Effect of polymer addition on the contact line crystallisation of paracetamol†
Jacqueline S.
Capes
*a and
Ruth E.
Cameron
b
aDepartment of Materials Science and Metallurgy, University of Cambridge, Pembroke Street, Cambridge, CB2 3QZ, UK. E-mail: jsc35@cam.ac.uk; Fax: +44 1223 334366; Tel: +44 1223 767059
bDepartment of Materials Science and Metallurgy, University of Cambridge, Pembroke Street, Cambridge, CB2 3QZ, UK. E-mail: rec11@cam.ac.uk; Fax: +44 1223 334567; Tel: +44 1223 334324
First published on 17th November 2006
Abstract
In an evaporating aqueous paracetamol solution, crystallisation of the metastable orthorhombic polymorph can occur at the solution–substrate contact line due to a favourable meniscus geometry and enhanced evaporation rates at the edges. Upon the addition of polymers or other common excipients to these systems, it was found that crystallisation of this form was suppressed. This was attributed to the polymer blocking the nucleation sites at the edge of the meniscus, altering the evaporation rate or disrupting the flow of solution to the edges. These results are in contrast to previous conclusions by other groups suggesting that the addition of certain polymers may aid in the selective crystallisation of the orthorhombic polymorph from solution via the formation of specific interactions.
Introduction
The use of metastable polymorphs of pharmaceuticals in formulations offers a potential route to improved properties such as faster dissolution, higher solubility and better compressibility. However, the use of the most thermodynamically stable crystal form is still preferred commercially to avoid changes in properties occurring during the product life cycle. The aim of much research has been to stabilise drugs in a thermodynamically metastable crystal structure to enable exploitation of the improved pharmaceutical properties. Several methods have been reported to selectively crystallise a particular polymorph,1–3 however, control of these processes is still a major issue and solution crystallisation remains the preferred production method, because of the ease of control and scale-up. One problem is that although kinetic factors such as a faster nucleation rate can cause a metastable polymorph to form first,4,5 it is then energetically favourable for it to undergo an uncontrolled solvent-mediated phase transformation to a more stable form,6 altering the resultant properties. This was observed experimentally by Ostwald7 and forms the basis of his Rule of Stages.One possible route to obtain and stabilise a less stable polymorph from solution is to use specific additives to either encourage the growth of the desired form or disrupt the growth of the other form.5,8 In many situations, heterogeneous nucleation is more energetically favourable than homogeneous nucleation and therefore tailor-made additives or substrates can be designed to manipulate the crystallisation process.
In nature, the crystal size, morphology, polymorph and orientation of inorganic biominerals are controlled by the presence of proteins or other macromolecules.9 In pharmaceutical crystallisation, it has been shown that polymers are able to influence the crystallisation rates10 and morphology of drug crystals,11 which ultimately alters their compaction properties12 and dissolution rates. However, few examples exist8,13,14 in which a different polymorphic form has been stabilised in this way because of the weak molecular interactions in organic crystals.
If the crystal structures of different polymorphic forms of a substance are well-characterised, solid substrates can be designed that will template a particular form. This approach has been shown to be applicable to a wide range of materials including minerals9 and semiconducting oxide films,15,16 as well as organic crystals.17,18 Interactions between the nucleating crystal and the substrate occur in a number of ways19,20 and it has been suggested20 that libraries of substrates could be utilised in the search for new polymorphic forms as well as to selectively template known polymorphs. It is also known that it is not only the chemistry of a substrate that is important in directing the crystallisation process. The topography of a surface can also direct the nucleation of a particular polymorph when designed to match the geometry of a particular crystal structure.21–23
The focus of the research in this paper is the crystallisation of the metastable, orthorhombic polymorph of paracetamol (acetaminophen).24 A number of studies have looked at a variety of additives and polymers that affect the nucleation and resultant morphology of crystals of the monoclinic, stable polymorph of paracetamol.25–30 In addition, the crystallisation of paracetamol from the melt in the presence of a range of polymers has been reported, to attempt to stabilise the metastable polymorphs.31 Currently, only two papers32,33 describe the use of solid polymer substrates to crystallise the orthorhombic polymorph of paracetamol from aqueous solution. Although the authors of both papers concluded that the ability to selectively crystallise one of the polymorphs was dependent on specific interactions with the polymer substrates, the exact nature of these interactions was not specified.
