Exploring the hydrogen-bond preference of N–H moieties in co-crystals assembled via O–H(acid)⋯N(py) intermolecular interactions
Christer B.
Aakeröy
a,
Izhar
Hussain
b,
Safiyyah
Forbes
a and
John
Desper
a
aDepartment of Chemistry, Kansas State University, Manhattan, KS 66506, USA
bDepartment of Pharmacy, University of Balochistan, Quetta, Pakistan
First published on 28th November 2006
Abstract
In order to establish the hydrogen-bond preference of an amide based N–H moiety faced with different C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or –OH hydrogen-bond acceptors, the crystal structures of several new co-crystals and salts were examined: 3-acetaminopyridine fumaric acid (2 : 1) 1, 4-(acetaminomethyl)pyridine fumaric acid (2 : 1) 2, 4-acetaminopyridine decanedioic (sebacic) acid (2 : 1) 3, 4-(acetaminomethyl)pyridine adipic acid (2 : 1) 4, 4-(acetaminomethyl)pyridine isophthalic acid (2 : 1) 5, 4-(acetaminomethyl)pyridinium 5-nitro-hydrogen isophthalate hydrate 6, 4-acetaminopyridinium hydrogenglutarate (1 : 1) 7. All co-crystals, 1–5, are constructed from an O–H(acid)⋯N(py) hydrogen bond and for the salts, 6–7, the primary synthon is the corresponding charge-assisted N–H+⋯−O interaction. The remaining N–H donor (on the amide moiety) shows a preference (4 out of 5) for the amide CO over the acid CO.
O or –OH hydrogen-bond acceptors, the crystal structures of several new co-crystals and salts were examined: 3-acetaminopyridine fumaric acid (2 : 1) 1, 4-(acetaminomethyl)pyridine fumaric acid (2 : 1) 2, 4-acetaminopyridine decanedioic (sebacic) acid (2 : 1) 3, 4-(acetaminomethyl)pyridine adipic acid (2 : 1) 4, 4-(acetaminomethyl)pyridine isophthalic acid (2 : 1) 5, 4-(acetaminomethyl)pyridinium 5-nitro-hydrogen isophthalate hydrate 6, 4-acetaminopyridinium hydrogenglutarate (1 : 1) 7. All co-crystals, 1–5, are constructed from an O–H(acid)⋯N(py) hydrogen bond and for the salts, 6–7, the primary synthon is the corresponding charge-assisted N–H+⋯−O interaction. The remaining N–H donor (on the amide moiety) shows a preference (4 out of 5) for the amide CO over the acid CO.
Introduction
The design and synthesis of co-crystals is currently receiving considerable attention1 partly because of fundamental interest in molecular-recognition driven assembly processes,2 and partly because of potential applications of co-crystals in all areas of functional solids, notably pharmaceuticals.3 Most supramolecular synthetic strategies employed in the deliberate design of binary co-crystals4,5 are based upon combinations of functional groups located on different molecules that prefer to interact with each other rather than with themselves,6–8 such that heteromeric recognition and binding (required for co-crystal formation) are favored over homomeric hydrogen bonds (which will lead to a simple recrystallization of the reactants). In this context, it is often helpful to utilize the Etter “rules”9 which suggest that the best hydrogen-bond donor will preferentially interact with the best hydrogen-bond acceptor and the second-best acceptor with the second-best donor etc. Empirically based guidelines like these have facilitated the deliberate construction of families of co-crystals with desired connectivity incorporating a wide range of acids, amides, and heterocycles.10In this particular study, we have employed a combination of two well-known and robust synthons, the heteromeric carboxylic acid⋯pyridine interaction and the N–H⋯O (amide⋯carbonyl) interaction in an attempt to prepare binary co-crystals. The initial assembly of the co-crystal will be achieved by locating a carboxylic acid and a pyridyl moiety on different molecular fragments. Once this interaction is locked into place, the resulting binary aggregates can be further organized through N–H⋯O interactions. However, since the N–H donor has a choice of three different potential hydrogen-bond acceptors, it is not obvious how the competition between those three oxygen atoms will play out, Scheme 1.
 | ||
| Scheme 1 Expected primary acid⋯pyridine synthon (I). Three possible interactions involving the N–H hydrogen-bond donor: with the amide CO (II), with the acid CO (III), with the acid O–H (IV). | ||
The goal of this study is to determine patterns of molecular recognition preferences of the N–H moiety based upon an analysis of several new co-crystals as well as of relevant data obtained from the CSD.11
Experimental
All chemicals were purchased from Aldrich and used without further purification. Melting points were determined on a Fisher-Johns melting point apparatus and are uncorrected.Preparations
O, s), 1533 cm−1 (Amide II, s).
