Cu(ii) sites in square planar geometry. Black/red lines are the 3-HYPA linker groups.](/image/article/2007/CE/b615696g/b615696g-f9.gif) | ||
| Fig. 9 3-Dimensional structure of [Cu3(C8H6NO2)6Nd2(NO3)6]. Purple polyhedra are discrete NdO9 units whereas blue squares are Cu(II) sites in square planar geometry. Black/red lines are the 3-HYPA linker groups. | ||
3.2 Heterometallic U(VI)–d systems
An example of this approach can be seen in the U–H3PDC system.83 A homonuclear uranyl ‘endmember’ may be constructed from this linker molecule in UO2(C5H2N2O4)(H2O) as shown in Fig. 10. Interestingly, this layered structure displays no HSAB specificity as the uranyl cation is bound to both carboxylic groups and the N–/O– sites, the latter of which may be understandable in the absence of a second metal center when one considers a possible chelate effect and formation of a five membered ring. Specificity with this linker is observed when Cu(II) centers are added (Fig. 10) and uranyl centers remain coordinated to O-donor sites exclusively as in (UO2)Cu(C5H2N2O4)2(H2O)2. As we have seen with the Eu(III)–Cu(II) system, luminescent behavior appears to be influenced by the Cu(II) center as UO2(C5H2N2O4)(H2O) is highly luminescent yet (UO2)Cu(C5H2N2O4)2(H2O)2 is not.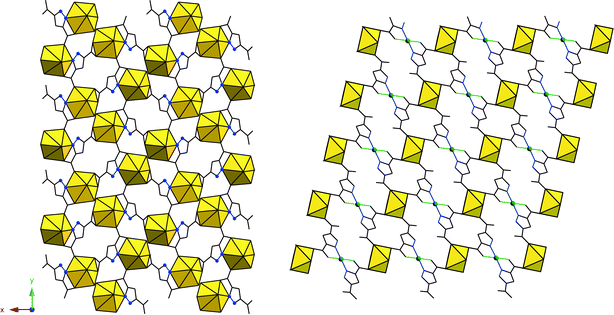 | ||
| Fig. 10 Left: UO2(C5H2N2O4)(H2O). Black lines and blue spheres are the H3PDC linker groups. Right: (UO2)Cu(C5H2N2O4)2(H2O)2. Cu(II) sites are shown as green spheres. Axial water molecules on these sites have been omitted. | ||
Related examples of this phenomenon are evident in the uranyl–4,5-imidazoledicarboxylic acid (4,5-idca) system (Fig. 11; see Experimental section). The uranyl ‘endmember,’ (UO2)(C5H2N2O4), displays similar coordination characteristics as that of the H3PDC system, that is, N–/O– coordination and chelation. Addition of Cu(II) to the hydrothermal reaction mixture results in the co-crystallization of two higher-dimensional phases: (UO2)2(C5N2O4H)2(C5N2O4H2)4Cu3(H2O)2·2H2O and (UO2)2(C5H2N2O4)2(OH)2Cu·2H2O (Fig. 12). The first structure contains two distinct coordination geometries for the Cu(II) sites as both square planar and octahedral coordination are observed. Polyhedra are isolated (e.g. no edge sharing) and the familiar pentagonal bipyramidal geometry of the uranyl center remains coordinated in the equatorial plane only. The latter structure may best be thought of as sheets of [(UO2)2O8] dimers (like those in the adipate, above) linked through dicarboxylate groups that are also coordinated to Cu(II) centers in square planar geometry. A 3-dimensional structure may be realized when one considers a possible bridging through the uranyl oxygen atoms to the Cu(II) centers. The uranyl and Cu(II)–O bond lengths are consistent with typical values (1.780 and 2.593 Å respectively) and may be considered an additional mode of connectivity between sheets. This is mentioned explicitly as coordination through the nominally terminal uranyl oxygen atoms is exceedingly rare and hence the disposition of uranyl containing compounds (especially oxides and minerals) to form layered structures. Similar connectivity is seen in the Cu(II) containing uranyl minerals metatorbernite and torbernite.84 We hope to capitalize on this observation and move toward harnessing the uranyl oxygen atoms as a binding site as these have long been considered ‘off-limits’ to additional coordination. Lastly, the mixed N–/O– coordination at the Cu(II) centers in these two examples has been common to all Cu(II) containing heterometallic structures presented herein. Synthesis optimization and fluorescent studies are in progress, yet we offer these structures at this juncture as further examples of heterometallic uranyl–d-metal coordination polymers.