In this work paracetamol has been crystallised from an evaporating aqueous solution in the presence of a similar range of polymers to establish the exact nature of the interactions that were causing the formation of a particular polymorph.
A mechanism by which the metastable, orthorhombic polymorph of paracetamol can be crystallised from solution without the use of additives has been proposed.34,35 In these studies, orthorhombic crystals were found to nucleate at the edge of a meniscus or drop during solvent evaporation. This was due to a favourable meniscus geometry leading to higher evaporation rates in the contact line region. The crystals were then unable to transform to the stable monoclinic polymorph because they were effectively removed from the solvent when they formed, as the solution level dropped through evaporation. This concept of selective contact line crystallisation is shown schematically in Fig. 1.
 | ||
| Fig. 1 Diagram showing how metastable polymorphs can be formed at the edge of a meniscus. | ||
This same method has now been used to study the effect of the addition of polymers at different stages of the crystallisation process, with particular reference to the amount of polymer added and its physical nature.
Experimental
Paracetamol (acetaminophen), 98% pure, was obtained from Sigma (UK). Galvanised steel sample dishes were the cap section of a capped washer and were purchased from Baker and Finnemore (UK). All sample dishes were cleaned with ethanol before use. The polymers used were from a Polymer Sample Kit purchased from Scientific Polymer Products Inc. (USA) and were the same as those used in previously reported studies.31 The polymers were used as received, and therefore were in several different physical forms including extruded pellets, powder and fibrous material. All the excipients were purchased from Sigma and were used as received. To aid in the analysis of the results, the polymers and excipients were divided into two categories, powders and non-powders.Effect of solution concentration and amount of polymer
An amount of polymer or excipient was placed into a galvanised steel sample dish and 0.3 ml of an aqueous paracetamol solution was added. The polymer and solution were heated at 80 °C, on a pre-heated block heater, for 4 min 40 s. Halfway through heating, 0.1 ml of distilled water was added to replace that lost through evaporation. The sample dishes were then cooled quickly by placing them on a cold metal block, and were left to evaporate at ambient temperature and humidity. Four initial paracetamol solution concentrations were tested, 16, 25, 33 and 42 mg ml−1, each with three different amounts of polymer or excipient. For the powder polymer samples three different masses (5, 12.5, 20 mg) were tested, to investigate if the quantity of polymer affected the crystallisation process. For many of the non-powder polymers, the mass of individual pieces was much more than 5 mg and so samples were created in which one, two or three polymer pellets or pieces were added.Effect of heating the polymer
The method described above was repeated, with an initial paracetamol solution concentration of 16 mg ml−1 and an arbitrary amount of polymer or excipient, within the limits of 5–20 mg or 1–3 pellets. In one experiment, the polymers were added before the solution heating step, and in a second experiment the polymers were added after heating, once the dishes had been removed to the cold metal block. Five repeats were carried out for each polymer in each heating regime.During the heating experiments, photographs of the samples were taken at the following time points:
• Polymer as received, before solution added
• At the start of the crystallisation process
• When crystallisation was complete
Raman spectroscopy