O), 1515 cm−1 (Amide II, s).
O, s), 1558 cm−1 (Amide II, s).
O acid, s), 1672 cm−1 (CO amide I, s), 1558 cm−1 (Amide II, s).
O acid, s), 1656 cm−1 (CO amide I, s), 1560 cm−1 (Amide II, s).
O acid, s), 1597 cm−1 (CO amide I, s), 1507 cm−1 (Amide II, s).
O acid, s), 1652 cm−1 (CO amide I, s), 1554 cm−1 (Amide II, s).
O acid, s), 1652 cm−1 (CO amide I, s), 1558 cm−1 (Amide II, s).
O acid, s), 1652 cm−1 (CO amide I, s), 1534 cm−1 (Amide II, s).
O acid, s), 1652 cm−1 (CO amide I, s), 1555 cm−1 (Amide II, s).
O acid, s), 1699 cm−1 (CO amide I, s), 1557 cm−1 (Amide II, s).
X-Ray crystallography
X-Ray data were collected on a Bruker SMART 1000 four-circle CCD diffractometer at 173 K (2, 4, 5, 6, and 7) or a SMART APEX CCD diffractometer at 100 K (1 and 3) using a fine-focus molybdenum Kα tube. Data were collected using SMART.14 Initial cell constants were found by small widely separated “matrix” runs. Generally, an entire hemisphere of reciprocal space was collected regardless of Laué symmetry. Scan speed and scan width were chosen based on scattering power and peak rocking curves.Unit cell constants and orientation matrix were improved by least-squares refinement of reflections thresholded from the entire dataset. Integration was performed with SAINT,15 using this improved unit cell as a starting point. Precise unit cell constants were calculated in SAINT from the final merged dataset. Lorenz and polarization corrections were applied. Laué symmetry, space group, and unit cell contents were found with XPREP.
Data were reduced with SHELXTL.16 The structures were solved in all cases by direct methods without incident, Table 1. In general, hydrogen atoms were assigned to idealized positions and were allowed to ride. Where possible, the coordinates of the amide hydrogen atoms were allowed to refine. Heavy atoms were refined with anisotropic thermal parameters. Unless otherwise noted, data were corrected for absorption.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| Formula moiety | (C7H8N2O)2 | (C8H10N2O)2 | (C7H8N2O)2 | (C8H10N2O)2 | (C8H10N2O)2 | (C8H10N2O) | (C7H8N2O) |
| (C4H4O4) | (C4H4O4) | (C10H18O4) | (C6H10O4) | (C8H6O4) | (C8H5NO6) | (C5H8O4) | |
| (H2O) | |||||||
| Empirical formula | C18H20N4O6 | C20H24N4O6 | C24H34N4O6 | C22H30N4O6 | C24H26N4O6 | C16H17N3O8 | C12H16N2O5 |
| Molecular weight | 388.38 | 416.43 | 474.55 | 446.50 | 466.49 | 379.33 | 268.27 |
| Color, habit | Colorless, plate | Colorless, plate | Colorless, irregular | Colorless, plate | Colorless, prism | Amber, prism | Colorless, prism |
| Crystal system | Orthorhombic | Triclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic | Triclinic |
| Space group, Z | Pca2(1), 4 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , 1 , 1 |
C2/c, 4 | P2(1)/c, 2 | Fdd2, 16 | P2(1)/c, 4 |
P, 2 |