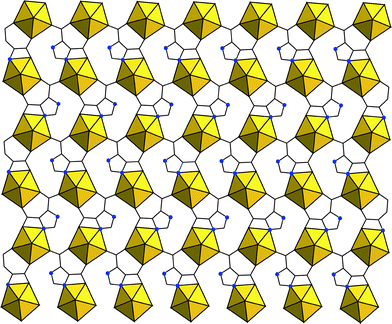 | ||
| Fig. 11 A single layer of 2-dimensional (UO2)(C5H2N2O4). | ||
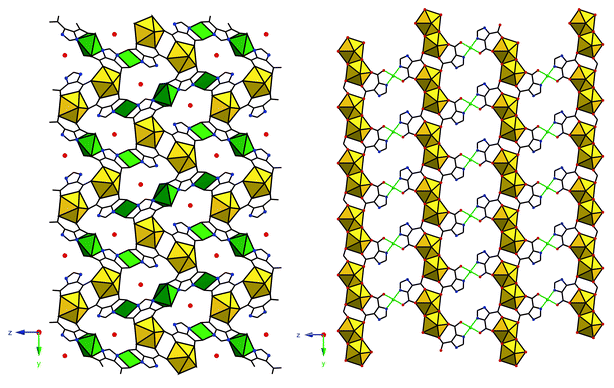 | ||
| Fig. 12 Left: A portion of the 3-dimensional (UO2)2(C5N2O4H)2(C5N2O4H2)4Cu3(H2O)2·2H2O. Cu(II) sites are shown as green polyhedra and guest water molecules as red spheres. Left: A single layer of (UO2)2(C5H2N2O4)2(OH)2Cu·2H2O. Subsequent stacking is through the axial positions of the Cu(II) sites (green spheres) and the uranyl oxygen atoms at the apices of the pentagonal bipyramids. | ||
Coupling the uranyl cation with Pb(II) centers has been successful through the use of 2,6-pyridinedicarboxylic acid and two 3-dimensional structures [(UO2)(C7H3NO4)2Pb2(C2O4)(H2O)2] and [(UO2)(C7H3NO4)2Pb·2H2O] are shown in Fig. 13.85 Both compounds display edge sharing of the uranyl and Pb(II) polyhedra. Such sharing was not present in previously described U(VI)–d systems and here results from the close proximity of the functional groups as well as the tridentate coordination to the uranyl centers; the latter provides open carboxylate oxygen atoms that serve to bridge the Pb(II) and uranyl sites. The first compound, [(UO2)(C7H3NO4)2Pb2(C2O4)(H2O)2], consists of uranyl hexagonal bipyramids edge shared to distorted Pb(II) octahedra to form trimers. These trimers are polymerized through the 2,6-pydc linkers to form sheets in the (104) plane. Interestingly, these sheets are connected through oxalate linkages, the presence of which resulted from in situ ligand synthesis, presumably from a decarboxylation of the pydc linker. In situ ligand synthesis is becoming an increasingly popular (although not always planned!) route to new materials and it is likely that more examples of this phenomenon will emerge in these systems.86 The second compound, [(UO2)(C7H3NO4)2Pb·2H2O], displays similar local geometry about the metal centers in that edge sharing of uranyl and Pb(II) polyhedra is observed. Further details of the syntheses and structures of these materials have been reported elsewhere.85
![Inset: 2,6-pyridinedicarboxylic acid. Top: (UO2)(C7H3NO4)2Pb2(C2O4)(H2O)2. Pb(ii) sites are shown as green polyhedra; oxalate groups that link sheets have been omitted. Bottom: [(UO2)(C7H3NO4)2Pb·2H2O]. Red spheres are water molecules.](/image/article/2007/CE/b615696g/b615696g-f13.gif) | ||
| Fig. 13 Inset: 2,6-pyridinedicarboxylic acid. Top: (UO2)(C7H3NO4)2Pb2(C2O4)(H2O)2. Pb(II) sites are shown as green polyhedra; oxalate groups that link sheets have been omitted. Bottom: [(UO2)(C7H3NO4)2Pb·2H2O]. Red spheres are water molecules. | ||
In addition to these compounds, several other pyridine carboxylates have been reported with both d- and f-metals. The coordination geometry in these uranyl–2,6-pydc systems is unique, however with respect to the tridentate geometry in the equatorial plane.51,87–94 Further, hydrothermal synthesis of solid state Pb–U(VI) oxide compounds has been explored and in these materials (e.g. without organic linkers), Pb(II) is often a charge balancing interlayer species.95
4. Conclusions and outlook