The paracetamol crystals that formed were analysed in situ using Raman spectroscopy. A Raman microscope was used with a ×50 long working distance lens. This produced a laser spot significantly less than 100 µm in diameter. An argon laser (wavelength 514.5 nm) was used. Spectra were collected over the extended range 50–3500 cm−1 in a period of 10 s using Renishaw WIRE software. Paracetamol polymorphs were identified by peaks in the ranges 375–515 cm−1 and 1210–1270 cm−1.36Results
In all experiments, any orthorhombic crystals that formed were found to lie on the sides of the dishes. This was consistent with the results from previous crystallisation experiments without polymer. Fig. 2 shows both orthorhombic and monoclinic paracetamol crystals formed without the addition of polymers or excipients. Although the use of galvanised steel dishes is unusual, they provided a convenient size and shape for these experiments. Other experiments34 conducted using other vessels confirmed that these results were not unique to these dishes. | ||
| Fig. 2 (a) Orthorhombic crystals on dish sides. (b) Monoclinic crystals on the dish base (scale: dish rim diameter is 20 mm). | ||
Results from the experiments in which both the amount of polymer and the solution concentration were varied are summarised in Table 1. The samples are divided both into powders and non-powders and also into hydrophobic and hydrophilic polymers/excipients. Where a polymer is included in the orthorhombic column, this polymorph was found to crystallise under at least one set of conditions. From these results there did not appear to be any significant dependence of the outcome of the crystallisation on the chemistry of the polymer, for example, monoclinic paracetamol crystallised in the presence of poly(diallyl isophthalate), while the orthorhombic polymorph formed with poly(diallyl phthalate). Comparing the hydrophilic and hydrophobic polymers and excipients, only 22% of the hydrophilic samples permitted the growth of the orthorhombic form, while this polymorph formed in 45% of the samples containing hydrophobic polymers. Therefore, although there may be some effect of the hydrophobicity of the polymer, in all cases the occurrence of orthorhombic paracetamol was less than 50%.
| Monoclinic | Orthorhombic | ||
|---|---|---|---|
| Powder | Hydrophilic | Alginic acid | Calcium carbonate |
| Carboxymethyl cellulose | Poly(diallyl phthalate) | ||
| Carrageenan | Polyglycolide-co-lactide | ||
| Corn starch | Sodium chloride | ||
| Ethyl cellulose | |||
| Hydroxypropyl cellulose | |||
| Hydroxypropyl methyl cellulose | |||
| Methyl cellulose | |||
| Polyacrylamide | |||
| Poly(acrylic acid) | |||
| Poly(diallyl isophthalate) | |||
| Poly(ethylene oxide) | |||
| Polyglycolide | |||
| Polyvinyl alcohol | |||
| Polyvinyl pyrrolidone | |||
| Potato starch | |||
| Pregelatinised starch | |||
| Sodium bicarbonate | |||
| Zein | |||
| Hydrophobic | Cellulose acetate | Microcrystalline cellulose | |
| Cellulose acetate butyrate | Poly(ethyl methacrylate) | ||
| Paraffin wax | Polypropylene | ||
| Polyethylene, 25% chlorinated | |||
| Polyethylene, 42% chlorinated | |||
| Polymethyl methacrylate | |||
| Polytetra fluoro ethylene | |||
| Polyvinyl chloride, 1.8% carboxylated | |||
| Non-powders | Hydrophilic | Chitosan | Nylon 6 |
| Sugar spheres | Poly(ethylene terephthalate) | ||
| Hydrophobic | Polyacetal | Cellulose propionate | |
| Polyethylene, chlorosulfonated | Cellulose triacetate | ||
| Polypropylene, chlorinated | Poly(1-butene) | ||
| Poly(vinyl acetate) | Polycaprolactone | ||
| Polycarbonate | |||
| Polyethylene, high density | |||
| Polystyrene | |||
There did, however, appear to be a significant effect when the physical form of the polymer was considered. Orthorhombic paracetamol crystallised in only 21% of the powder samples, independent of the degree of hydrophobicity, while this polymorph formed in 60% of the samples in which larger pellets or chunks of polymer were present.