| a/Å | 17.930(4) | 4.9718(10) | 28.532(3) | 4.7523(7) | 34.784(6) | 18.5868(12) | 7.1730(8) |
| b/Å | 3.9380(10) | 7.0047(11) | 8.2965(8) | 24.295(3) | 57.476(12) | 6.8213(5) | 7.6117(8) |
| c/Å | 25.723(6) | 15.101(2) | 10.6120(10) | 10.0355(12) | 4.7674(9) | 14.0335(10) | 11.9194(13) |
| α/ | 77.032(10) | 80.462(7) | |||||
| β/° | 89.414(12) | 101.987(2) | 97.944(10) | 103.406(4) | 75.807(7) | ||
| γ/° | 82.324(12) | 87.896(7) | |||||
| Volume/Å3 | 1816.2(8) | 507.80(15) | 2457.2(4) | 1147.6(3) | 9531(3) | 1730.8(2) | 622.19(12) |
| Density/g cm−3 | 1.420 | 1.362 | 1.283 | 1.292 | 1.300 | 1.456 | 1.432 |
| T/K | 100(2) | 173(2) | 100(2) | 173(2) | 153(2) | 173(2) | 173(2) |
| X-Ray wavelength | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| μ/m−1 | 0.109 | 0.102 | 0.093 | 0.095 | 0.095 | 0.119 | 0.112 |
| Θ min/° | 1.58 | 1.38 | 2.56 | 1.68 | 1.37 | 2.25 | 1.79 |
| Θ max/° | 28.22 | 28.23 | 30.53 | 28.24 | 28.43 | 28.30 | 26.37 |
| Reflections collected | 11419 | 3376 | 14481 | 8215 | 16779 | 21273 | 3381 |
| independent | 2172 | 2078 | 3740 | 2669 | 3203 | 4088 | 2213 |
| observed | 1612 | 1054 | 3007 | 1557 | 1325 | 2360 | 1710 |
| Threshold expression | >2σ(I) | >2σ(I) | >2σ(I) | >2σ(I) | >2σ(I) | >2σ(I) | >2σ(I) |
| R 1 (observed) | 0.0861 | 0.0883 | 0.0555 | 0.0620 | 0.0699 | 0.0683 | 0.0542 |
| wR 2 (all) | 0.2264 | 0.2920 | 0.1600 | 0.1486 | 0.1904 | 0.2112 | 0.1483 |
| GooF | 1.110 | 0.941 | 1.066 | 0.963 | 0.823 | 1.032 | 1.000 |
CCDC reference numbers 624060–624066. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b614984g
Results
The crystal structure of 1 contains two molecules of 3-acetaminopyridine and one molecule of fumaric acid in the asymmetric unit. The two unique hydrogen bonds O–H⋯N are formed by the O–H group, from each end of the dicarboxylic acid and the pyridine N atom of the 3-acetaminopyridine molecules, (O31⋯N21, 2.597(7) Å and O34⋯N11, 2.625(7) Å), Table 2. Each trimeric unit resulting from primary hydrogen bonding is coplanar and the two pyridine moieties are arranged in an up–down manner with respect to each other Fig. 1. | ||
| Fig. 1 Thermal ellipsoids (50%) and labeling scheme of the supramolecular 2 : 1 trimer in 1. | ||
| Compound | D–H⋯A | d(D–H)/Å | d(H⋯A)/Å | d(D⋯A)/Å | <(DHA)/° |
|---|---|---|---|---|---|
| Symmetry codes: #1 1 − x, 1 − y, −0.5 + z; #2 1.5 − x, −2 + y, 0.5 + z; #3 1 + x, y, z; #4 −1 + x, y, z; #5 x, y, −1 + z; #6 x, y, 1 + z; #7 x, 0.5 − y, 0.5 + z; #8 x, −0.5 − y, −0.5 + z; #9 −x, −y, 2 − z; #10 1 + x, 1 + y, −1 + z. | |||||
| 1 | O(31)–H(31)⋯N(21) | 0.84 | 1.76 | 2.597(7) | 173.8 |
| N(23)–H(23)⋯O(17)#1 | 0.88 | 2.02 | 2.847(7) | 157.0 | |
| O(34)–H(34)⋯N(11) | 0.84 | 1.79 | 2.623(7) | 174.7 | |
| N(13)–H(13)⋯O(27)#2 | 0.88 | 2.02 | 2.872(7) | 163.2 | |
| 2 | O(31)–H(31)⋯N(11) | 0.84 | 1.78 | 2.614(5) | 176.0 |