The purpose of this Highlight was to demonstrate, by way of example, some of the unique structural features of f-metal based coordination polymers and metal–organic frameworks. Much of the (structural) appeal of these materials stems from the local geometry of the Ln or uranyl centers and how they ultimately complete their coordination spheres. These include polymerization of primary building units to form secondary species, coordination specificity based on hard–soft/acid–base considerations and steric influences based on functional group location. We touched on properties including guest sensitized luminescence within the Ln–MOF materials and linker-sensitized behavior in the uranyl systems. The latter of which confers the possibility of tuning such emission by the presence of secondary metal centers. The potential applications of these materials however, in particular sensing, catalysis and luminescent technologies will begin to be realized as advances are made in targeting and tuning structures of interest. Templating extended topologies, use of multiple linker molecules and expanding the catalogue of heterometallic systems are likely routes to this end. Although only mentioned briefly in the last section, in situ ligand synthesis is likely to be a strong contributor to new materials once more mechanistic information and ultimately, control in such systems is achieved. The frequency of these reports in the literature is increasing and we look forward to witnessing and participating in this development.Experimental
[Nd2(C6H8O4)3(H2O)2]·(C13H14N2) (GWMOF-9)
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), Z
= 2, μ(Mo-Kα)
= 2.988 mm−1, 35281 reflections measured, 7145 unique (Rint
= 0.1064) which were used in all calculations. The final wR(F2) was 0.0651 (observed data). CCDC reference number 625116.
(no. 2), Z
= 2, μ(Mo-Kα)
= 2.988 mm−1, 35281 reflections measured, 7145 unique (Rint
= 0.1064) which were used in all calculations. The final wR(F2) was 0.0651 (observed data). CCDC reference number 625116.[Pr2(C8H12O4)3(H2O)2]·(C10H8N2) (GWMOF-10)
(no. 2), Z
= 2, μ(Mo-Kα)
= 2.663 mm−1, 13391 reflections measured, 3757 unique (Rint
= 0.0949) which were used in all calculations. The final wR(F2) was 0.0885 (observed data). CCDC reference number 625117.(UO2)(C5H2N2O4)
(UO2)2(C5H2N2O4)2(C5N2O4H)2Cu3(H2O)2·2H2O
(UO2)2(C5H2N2O4)2(OH)2Cu·2H2O
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b615696g
Acknowledgements
Research efforts in the Cahill group are supported by the National Science Foundation (DMR-0348982 and DMR-0419754) and the United States Department of Energy (DE-FG02-05ER15736). DTD is grateful to the ARCS Foundation of Metro Washington for a tuition scholarship. Undergraduate researchers Daniel Bozzuto and Deepak Chander assisted with the syntheses of GWMOF-9 and -10.References
- C. J. Kepert and M. J. Rosseinsky, Chem. Commun., 1999, 375 RSC . As this reference will indicate however, a 3-dimensional structure is not a pre-requisite for accessible void space.
- H. C. Aspinall, Chemistry of the f-Block Elements (Advanced Chemistry Texts), ed. D. Phillips, P. O'Brien, and S. Roberts, Gordon and Breach Science Publishers, Amsterdam, 2001 Search PubMed.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC.
- S. Kitagawa, S.-i. Noro and T. Nakamura, Chem. Commun., 2006, 701 RSC.
- K. Uemura, R. Matsuda and S. Kitagawa, J. Solid State Chem., 2005, 178, 2420 CrossRef CAS.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC.