The effect of changing the paracetamol solution concentration independent of the amount of polymer present is shown in Table 2. The orthorhombic polymorph crystallised in a greater number of the samples with a lower initial concentration, and only monoclinic paracetamol formed in solutions of the highest concentration. It must be noted that at all concentrations only a small number of samples contained the metastable polymorph.
| Initial concentration/mg ml−1 | 17 | 25 | 33 | 42 |
| Number of samples | 132 | 141 | 139 | 140 |
| Occurrence of the orthorhombic polymorph (%) | 13 | 6 | 2 | 0 |
To investigate the effect of the amount of polymer or excipient present, independent of the paracetamol solution concentration, the samples were first split into powders and non-powders. The results were then calculated for 5, 12.5 and 20 mg of powder and for 1, 2 or 3 pellets (shown in Table 3). From these results there did not appear to be a strong relationship between the amount of polymer present and the formation of the orthorhombic polymorph, and once again the metastable, orthorhombic polymorph formed in only a small number of the samples.
| Amount of powder/mg | Number of pellets | |||||
|---|---|---|---|---|---|---|
| 5 | 12.5 | 20 | 1 | 2 | 3 | |
| Number of samples | 131 | 130 | 127 | 56 | 54 | 54 |
| Occurrence of the orthorhombic polymorph (%) | 9 | 6 | 11 | 7 | 3 | 2 |
To further investigate the effect of the chemistry and the physical form of the added polymers, two experiments were conducted to establish if heating the polymers changed the outcome of the crystallisation. In the first experiment the polymers were heated in the paracetamol solution, while in the second experiment the polymers were added to a hot paracetamol solution. It is likely that a small amount of heating of the polymer would have occurred when they were first added, but this was not thought to be significant. Results from these experiments are shown in Fig. 3 along with the results from a control experiment in which paracetamol was crystallised from a solution of concentration 16 mg ml−1 without any additional polymers or excipients. There appeared to be little dependence of the chemical functionality of the polymer on the crystallisation outcome, and it must be noted that comparing the results in Fig. 2 and 3, the results are not consistent for some of the polymers. For example, only monoclinic paracetamol formed in the cellulose acetate butyrate, polytetrafluoroethylene and poly(diallyl isophthalate) samples in the first experiment, but a significant proportion of these samples contained the orthorhombic polymorph in the second two experiments.
 | ||
| Fig. 3 The effect of heating the polymer in the solution (black) or adding the polymer to the heated solution (light grey) on the occurrence of the orthorhombic polymorph during crystallisation. | ||
Variations in the paracetamol polymorph that crystallised were also seen for some polymers and excipients depending on whether the polymer had been heated in the solution, for example corn starch, poly(diallyl phthalate) and polyacetal. To understand the differences in the samples that were causing a change in the crystallisation conditions, photographs were taken at various times during these experiments. These showed how the physical form of the polymers and excipients changed upon heating and how the polymers interacted with the evaporating solution.
Discussion
Results of previous experiments (reproduced in Fig. 4) showed that crystallisation of the orthorhombic polymorph occurred most reliably with post-heating paracetamol solution concentrations in the range 16–28 mg ml−1. This is because, at this concentration, the solution lies just beyond the solubility line at room temperature. In this region the solution is within the metastable zone and there is a greater likelihood of crystallising the orthorhombic form. In this work, it was found that with the addition of polymers, the growth of the orthorhombic polymorph was still dependent on the solution concentration. The smallest amount of paracetamol (16 mg ml−1) corresponded to a post-heating concentration of 25 mg ml−1, increasing the chances of crystallising the orthorhombic polymorph. At the higher concentrations, the solutions were well within the supersaturated region and so the monoclinic polymorph formed very quickly and more homogeneously.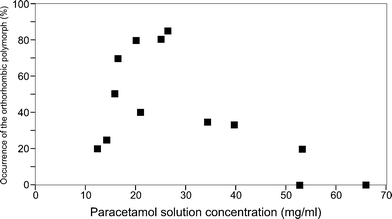 | ||
| Fig. 4 The dependence of the formation of the orthorhombic polymorph on the post-heating solution concentration, without the addition of polymers. | ||
At all of the solution concentrations, however, even at those that more strongly favoured the growth of the metastable polymorph, the orthorhombic form crystallised in very few of the samples. It was likely that the addition of the polymers to the dishes was acting to suppress crystallisation of this form. Investigating the effect of adding different amounts of polymer independent of the solution concentration showed that for polymers in the form of powders, the quantity of polymer had little effect on the occurrence of the orthorhombic polymorph. When considering the non-powders, there appeared to be a slight increase in the occurrence of the orthorhombic polymorph when only one pellet or piece of polymer was added, although the percentage occurrence was still less than that found with larger amounts of polymer powders.
From these results it may be concluded that the presence of the polymers or excipients was suppressing crystallisation of the orthorhombic polymorph. Studying the form of each of the polymers and excipients, and the changes that occurred on heating, it was hypothesised that this was due to the polymers either blocking nucleation at the contact line or disrupting the flow of solution to these regions. The physical form of the polymers, their interaction with water and whether the polymer melted or absorbed more water on heating were all identified as important factors in determining the outcome of the crystallisation. By studying the photographs taken during the experiments four different effects of the addition of polymer on the crystallisation process were established.
1. Formation of a gel
Polymer powders that were highly hydrophilic formed a gel and decreased the water mobility in the samples. This prevented the formation of orthorhombic crystals at the contact line by altering the evaporation rate and disrupting the flow of solution to the edge of the meniscus. Examples of materials exhibiting this effect were corn starch, potato starch and hydroxypropyl cellulose (Fig. 5). In the case of corn starch, the crystallisation of the orthorhombic polymorph depended on the experimental method that was used. When the starch was heated in the solution a gel formed and only monoclinic crystals grew. When the starch was not heated a gel did not form and over half of the samples contained orthorhombic crystals. In contrast, potato starch and hydroxypropyl cellulose formed a gel whether or not they were heated and the orthorhombic form never crystallised. | ||
| Fig. 5 Photographs showing appearance of samples containing gel-forming polymers at the beginning and end of crystallisation. Corn starch: gel formed upon heating and only monoclinic crystals formed, starch powder remained on the base of the dish when not heated and this allowed growth of orthorhombic crystals (indicated by arrow). Hydroxypropyl cellulose: gel formed whether heated or not and the orthorhombic polymorph did not crystallise. | ||
2. Surface disruption
Slightly hydrophilic polymers and excipients, in the form of powders, had a tendency to spread over the surface of the solution (Fig. 6) and disrupt any flow of solvent to the contact line that may have occurred through surface tension effects. Surface disruption was also an issue for physical forms of polymers with large surface areas, such as fibrous material like chitosan. In addition, the presence of polymer or excipient at the edge of the meniscus may have disrupted the nucleation of the paracetamol crystals at this point. Examples of polymers of this nature were cellulose acetate and ethyl cellulose. | ||
| Fig. 6 Photographs showing the appearance of samples containing powders that spread over the surface of the solutions, disrupting the evaporation. | ||
An interesting result was seen for paraffin wax, due to its melting point being below 80 °C. When heated in the solution, the wax melted and formed a layer over the surface. This disrupted the solvent evaporation and the orthorhombic form crystallised in only 1 out of 5 of the samples. When the wax was not heated and it did not melt, again the metastable polymorph only crystallised in 1 out of 5 samples. In this case, even though the wax particles were hydrophobic, they remained on the surface of the solution and disrupted the evaporation in a similar way to the melted layer.
3. Evidence of adsorption
In some samples, in which the polymers were in the form of pellets, paracetamol crystallised on the sides or ends of the pellets (Fig. 7), rather than on the sides or base of the dish. These crystals were always found to be monoclinic. For these polymers there may have been some degree of affinity between the monoclinic crystals and the material or the crystals may have nucleated on the polymer pellet due to another contact line forming in this region. It is not known from these experiments whether templating was occurring, but because crystals did not form on the pellets in all of the samples, it was unlikely, even if this were the case, that templating was a strong effect. Examples of this behaviour were seen for polyacetal, nylon 6 and polystyrene | ||
| Fig. 7 Photographs showing the crystallisation of monoclinic paracetamol on polymer pellets. Arrows indicate positions of crystals. | ||
4. Deposition around contact line
Sodium chloride and sodium bicarbonate both dissolved in the paracetamol solution. As the water evaporated they crystallised at the same time as, or slightly earlier than, the paracetamol. As expected,37 they also crystallised at the contact line (Fig. 8) and, therefore, were likely to have disrupted nucleation of the orthorhombic polymorph of paracetamol in this region. | ||
| Fig. 8 Photographs showing the crystallisation of salts around the contact line, disrupting the nucleation of paracetamol crystals in this region. | ||
Conclusions
The metastable, orthorhombic polymorph of paracetamol can be obtained from an evaporating aqueous solution via the process of contact line crystallisation. The addition of polymers or excipients to the solution generally suppressed the growth of this polymorph and monoclinic crystals predominantly formed. The extent to which the polymer or excipient disrupted the crystallisation process was dependent partly on its chemistry, specifically its hydrophilicity, but mostly on its physical form, e.g. powder, solid pellet or fibrous material. Four distinct results were identified,• The polymer formed a gel, disrupting evaporation and solution flows
• Polymer remained on the surface of the solution affecting evaporation and nucleation at the contact line
• Adsorption on polymer pellets occurred either due to affinity or the presence of a second contact line
• Excipient deposited at the contact line prevented or disrupted paracetamol nucleation
Acknowledgements
The authors thank Howard Ando (Pfizer) for useful discussions. This work was funded by Pfizer.References
- G. W. Stowell, R. J. Behme, S. M. Denton, I. Pfeiffer, F. D. Sancilio, L. B. Whittall and R. R. Whittle, J. Pharm. Sci., 2002, 91, 2481–2488 CrossRef CAS.
- U. J. Griesser, A. Burger and K. Mereiter, J. Pharm. Sci., 1997, 86, 352–358 CrossRef CAS.
- W. Beckmann and W. H. Otto, Chem. Eng. Res. Des., 1996, 74, 750–757 CAS.
- J. W. Schroer and K. M. Ng, Ind. Eng. Chem. Res., 2003, 42, 2230–2244 CrossRef CAS.
- R. J. Davey, N. Blagden, G. D. Potts and R. Docherty, J. Am. Chem. Soc., 1997, 119, 1767–1772 CrossRef CAS.
- F. Wang, J. A. Wachter, F. J. Antosz and K. A. Berglund, Org. Process Res. Dev., 2000, 4, 391–395 Search PubMed.
- W. Ostwald, Z. Phys. Chem., 1897, 22, 289 Search PubMed.
- C.-H. Gu, K. Chatterjee, V. Young, Jr. and D. J. W. Grant, J. Cryst. Growth, 2002, 235, 471–481 CrossRef CAS.
- K. Naka and Y. Chujo, Chem. Mater., 2001, 13, 3245–3259 CrossRef CAS.
- Y. Aso, S. Yoshioka and S. Kojima, Chem. Pharm. Bull., 1996, 44, 1065–1067 CAS.
- A. B. Rizzuto, A. C. Chen and M. F. Veiga, Pharm. Technol., 1984, 8, 32–39 Search PubMed.
- M. R. Whelan, J. L. Ford and M. W. Powell, J. Pharm. Biomed. Anal., 2002, 30, 1355–1359 CrossRef CAS.
- A. P. Simonelli, S. C. Mehta and W. I. Higuchi, J. Pharm. Sci., 1969, 58, 538–549 CrossRef CAS.
- A. P. Simonelli, S. C. Mehta and W. I. Higuchi, J. Pharm. Sci., 1976, 65, 355–361 CrossRef CAS.
- B.-S. Jeong, J. D. Budai and D. P. Norton, Thin Solid Films, 2002, 422, 166–169 CrossRef CAS.
- O. Y. Gorbenko, S. V. Samoilenkov, I. E. Graboy and A. R. Kaul, Chem. Mater., 2002, 14, 4026–4043 CrossRef CAS.
- A. Hoshino, S. Isoda and T. Kobayashi, J. Cryst. Growth, 1991, 115, 826–830 CrossRef CAS.
- M. C. Frincu, R. E. Sharpe and J. A. Swift, Cryst. Growth Des. Search PubMed.
- D. E. Hooks, T. Fritz and M. D. Ward, Adv. Mater., 2001, 13, 227–241 CrossRef CAS.
- C. A. Mitchell, L. Yu and M. D. Ward, J. Am. Chem. Soc., 2001, 123, 10830–10839 CrossRef CAS.
- J. Zhang, D. Yang, A. Thierry, J. C. Wittmann and B. Lotz, Macromolecules, 2001, 34, 6261–6267 CrossRef CAS.
- P. W. Carter, L. M. Frostman, A. C. Hillier and M. D. Ward, in Interfacial Design and Chemical Sensing, ed. Thomas E. Mallouk and D. Jed Harrison, American Chemical Society, 1994, ch. 17 Search PubMed.
- S. J. Bonafede and M. D. Ward, J. Am. Chem. Soc., 1995, 117, 7853–7861 CrossRef CAS.
- G. Nichols and C. S. Frampton, J. Pharm. Sci., 1998, 87, 684–693 CrossRef CAS.
- B. A. Hendriksen, D. J. W. Grant, P. Meenan and D. A. Green, J. Cryst. Growth, 1998, 183, 629–640 CrossRef CAS.
- B. Y. Shekunov, D. J. W. Grant, R. J. Latham and J. N. Sherwood, J. Phys. Chem. B, 1997, 101, 9107–9112 CrossRef CAS.
- K. V. R. Prasad, R. I. Ristic, D. B. Sheen and J. N. Sherwood, Int. J. Pharm., 2001, 215, 29–44 CrossRef CAS.
- K. Kachrimanis and S. Malamatris, J. Pharm. Pharmacol., 1999, 51, 1219–1227 CrossRef CAS.
- H. A. Garekani, J. L. Ford, M. H. Rubinstein and A. R. Rajabi-Siahboomi, Int. J. Pharm., 2000, 208, 87–99 CrossRef CAS.
- F. Giordano, A. Rossi, R. Bettini, A. Savioli, A. Gazzaniga and C. Novak, J. Therm. Anal. Calorim., 2002, 68, 575–590 CrossRef CAS.
- A. Rossi, A. Savioli, M. Bini, D. Capsoni, V. Massarotti, R. Bettini, A. Gazzaniga, M. E. Sangalli and F. Giordano, Thermochim. Acta, 2003, 406, 55–67 CrossRef CAS.
- M. Lang, A. L. Grzesiak and A. J. Matzger, J. Am. Chem. Soc., 2002, 124, 14834–14835 CrossRef CAS.
- C. P. Price, A. L. Grzesiak and A. J. Matzger, J. Am. Chem. Soc., 2005, 127, 5512–5517 CrossRef CAS.
- J. S. Capes, PhD Thesis, University of Cambridge, Cambridge, UK, 2006.
- J. S. Capes and R. E. Cameron, J. Cryst. Growth Des. Search PubMed Submitted.
- N. Al-Zoubi, J. E. Koundourellis and S. Malamataris, J. Pharm. Biomed. Anal., 2002, 29, 459–467 CrossRef CAS.
- J. Satterly, Am. J. Phys., 1956, 24, 529–530 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Enlarged images of Fig. 5–8. See DOI: 10.1039/b613663j |
| This journal is © The Royal Society of Chemistry 2007 |