| N(17)–H(17)⋯O(21)#3 | 0.88 | 2.04 | 2.869(4) | 157.2 | |
| 3 | O(21)–H(21)⋯N(11) | 1.136(19) | 1.421(19) | 2.5516(15) | 172.5(17) |
| N(17)–H(17)⋯O(22) | 0.845(19) | 2.012(19) | 2.8494(15) | 170.8(17) | |
| 4 | O(31)–H(31)⋯N(11A) | 0.95(2) | 1.78(2) | 2.731(11) | 172(2) |
| O(31)–H(31)⋯N(11B) | 0.95(2) | 1.66(2) | 2.610(9) | 175(2) | |
| N(21)–H(21)⋯O(22)#4 | 0.90(2) | 1.86(2) | 2.759(2) | 173.1(19) | |
| 5 | O(31)–H(31)⋯N(11) | 0.84 | 1.79 | 2.632(6) | 176.9 |
| N(17)–H(17)⋯O(18)#5 | 0.88 | 1.89 | 2.762(6) | 172.3 | |
| O(33)–H(33)⋯N(21) | 0.84 | 1.76 | 2.582(6) | 164.7 | |
| N(27)–H(27)⋯O(28)#6 | 0.88 | 1.88 | 2.740(6) | 165.7 | |
| 6 | O(23)–H(23)⋯O(22) | 0.87(3) | 1.74(3) | 2.602(3) | 171(3) |
| N(11)–H(11)⋯O(22) | 1.09(3) | 1.55(3) | 2.635(3) | 173(3) | |
| O(1A)–H(1)⋯O(21) | 1.09(4) | 2.48(7) | 2.743(3) | 92(4) | |
| N(17)–H(17)⋯O(1A)#7 | 0.88(3) | 2.01(3) | 2.833(4) | 156(3) | |
| O(1A)–H(1)⋯O(18)#8 | 1.12(4) | 1.80(5) | 2.790(3) | 145(5) | |
| 7 | O(25)–H(25)⋯O(21)#9 | 0.96(3) | 1.58(3) | 2.5309(19) | 170(2) |
| N(11)–H(11)⋯O(21) | 0.88(3) | 1.79(3) | 2.677(2) | 177(2) | |
| N(14)–H(14)⋯O(26)#10 | 0.86(2) | 1.97(2) | 2.822(2) | 177(2) | |
In addition to the hydrogen bonding within the trimer, each N–H donor (amide) forms a hydrogen bond with an adjacent amide CO moiety (mode II, Scheme 1), N13⋯O27(#1), 2.874(7) Å and N23⋯O17(#2), 2.851(7) Å, Fig. 2.
 | ||
| Fig. 2 Alignment of trimers by N–H⋯O (amide) hydrogen bonds in 1. | ||
The asymmetric unit of 2 contains one molecule of 4-acetaminomethylpyridine and half a molecule of fumaric acid. Two symmetry related O–H⋯N hydrogen bonds are formed by the two O–H groups of the dicarboxylic acid and pyridine nitrogen atoms (O31⋯N11, 2.611(5) Å), Table 2. The two pyridine rings in the trimer are coplanar with respect to one another as well as with the plane of fumaric acid but the amide groups at both ends are positioned in an up–down arrangement, Fig. 3.
 | ||
| Fig. 3 Thermal ellipsoids (50%) and labeling scheme of the supramolecular 2 : 1 trimer in 2. | ||
Adjacent trimeric supermolecules produced from the primary hydrogen bonds are connected to one another via symmetry related hydrogen bonds involving H17 from the amide N–H group of one molecule and O21(#2) to an adjacent amide CO group, N17⋯O21(#2), 2.868(4) Å (mode II, Scheme 1). The overall result is an infinite 1-D ladder of trimers, Fig. 4.
 | ||
| Fig. 4 Alignment of trimers by N–H⋯O hydrogen bonds resulting in infinite 1-D ladders in 2. | ||
The crystal structure of 3 has one molecule of 4-acetaminopyridine and half a molecule of sebacic acid in the asymmetric unit. The trimeric supermolecule is constructed from symmetry related O–H⋯N hydrogen bonds between the O–H of the dicarboxylic acid and the pyridine nitrogen atom (O21⋯N11, 2.5516(15) Å), Fig. 5, Table 2.
 | ||
| Fig. 5 Thermal ellipsoids (50%) and labeling scheme of the supramolecular 2 : 1 trimer in 3. | ||
Adjacent trimers are interconnected via an amide–carbonyl hydrogen bond but this time the acceptor moiety is the CO moiety located on the carboxylic acid (mode III, Scheme 1). The result of this interaction is an infinite 2-D layer of orthogonal supermolecules, Fig. 6.
 | ||
| Fig. 6 Orientation of adjacent supramolecular trimers in 3. | ||
The crystal structure of 4 contains one molecule of 4-acetaminomethylpyridine and half a molecule of adipic acid in the asymmetric unit. A trimeric supermolecule is constructed from the two O–H groups of the dicarboxylic acid and pyridine nitrogen atoms, O31⋯N(11A), 2.731(11) Å (Table 2; for a description of the disorder of the pyridine ring, see the Experimental section). Both pyridine rings in the trimeric unit are coplanar with respect to each other but perpendicular to the plane of the COOH groups, and the two amide groups are organized in an up–down manner, Fig. 7.
 | ||
| Fig. 7 Thermal ellipsoids (50%) and labeling scheme of the supramolecular 2 : 1 trimer in 4. | ||
Each 4-acetaminomethylpyridine molecule forms an N–H hydrogen bond with a neighboring CO (amide) moiety, N21⋯O22(#2), 2.759(2) Å (mode II, Scheme 1) resulting in infinite 1-D ribbons of trimers, Fig. 8.
 | ||
| Fig. 8 Orientation of adjacent supramolecular trimers in 4 affected by N–H⋯OC (amide) hydrogen bonds (pyridine moieties are disordered). | ||
In the crystal structure of 5, the asymmetric unit comprises two molecules of 4-acetaminomethylpyridine and one molecule of isophthalic acid. The primary synthons in this structure are the two unique O–H⋯N hydrogen bonds resulting from the interactions between the diacid and two 4-acetaminomethylpyridine molecules (O31⋯N11, 2.620(6) Å; and O33⋯N21, 2.580(6) Å), Fig. 9, Table 2.
 | ||
| Fig. 9 Thermal ellipsoids (50%) and labeling scheme of the supramolecular 2 : 1 trimer in 5. | ||
Adjacent trimeric supermolecules produced from the primary hydrogen bonds are further connected with one another (mode II, Scheme 1) via two unique hydrogen bonds, involving H17 from the amide N–H group and O18 on the amide CO group and H27 from the amide N–H group and O28 on the amide carbonyl group (N17⋯O18(#1), 2.760(6) Å and N27(#1)⋯O28, 2.739(6) Å), resulting in an infinite 1-D ladder, Fig. 10.
 | ||
| Fig. 10 Orientation of adjacent supramolecular trimers in 5 affected by N–H⋯OC (amide) hydrogen bonds. | ||
The crystal structure of 6 contains one 4-acetaminomethylpyridinium cation, one monoanion of 5-nitro-isophthalic acid and one disordered water molecule in the asymmetric unit. A charge-assisted N–H⋯O interaction forms as a consequence of proton transfer between one of the carboxyl moieties in 5-nitro-isophthalic acid and the nitrogen atom in the pyridine ring (N11⋯O22, 2.635(3) Å), Fig. 11, Table 2.
 | ||
| Fig. 11 Thermal ellipsoids (50%) and labeling scheme for 6. | ||
The same oxygen atom of the carboxylate group (O22) is bifurcated as it also engages in an O–H⋯O hydrogen bond with the hydroxyl group of the remaining carboxylic acid moiety of the anion, O23#1⋯O22, 2.602(3) Å, producing an infinite 1-D helical chain, Fig. 12.
 | ||
| Fig. 12 1-D helical chain of 6 obtained through the formation of hydrogen bonds between the carboxylate groups and carboxylic acid groups of neighboring asymmetric units (water molecules left out for clarity). | ||
The second oxygen atom of the carboxylate group forms an O–H⋯O hydrogen bond with a water molecule (O1A⋯O21, 2.744 Å). This disordered water molecule further forms hydrogen bonds with a neighboring amide CO group (O1A⋯O18#2, 2.790(3) Å) and an N–H donor from a second cation (O1A⋯N17#1, 2.834(3) Å), Table 2; the combination of these interactions produce an infinite 1-D chain parallel to that of the 1-D helical backbone.
The crystal structure determination of 7 showed that a salt had formed resulting from proton transfer from one of acid moieties of glutaric acid to the pyridine ring leading to a charge assisted N–H+⋯O− hydrogen bond as the driving force for the formation of the salt, Fig. 13.
 | ||
| Fig. 13 Thermal ellipsoids (50%) and labeling scheme for 7. | ||
As was the case in 6, one oxygen atom is bifurcated and is participating in an O–H⋯O hydrogen bond with the remaining neutral carboxylic acid moiety resulting in a tetrameric pair of ions, Fig. 14.
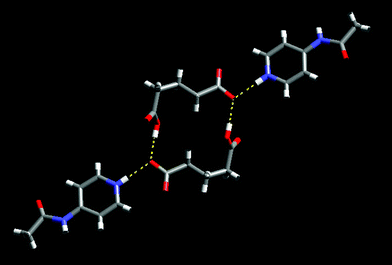 | ||
| Fig. 14 Two ion pairs connected into a tetramer in 7. | ||
The N–H amide moiety forms a hydrogen bond with a CO moiety at the neutral end of the anion (N14–H14⋯O26, 2.823 Å) corresponding to mode III, Scheme 1. These interactions connect adjacent tetramers into an infinite chain.
Discussion
The infrared spectra for compounds 1–8 reported in the present study indicate that reactions between acetaminopyridines and dicarboxylic acids produced molecular co-crystals in six (1–5, and 8) of the eight cases. Broad stretches near 2450 cm−1 and 1900 cm−1, which are characteristic of O–H⋯N(py) hydrogen bonds, appeared in the vibrational spectra of all of these six compounds suggesting the presence of neutral COOH⋯py intermolecular interactions thus eliminating the possibility of salt formation by conversion to the carboxylate anion (COO−). In the vibrational spectra of compounds 6–7, however, the 1900 cm−1 band is shifted considerably towards higher wavenumbers, about 2150 cm−1, suggesting the presence of COO− moieties.The single-crystal structure determinations carried out on 1–7 confirmed the assignments made on the basis of the vibrational spectroscopy. Structures 1–5 contain similar primary trimeric supermolecules constructed through the expected intermolecular O–H⋯N synthon between the dicarboxylic acid and pyridine. No proton transfer from acid to the base was observed in any of these five structures confirming the formation of co-crystals. The distinction between salt and co-crystal can also be made based upon select intermolecular distances and angles.
In the crystal structures of compounds 1–5 each carboxylic acid contains distinctly different C–O bond distances corresponding to the CO (1.187–1.216 Å) and C–O(H) (1.300–1.323 Å) covalent bonds, and the C–N–C endocyclic bond angle of the heterocyclic moieties fall in the range 114.4–119.6° which is indicative of a non-ionized pyridine unit. In contrast, in the structures of 6 and 7 the carboxylate C–O bond distances are more similar (1.27/1.23 Å, and 1.29/1.22 for 6 and 7, respectively). Furthermore, the C–N–C endocyclic bond angles of the heterocyclic moieties in 6 and 7 are 121.8° and 121.1°, respectively, which is typical for a pyridinium cation.17
In addition to the primary hydrogen bonding within each trimer, the N–H moiety in each acetamino group is further involved in the formation of hydrogen bonds with a neighboring molecule; in four out of these five cases, the amide CO moiety acts as the hydrogen-bond acceptor and only in 3 is this motif broken in favor of a CO acid site. There is no obvious reason as to why the acid–based CO is the preferred binding site for the N–H donor is this case since the ladder-type arrangement seen in 2, 4, and 5, would seem attainable even with a longer dicarboxylic acid such as sebacic acid (which is present in 3).
It is difficult to make direct comparisons between the co-crystals 1–5, and the salts, 6–7, but both salts display charge assisted carboxylate⋯pyridinium interactions. The N–H moiety in 6 forms a hydrogen bond with a water molecule (not a predictable option) and in 7 the acid CO moiety is the acceptor.
A search for relevant acetaminopyridine⋯carboxylic acid co-crystals in the CSD produced 46 hits. Eighteen of those hits contained an amide moiety adjacent to the nitrogen atom of the heterocycle, whereas the remaining 28 structures contain pyridine substituted in the 3- or 4 position; we will only discuss structures in the latter category, as they are most relevant to this study.
First of all, every one of the 28 structures contains an O–H(acid)⋯N(py) hydrogen bond; this is undoubtedly the primary driving force for the formation of the co-crystals in this family of compounds. In 18 out of 28 hits, the amide N–H donor is forming a hydrogen bond with a CO amide moiety (mode II, Scheme 1). In 8 out of 28 hits, the N–H donor is, instead, interacting with the CO group from a carboxylic acid (mode III, Scheme 1). In the remaining two structures, the amide N–H donor is (a) structurally inactive, (b) using another py nitrogen atom as an acceptor, respectively.
The self-complementarity of monoacetylated amides is, generally, a reliable motif with the combination of functionalities present in 1–5, despite the potential competition from a CO moiety located on a carboxylic acid. This intermolecular compatibility of the amide N–H and the amide CO may therefore be employed in subsequent supramolecular synthetic strategies based upon a hierarchy of hydrogen-bond interactions even in the presence of potentially competitive hydrogen-bond acceptors. Furthermore, the carbonyl moiety located on the neutral carboxylic acid is not utilized as a hydrogen-bond acceptor in 1–5, and it may therefore provide a binding site for a third molecular component in the design of ternary co-crystals.
Acknowledgements
We are grateful for financial support from NSF (CHE-0316479), the Terry C. Johnson Center for Basic Cancer Research, and for a Fulbright Fellowship (to I.H.) by the U.S. Department of State Bureau of Educational and Cultural Affairs and the Council for International Exchange of Scholars.References
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439 RSC; B. R. Bhogala, S. Basavoju and A. Nangia, Cryst. Growth Des., 2005, 5, 1683 CrossRef CAS; A. V. Trask, J. van de Streek, S. W. D. Motherwell and W. Jones, Cryst. Growth Des., 2005, 6, 2233 CrossRef; B. K. Saha, A. Nangia and M. Jaskolski, CrystEngComm, 2005, 7, 355 RSC; P. Vishweshwar, J. A. McMahon, M. L. Peterson, M. B. Hickey, T. R. Shattock and M. J. Zaworotko, Chem. Commun., 2005, 4601 RSC; M. W. Hosseini, CrystEngComm, 2004, 6, 318 RSC; G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef CAS; T. R. Shattock, P. Vishweshwar, Z. Wang and M. J. Zaworotko, Cryst. Growth Des., 2005, 6, 2046 CrossRef; C. B. Aakeröy, J. Desper, D. J. Salmon and M. M. Smith, Cryst. Growth Des., 2006, 4, 1033 CrossRef; V. R. Pedireddi, J. PrakashaReddy and K. K. Arora, Tetrahedron Lett., 2003, 44, 4857 CrossRef CAS; S. L. Childs, L. J. Chyall, J. T. Dunlap, V. N. Smolenskaya, B. C. Stahly and G. P. Stahly, J. Am. Chem. Soc., 2004, 126, 13335 CrossRef CAS.
- C. B. Aakeröy, J. Desper and J. F. Urbina, Chem. Commun., 2005, 2820 RSC; J. Pansanel, A. Jouaiti, S. Ferlay, M. W. Hosseini, J. Planeix and N. Kyritsakas, New J. Chem., 2006, 30, 683 RSC; J. D. Wuest, Chem. Commun., 2005, 5830 RSC.
- Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889 RSC; P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499 CrossRef; J. F. Remenar, S. L. Morissette, M. L. Peterson, B. Moulton, J. M. MacPhee, H. R. Guzman and Ö. Almarsson, J. Am. Chem. Soc., 2003, 125, 8456 CrossRef CAS.
- S. Shan, E. Batchelor and W. Jones, Tetrahedron Lett., 2002, 43, 8721–8725 CrossRef CAS; L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817–7818 CrossRef CAS; P. Vishweshwar, R. Thaimattam, M. Jaskolski and G. R. Desiraju, Chem. Commun., 2002, 1830–1831 RSC; J. A. Zerkowski, J. C. MacDonald and G. M. Whitesides, Chem. Mater., 1997, 9, 1933–1941 CrossRef CAS; J. J. Kane, R.-F. Liao, J. W. Lauher and F. W. Fowler, J. Am. Chem. Soc., 1995, 117, 12003–12004 CrossRef CAS; J.-M. Lehn, M. Mascal, A. DeCian and J. Fischer, J. Chem. Soc., Chem. Commun., 1990, 479–480 RSC; P. Vishweshwar, A. Nangia and V. M. Lynch, CrystEngComm, 2003, 5 Search PubMed; Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889–1896 RSC.