- M.-L. Tong, S. Hu, J. Wang, S. Kitagawa and S. W. Ng, Cryst. Growth Des., 2005, 5, 837 CrossRef CAS.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS.
- J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239 CrossRef CAS.
- N. Ockwig, W. O. Delgado-Friedrichs, M. O'Keeffe and M. Yaghi Omar, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS.
- B. Moulton and M. J. Zaworotko, Curr. Opin. Solid State Mater. Sci., 2002, 6, 117 CrossRef CAS.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS.
- C. Janiak, Dalton Trans., 2003, 2781 RSC.
- C. J. Kepert, Chem. Commun., 2006, 695 RSC.
- J. D. Wuest, Chem. Commun., 2005, 5830 RSC.
- C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS.
- G. Férey, Chem. Mater., 2001, 13, 3084 CrossRef CAS.
- G. Férey, J. Solid State Chem., 2000, 152, 37 CrossRef CAS.
- B.-H. Ye, M.-L. Tong and X.-M. Chen, Coord. Chem. Rev., 2005, 249, 545 CrossRef CAS.
- K. Biradha, M. Sarkar and L. Rajput, Chem. Commun., 2006, 4169 RSC.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS.
- R. J. Hill, D.-L. Long, P. Hubberstey, M. Schroder and N. R. Champness, J. Solid State Chem., 2005, 178, 2414 CrossRef.
- Y. Kim, M. Suh and D.-Y. Jung, Inorg. Chem., 2004, 43, 245 CrossRef CAS.
- J.-Y. Wu, T.-T. Yeh, Y.-S. Wen, J. Twu and K.-L. Lu, Cryst. Growth Des., 2006, 6, 467 CrossRef CAS.
- G.-F. Liu, Z.-P. Qiao, H.-Z. Wang, X.-M. Chen and G. Yang, New J. Chem., 2002, 26, 791 RSC.
- L. Ma, O. R. Evans, B. M. Foxman and W. Lin, Inorg. Chem., 1999, 38, 5837 CrossRef CAS.
- T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651 CrossRef CAS.
- Y.-Q. Sun, J. Zhang and G.-Y. Yang, Chem. Commun., 2006, 1947 RSC.
- X. Guo, G. Zhu, Q. Fang, M. Xue, G. Tian, J. Sun, X. Li and S. Qiu, Inorg. Chem., 2005, 44, 3850 CrossRef CAS.
- S. Mondal, M. Mukherjee, S. Chakraborty and A. K. Mukherjee, Cryst. Growth Des., 2006, 6, 940 CrossRef CAS.
- A. de Bettencourt-Dias, Inorg. Chem., 2005, 44, 2734 CrossRef CAS.
- B. D. Chandler, D. T. Cramb and G. K. H. Shimizu, J. Am. Chem. Soc., 2006, 128, 10403 CrossRef CAS.
- J.-m. Shi, W. Xu, Q.-y. Liu, F.-l. Liu, Z.-l. Huang, H. Lei, W.-t. Yu and Q. Fang, Chem. Commun., 2002, 756 RSC.
- R. Vaidhyanathan, S. Natarajan and C. N. R. Rao, J. Solid State Chem., 2004, 177, 1444 CrossRef CAS.
- Z. He, Z.-M. Wang and C.-H. Yan, CrystEngComm, 2005, 7, 143 RSC.
- Z. Wang, M. Strobele, K.-L. Zhang, H.-J. Meyer, X.-Z. You and Z. Yu, Inorg. Chem. Commun., 2002, 5, 230 CrossRef CAS.
- Y. Wan, L. Zhang, L. Jin, S. Gao and S. Lu, Inorg. Chem., 2003, 42, 4985 CrossRef CAS.
- J. Legendziewicz, B. Keller, I. Turowska-Tyrk and W. Wojciechowski, New J. Chem., 1999, 23, 1097 RSC.
- E. Galdecka, Z. Galdecki, P. Gawryszewska and J. Legendziewicz, New J. Chem., 2000, 24, 387 RSC.
- A. Michaelides, S. Skoulika, E. G. Bakalbassis and J. Mrozinski, Cryst. Growth Des., 2003, 3, 487 CrossRef CAS.
- C. Serre and G. Ferey, J. Mater. Chem., 2002, 12, 3053 RSC.
- L. Pan, E. B. Woodlock, X. Wang and C. Zheng, Inorg. Chem., 2000, 39, 4174 CrossRef.
- C. Daiguebonne, N. Kerbellec, K. Bernot, Y. Gerault, A. Deluzet and O. Guillou, Inorg. Chem., 2006, 45, 5399 CrossRef CAS.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504 CrossRef CAS.
- L. Pan, K. M. Adams, H. E. Hernandez, X. Wang, C. Zheng, Y. Hattori and K. Kaneko, J. Am. Chem. Soc., 2003, 125, 3062 CrossRef CAS.
- F. Serpaggi, T. Luxbacher, A. K. Cheetham and G. Ferey, J. Solid State Chem., 1999, 145, 580 CrossRef CAS.
- R. Koner, M. Nayak, G. Ferguson, J. N. Low, C. Glidewell, P. Misra and S. Mohanta, CrystEngComm, 2005, 7, 129 RSC.
- H. Masu, M. Tominaga, K. Katagiri, T. Kato and I. Azumaya, CrystEngComm, 2006, 8, 578 RSC.
- W. Chen, H.-M. Yuan, J.-Y. Wang, Z.-Y. Liu, J.-J. Xu, M. Yang and J.-S. Chen, J. Am. Chem. Soc., 2003, 125, 9266 CrossRef CAS.
- Y.-S. Jiang, Z.-T. Yu, Z.-L. Liao, G.-H. Li and J.-S. Chen, Polyhedron, 2006, 25, 1359 CrossRef CAS.
- Y.-Z. Zheng, M.-L. Tong and X.-M. Chen, Eur. J. Inorg. Chem., 2005, 4109 CrossRef.
- D. Wittberger, C. Berens, C. Hammann, E. Westhof and R. Schroeder, J. Mol. Biol., 2000, 300, 339 CrossRef CAS.
- H. D. Burrows and T. J. Kemp, Chem. Soc. Rev., 1974, 3, 139 RSC.
- S. L. Suib and K. A. Carrado, Inorg. Chem., 1985, 24, 863 CrossRef CAS.
- S. L. Suib, A. Kostapapas and D. Psaras, J. Am. Chem. Soc., 1984, 106, 1614 CrossRef CAS.
- Z.-T. Yu, Z.-L. Liao, Y.-S. Jiang, G.-H. Li and J.-S. Chen, Chem.–Eur. J., 2005, 11, 2642 CrossRef CAS.
- A. E. V. Gorden, J. Xu, N. Raymond Kenneth and P. Durbin, Chem. Rev., 2003, 103, 4207 CrossRef CAS.
- R. C. Ewing, Can. Mineral., 2001, 39, 697 CrossRef CAS.
- P. Prapaipong, E. L. Shock and C. M. Koretsky, Geochim. Cosmochim. Acta, 1999, 63, 2547 CrossRef CAS.
- L. A. Borkowski and C. L. Cahill, Inorg. Chem. Commun., 2004, 7, 725 CrossRef CAS.
- D. T. de Lill, N. S. Gunning and C. L. Cahill, Inorg. Chem., 2005, 44, 258 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581 CrossRef CAS.
- D. T. de Lill, D. J. Bozzuto and C. L. Cahill, Dalton Trans., 2005, 2111 RSC.
- Y.-B. Dong, H.-X. Xu, J.-P. Ma and R.-Q. Huang, Inorg. Chem., 2006, 45, 3325 CrossRef CAS.
- Q.-R. Fang, G.-S. Zhu, M. Xue, J.-Y. Sun and S.-L. Qiu, Dalton Trans., 2006, 20, 2399 RSC.
- M. Du, S.-T. Chen, X.-H. Bu and J. Ribas, Inorg. Chem. Commun., 2002, 5, 1003 CrossRef CAS.
- C. Inman, J. M. Knaust and S. W. Keller, Chem. Commun., 2002, 156 RSC.
- C.-B. Liu, X.-J. Zheng, Y.-Y. Yang and L.-P. Jin, Inorg. Chem. Commun., 2005, 8, 1045–1048 CrossRef CAS.