- G. R. Desiraju and J. A. R. P. Sarma, J. Chem. Soc., Chem. Commun., 1983, 45–6 RSC; C. Huang, L. Leiserowitz and G. M. Schmidt, J. Chem. Soc., Perkin Trans. 2, 1973, 503 RSC; F. Pan, W. S. Wong, V. Gramlich, C. Bosshard and P. Gunter, Chem. Commun., 1996, 2 Search PubMed; V. R. Pedireddi, W. Jones, A. P. Chorlton and R. Docherty, Chem. Commun., 1996, 997 RSC; P. Vishweshwar, A. Nangia and V. M. Lynch, J. Org. Chem., 2002, 67, 556 CrossRef CAS; C. B. Aakeröy, A. M. Beatty, M. Nieuwenhuyzen and M. Zou, Tetrahedron, 2000, 56, 6693–6699 CrossRef; S. H. Dale, M. R. J. Elsegood, M. Hemmings and A. L. Wilkinson, CrystEngComm, 2004, 6, 207–214 RSC; V. R. Pedireddi, J. PrakashaReddy and K. K. Arora, Tetrahedron Lett., 2003, 44, 4857–4860 CrossRef CAS.
- C. B. Aakeröy, J. Desper and B. A. Helfrich, CrystEngComm, 2004, 6, 19–24 RSC; C. B. Aakeröy, J. Desper and B. M. T. Scott, Chem. Commun., 2006, 1445 RSC; C. B. Aakeröy, N. Schultheiss, J. Desper and C. Moore, New J. Chem., 2006, 30, 1452 RSC; C. B. Aakeröy, M. E. Fasulo and J. Desper, CrystEngComm, 2006, 8, 586 RSC.
- Approaches such as this are also predicated upon the idea that a small number of specific intermolecular interactions can provide a significant part of the stabilization energy of molecular crystals: P. Dauber and A. T. Hagler, Acc. Chem. Res., 1980, 13, 105–112 Search PubMed.
- C. B. Aakeröy, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 569 CrossRef; B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef CAS; M. W. Hosseini, CrystEngComm, 2004, 6, 318 RSC; L. R. MacGillivray, CrystEngComm, 2004, 6, 77 RSC; D. Braga, Chem. Commun., 2003, 2751 RSC; L. Brammer, Chem. Soc. Rev., 2004, 33, 476 RSC; C. B. Aakeröy and A. M. Beatty, Aust. J. Chem., 2001, 54, 409 CrossRef CAS; C. B. Aakeröy, J. Desper and B. M. T. Scott, Chem. Commun., 2006, 1445 RSC C. B..
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef CAS.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, Angew. Chem., Int. Ed., 2001, 40, 3240 CrossRef CAS; C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, J. Am. Chem. Soc., 2002, 124, 14425 CrossRef; C. B. Aakeröy, J. Desper and B. A. Helfrich, CrystEngComm, 2004, 6, 19 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- C. Li, L. S. Rittmann, A. S. Tsiftsoglou, K. K. Bhargava and A. C. Sartorelli, J. Med. Chem., 1978, 21, 874–877 CrossRef CAS.
- T. Singh, R. G. Stein and J. H. Biel, J. Med. Chem., 1969, 12, 949–950 CrossRef CAS.
- SMART v5.060, Bruker Analytical X-ray Systems, Madison, WI, 1997–1999 Search PubMed.
- SAINT v6.02, Bruker Analytical X-ray Systems, Madison, WI, 1997–1999 Search PubMed.
- SHELXTL v5.10, Bruker Analytical X-ray Systems, Madison, WI, 1997 Search PubMed.
- J. A. Cowan, J. A. K. Howard, G. J. McIntyre, S. M.-F. Lo and I. D. Williams, Acta Crystallogr., Sect. B: Struct. Sci., 59, 794 Search PubMed.
| This journal is © The Royal Society of Chemistry 2007 |