- K.-T. Youm, H. C. Kang, G.-b. Lee, H. K. Woo, Y. J. Park, N.-k. Lee, J. Ko and M.-J. Jun, Polyhedron, 2006, 25, 2318 CrossRef CAS.
- Z. Wang, B. Zhang, T. Otsuka, K. Inoue, H. Kobayashi and M. Kurmoo, Dalton Trans., 2004, 2209 RSC.
- A. Mohanu, C. Brouca-Cabarrecq and J. C. Trombe, J. Solid State Chem., 2006, 179, 3 CrossRef CAS.
- X.-J. Zheng, L.-P. Jin, S. Gaob and S.-Z. Luc, New J. Chem., 2005, 29, 798 RSC.
- S. V. Ganesan, P. Lightfoot and S. Natarajan, Solid State Sci., 2004, 6, 757 CrossRef CAS.
- A. D. Burrows, K. Cassar, R. M. W. Friend, M. F. Mahon, S. P. Rigby and J. E. Warren, CrystEngComm, 2005, 7, 548 RSC.
- C.-B. Ma, C.-N. Chen and Q.-T. Liu, CrystEngComm, 2005, 7, 650 RSC.
- E.-Y. Choi, K. Park, C.-M. Yang, H. Kim, J.-H. Son, S. W. Lee, Y. H. Lee, D. Min and Y.-U. Kwon, Chem.–Eur. J., 2004, 10, 5535 CrossRef CAS.
- Z. Shi, G. Li, L. Wang, L. Gao, X. Chen, J. Hua and S. Feng, Cryst. Growth Des., 2004, 4, 25 CrossRef CAS.
- M. Du and X.-J. Zhao, Inorg. Chem. Commun., 2004, 7, 1056 CrossRef CAS.
- L. A. Borkowski and C. L. Cahill, Inorg. Chem., 2003, 42, 7041 CrossRef CAS.
- L. A. Borkowski and C. L. Cahill, Cryst. Growth Des., 2006, 6, 2248 CrossRef CAS.
- L. A. Borkowski and C. L. Cahill, Cryst. Growth Des., 2006, 6, 2241 CrossRef CAS.
- N. S. Gunning and C. L. Cahill, Dalton Trans., 2005, 2788 RSC.
- M. Frisch and C. L. Cahill, Dalton Trans., 2005, 1518 RSC.
- A. J. Locock and P. C. Burns, Can. Mineral., 2003, 41, 489 CrossRef CAS.
- M. Frisch and C. L. Cahill, Dalton Trans., 2006, 4679 RSC.
- X.-M. Zhang, Coord. Chem. Rev., 2005, 249, 1201 CrossRef CAS.
- Y.-S. Jiang, G.-H. Li, Y. Tian, Z.-L. Liao and J.-S. Chen, Inorg. Chem. Commun., 2006, 9, 595 CrossRef CAS.
- B. O. Patrick, C. L. Stevens, A. Storr and R. C. Thompson, Polyhedron, 2005, 24, 2242 CrossRef CAS.
- B. Masci and P. Thuery, Polyhedron, 2005, 24, 229 CrossRef CAS.
- B. Zhao, X.-Y. Chen, P. Cheng, D.-Z. Liao, S.-P. Yan and Z.-H. Jiang, J. Am. Chem. Soc., 2004, 126, 15394 CrossRef CAS.
- D. Min and S. W. Lee, Inorg. Chem. Commun., 2002, 5, 978 CrossRef CAS.
- Y. C. Liang, R. Cao, M. C. Hong, D. F. Sun, Y. J. Zhao, J. B. Weng and R. H. Wang, Inorg. Chem. Commun., 2002, 5, 366 CrossRef CAS.
- G. B. Andreev, M. Y. Antipin, A. M. Fedoseev and N. A. Budantseva, Dokl. Akad. Nauk, 2000, 374, 343 CAS.
- J. M. Harrowfield, N. Lugan, G. H. Shahverdizadeh, A. A. Soudi and P. Thuery, Eur. J. Inorg. Chem., 2006, 389 CrossRef CAS.
- P. C. Burns, Can. Mineral., 2005, 43, 1839 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |