Organic crystal hydrates: what are the important factors for formation†
Lourdes
Infantes
*a,
László
Fábián
b and
W. D. Sam
Motherwell
c
aInstituto de Química-Física ‘Rocasolano’, CSIC, Serrano 119, E-28006, Madrid, Spain
bPfizer Institute for Pharmaceutical Materials Science, Cambridge Crystallographic Data Centre, 12 Union Rd, Cambridge, UK CB2 1EZ
cCambridge Crystallographic Data Centre, 12 Union Rd, Cambridge, UK CB2 1EZ. E-mail: xlourdes@iqfr.csic.es
First published on 14th November 2006
Abstract
A database study of 34 770 accurate organic crystal structures reveals that the most important factor determining a higher frequency of hydrates is the sum of the average donor and acceptor counts for the functional groups. Different parameter forms for donor/acceptor properties were investigated for correlation with formation of hydrates, and are discussed. We did not find that the donor/acceptor ratio or the molecular weight significantly affects the hydrate formation. We also examined a wide range of calculated molecular properties and found that increasing polar surface is correlated with increasing hydrate formation. The water environment pattern of donor and acceptor hydrogen bonds in the crystal is influenced by the donor/acceptor ratio of the molecule.
1. Introduction
Why are crystalline hydrates formed for organic molecules? Although there have been several studies1,2 of the occurrence of hydrates in the Cambridge Structural Database (CSD),3 there is no firm rule for prediction of hydrate formation. In our previous work on the structures of hydrogen bonded water patterns4 we did consider the question of a predictive rule, but could not reach a firm conclusion. However, an important observation was made that presence of some chemical functional groups causes a significantly higher frequency of hydrated structures. For example, if we count structures where a certain group, say COOH, is present and could therefore possibly form a hydrate, Nposs, and then count how many of these structures contain water, Nhydr, we define the ratio Nhydr/Nposs as the ‘water affinity’ of each group, for COOH the value is 15%. Nhydr, then, is the number of structures containing water and the group, and Nposs is the number of structures containing the group within the given CSD sample. Not surprisingly, the more polar the groups the higher the water affinity, with charged groups showing the higher values e.g. Ca2+ 62% hydrates, COO− 35%, compared to the total CSD frequency of hydrates of 6.6%. It was further observed that if a group with high water affinity occurs several times in the molecular formula of the compound then the percentage of hydrates increases steadily with the number of groups, e.g. for 3 COO− in the formula we have 72% hydrates, and this increase is found without any restriction on the presence of other functional groups.This paper investigates the percentage hydrate formation in relation to the total number of donor and acceptor groups present in the compound. We test various parametric descriptors with a view to giving some probability of formation of hydrates for a given molecule.
2. Methodology
2.1 CSDHBond database
Version 5.26 of the CSD (November 2004) contains 101 244 organic entries with no metals, no error, no disorder, not polymeric, R factor less than 10% and 3-D coordinates determined. Of these entries only 6558 (6.5%) are hydrates. Since hydrate formation in the absence of a hydrogen bond donor is very rare we decided to study only the subsets which contain at least one hydrogen bond donor, OH, NH, or SH, giving 41 606 entries. In order to study the formation of hydrates we required some confidence in the hydrogen positions, which are often missing for water molecules, and so we reduced the sample to the more accurate structures, by requiring that all hydrogen atoms given in the chemical formula are present in the 3-D coordinates,5 namely 34 770 structures, of which 3258 (9.4%) are hydrates. We processed these entries using RPluto6 to generate all inter- and intramolecular contacts with a distance less than the sum of the van der Waals radii +0.1 Å. Inter- and intramolecular contacts between atoms X⋯Y were categorised as donor (D) XH⋯Y, acceptor (A) X⋯HY, non-hydrogen-bonds (X), and uncertain (U). We apply a test on the angle X–H⋯Y which must be greater or equal to 115° to be classified as D or A. Category X is assigned when neither atom X or Y has a hydrogen atom attached. Category U is assigned if the angle X–H⋯Y is less than 115°. The data were output from the RPluto program into tables, with atoms assigned an atom code for chemical environment (e.g. O in OH, O in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and belonging to a list of 113 common chemical groups (e.g. carboxylic COOH, hydroxyl C–OH, oxime CN–OH, etc.). To facilitate effective searching and analysis of the contact data, a relational database using Microsoft Access was built from the output tables from RPluto as in our previous work.7,8
O), and belonging to a list of 113 common chemical groups (e.g. carboxylic COOH, hydroxyl C–OH, oxime CN–OH, etc.). To facilitate effective searching and analysis of the contact data, a relational database using Microsoft Access was built from the output tables from RPluto as in our previous work.7,8
The new database differs from the previously reported CSDContact in the following aspects: (a) only those compounds with all the atoms present in 3-D coordinates (including H); (b) only donor (D) and acceptor (A) contacts have been included; (c) we have improved the classification of atom types by using a combination of the atom code and group code to which it belongs, called the atom-group code. For example, the oxygen belonging to OH in a COOH group has a different code to OH in a CN–OH group. We can easily select subsets of structures on any combination and stoichiometry of such atom-group codes. A table of the atom-group codes observed more than 100 times in our sample is given in the ESI.†
Thus we have a computational tool for studying hydrates and anhydrous compounds. The main objective of this work is to study the frequency of hydrates for different subsets of the sample subdivided by ranges of various descriptive parameters, which we now proceed to define.
2.2 Donor acceptor property calculation
It is important to carefully define how to calculate the donor–acceptor property of atoms in functional groups. The counting of donor or acceptor bonds from each atom is easily done, as these have been categorised in the Rpluto output, by letters D or A. Thus any particular instance of an atom in a group, say the oxygen of an aliphatic C–OH group, has a recorded number of donor bonds, ND, and acceptor bonds, NA; examination of all C–OH groups show that ND = 1, NA = 1 is quite common as expected, but there are a significant number of occurrences of ND = 1, NA = 0, and also ND = 0, NA = 1. Thus, a method of assigning atoms by a simple fixed integer values of ND, NA in advance of any analysis has its difficulties. Donors are relatively straightforward, as such groups nearly always donate one hydrogen bond per H atom, so the C–NH3+ group would have a donor count of 3 and an acceptor count of 0. However when one considers groups such as a primary amine, C–NH2, as having a donor count of 2 and acceptor count of 1 we know that although it is possible for the N to accept hydrogen bonds this happens relatively infrequently compared to the donor hydrogen bonds from N. Another example of how all N and O atoms can not reasonably be treated as equal in accepting frequency is the nitro group C–NO2; it is unrealistic to count this as an acceptor count of NA = 3 compared to a keto group C–CO–C with NA = 1. We therefore devised and tested some parameters to reflect the observed frequency of formation of donor and acceptor bonds per atom-group code.Intramolecular contacts are also included in the contact data, and we have the option to include these in the parameters defined above, NDavg, NAavg, NDmax, NAmax, or exclude intramolecular contacts, so that we examine intermolecular hydrogen bonds alone. This second option might be considered relevant to hydrate formation. We define these intermolecular-only parameters with the names:
NDavg,inter, NAavg,inter
NDmax,inter, NAmax,inter
We require an estimation of the potential number of donor and acceptor bonds per molecular residue. For this purpose we formed the arithmetic sum of the atomic donor acceptor property over all atoms in each structure, for each molecular residue and the total structure, thus:
| SDavg = ∑NDavg | SAavg = ∑NAavg |
| SDavg,inter = ∑NDavg,inter | SAavg-inter = ∑NAavg,inter |
| SDmax = ∑NDmax | SAmax = ∑NAmax |
| SDmax,inter = ∑NDmax,inter | SAmax,inter = ∑NAmax,inter |
To test for correlation of these sum parameters with hydrate formation, we have combined these sum-values SD, SA in different ways:
We thus have 12 different donor–acceptor parameters which we examined in detail for effective correlation with frequency of hydrate formation; only the most relevant results will be discussed.
2.3 Calculated molecular properties
We used the QSAR module of the Sybyl program9 to calculate a range of molecular properties and to check if any of them can be correlated with the tendency to hydrate formation. These properties describe molecules rather than crystal structures, so only the largest residue was used from each crystal structure. (If more than one chemically different residue had the same number of heavy atoms then all of them were used.) Using the sample list of reference codes, 34 770, the hydrogen normalised coordinates of 35 175 molecules were exported from Conquest to mol2 files. The calculation of some properties requires partial atomic charges, which were computed using the method of Gasteiger and Marsili10 as implemented in Sybyl. The calculation of partial charges failed for 116 hydrates forming and for 883 non-hydrate molecules, which were excluded from the analysis. The calculated properties included atom and bond counts (e.g. number of halogen atoms, number of rotatable bonds), simple charge-derived properties (dipole moment, minimum/maximum/mean partial charges), and molecular volume or surface area related properties (including various charge weighted volume and area descriptors).The average value and e.s.d. of each property were calculated separately for the molecules extracted from hydrate and non-hydrate crystal structures. The difference between the average values for hydrates and non-hydrates relative to the e.s.d. was used to select properties for more detailed analysis.
3. Results and discussion
We show in Fig. 1 an example of an anhydrous structure (ACYHXA01) and a hydrate structure (ABCOCX) where the molecules have the same functional groups. This serves to illustrate that although the functional group counts for molecules may be same, and even their mutual geometrical disposition is the same, one molecule adopts the anhydrate structure and the other a hydrate. We will return to this example in discussing our donor–acceptor parameters below. | ||
| Fig. 1 Example of an anhydrous structure (ACYHXA01, left) and a hydrate (ABCOCX, right), showing hydrogen bonded contacts O⋯O and O⋯N less than the sum of the van der Waals radii + 0.1 Å. | ||
3.1 Hydrate frequency correlation with the donor–acceptor parameters
Desiraju2 in 1991 proposed the idea that the formation of organic hydrates is favoured when the number of hydrogen-bond acceptor groups increases with respect to the number of donor groups in the molecule. In this theory the water is included to compensate the donor–acceptor imbalance in the molecular formula. However he did not compare his studies with the non-hydrate compounds. It is already known11 that water molecules in the CSD prefer to form the pattern of two donor hydrogen bonds and 1 acceptor, and thus when water is included in the crystals it tends to compensate for a lack of donors in the parent molecules. By this theory one might expect that molecules with a lack of donors, i.e. small ratio of donors/acceptors, would form hydrates to balance the ratio more closely towards unity. We tested this idea using our ratio parameter, RDA. | ||
| Fig. 2 Hydrate (blue) and anhydrate (red) frequency for donor/acceptor ratio ranges. The donor/acceptor ratio has been calculated as (a) RDA = SDavg/SAavg and (b) RDAmax = SDmax/SAmax. The numbers at the top of the bars represent the total number of observations in that interval (hydrates + anhydrates). | ||
If we examine the histogram in Fig. 2 for percentage hydrates for ranges of RDA, we see a distribution with no trend to higher hydrate formation at high RDA values. The histogram value at 0.0 requires some explanation—this is an artefact of the sample selection which insisted on at least one NH, OH, or SH present in the CSD entry, which of course includes hydrates of molecules which themselves have no donors giving a RDA value of zero. This is the reason that most of the CSD compounds in this low RDA range are hydrates, 97%. The remaining 3% correspond to compounds with SH donor groups with very low donor capacity, ND often <0.05.
 | ||
| Fig. 3 Hydrate (blue) and anhydrate (red) frequency for ranges of absolute donor–acceptor difference, (a) DAdiff = |NDavg-NAavg|, (b) DAdiff,inter = |NDavg,inter-NAavg,inter|, (c) DAdiff,max = |NDmax-NAmax|. | ||
 | ||
| Fig. 4 Hydrate (blue) and anhydrate (red) frequency for different donor + acceptor sum ranges. The donor + acceptor sum has been calculated as (a) SDA = SDavg + SAavg, (b) SDAmax = SDmax + SAmax. | ||
By visual inspection of Fig. 4 we can see that the most consistent trend is for the sum-property SDA (there was little difference in distribution for SDA or SDAinter).
3.2 Number of independent water molecules per structure
It is interesting to note that when hydrates occur, the number of independent water molecules per structure is affected by the sum-property SDA, see Fig. 5. We divide each sample for an SDA range into four categories based on the number of independent water molecules NW = 1, 2, 3, and greater or equal to 4. These are plotted as a histogram where we see a significant decline in the NW = 1 population as we increase the SDA value. In other words, higher SDA tends to give a larger frequency of multi-hydrates. (We note that when water molecules sit on a special position in the crystal their contribution to the SDA for the asymmetric unit is lessened, but we feel that this comparatively rare event does not affect the qualitative trend observed.) | ||
| Fig. 5 Frequency histogram for NW, the number of independent water molecules per structure, plotted for ranges of SDA, for the total sample of 3258 hydrate structures. We show categories NW = 1, 2, 3, and ≥4. | ||
3.3 Molecular properties and hydrate frequency
There is a prevalent idea that bulky molecules are more favourable for hydrate formation, because all molecules must leave empty space in the crystal, the average packing coefficient being around 0.70, so as molecules become larger the voids between them should also increase, and accommodate water molecules primarily as a space-filler. A simple way of testing this hypothesis is to calculate the hydrate frequency for ranges of the molecular weight of the largest molecular residue per structure, for the hydrate and the anhydrate samples, Fig. 6. We see that there is not a simple linear dependence of hydrate frequency on molecular weight. Similar results are obtained if hydrate formation frequency is plotted against molecular volume or molecular surface area. | ||
| Fig. 6 Plot of frequency of hydrate formation versus the molecular weight of the largest molecular residue. | ||
Another property often linked to hydrate formation is molecular polarity. The simplest quantity related to molecular polarity is dipole moment. In our data set the frequency of hydrate formation is higher for dipole moments above 15 Debye than for those bellow it (Fig. 7). Polar surface area and polar volume are further descriptors of molecular polarity. They are defined as the surface area or volume of the molecule that belongs to N, O or S atoms or H atoms covalently bonded to it. Polar surface area (PSA) was found to be strongly correlated with the frequency of hydrate formation (Fig. 8). The tendency to form hydrate increases gradually with increasing PSA from 5–6% to above 30%. As expected from its definition, PSA is strongly correlated with SDA (r = 0.487), but differs from it in that PSA is related also to the surface accessibility of the polar groups. Polar volume, which does not contain accessibility information, was found to be a much weaker indicator of hydrate formation probabilities. (Observed hydrate formation frequencies range from 6 to 20% as a function of polar volume, and only 130 observations are available in the data range that corresponds to frequencies above 15%.) Polar surface area and polar volume were also expressed as a fraction of the total molecular surface and volume (i.e. as PSA/surface area), but the use of such fractional descriptors did not give a better description of hydrate formation. The variation of hydrate formation frequency displays a more smooth dependence on PSA than on PSA/surface area.
 | ||
| Fig. 7 Plot of frequency of hydrate formation versus the dipole moment of the largest molecular residue. | ||
 | ||
| Fig. 8 Plot of frequency of hydrate formation versus the polar surface area of the largest molecular residue. | ||
3.4 Water hydrogen bonded environment patterns
We have carried out a study of the hydrogen bonded environment of all the water molecules in the hydrate sample, 4616 molecules in 3258 structures, see Fig. 9 and 10. Because we have used an accurate sample of the CSD with all the hydrogen atoms present as 3-D coordinates, we are reasonably confident in our identification of donor bonds OH⋯X and acceptor bonds O⋯ HX, classified as donor (D) and acceptor (A). We show the distribution of the total number of hydrogen bonds per water molecule, NHB, in Fig. 9, where we see the previously observed result4,11 that the coordination three (NHB = 3) is the most commonly observed, 2390 observations (52%). We notice that ∼1% of water molecules do not show any hydrogen bonds, which are truly ‘inclusion compounds’, filling in space between molecules. | ||
| Fig. 9 Distribution of the total number of hydrogen bonds per water molecule NHB in the sample of 3258 structures. | ||
 | ||
| Fig. 10 Patterns of hydrogen bonds around water molecules as a frequency (%) for each observed environment, described by letters, D signifying donor-bonds, and A acceptor-bonds, ( Scheme 1). | ||
We find a wide variety of hydrogen bond patterns for the water molecules, which are shown using the D and A notation for the hydrogen bonds, in Fig. 10. The most common environment is two donor-bonds and one acceptor-bond, DDA 50%, then DDAA at 17%, DD at 14%, and DA at 7%, these four patterns comprising 88% of the environments, Scheme 1 and Fig. 9, and CSD reference codes are given in the ESI. We also note that other environments are seen at very low frequencies, <5%.
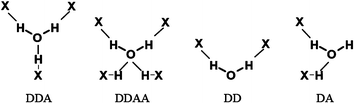 | ||
| Scheme 1 Most common hydrogen bond patterns for the water molecules (X means any possible donor/acceptor atom) | ||
We saw above that the ratio-parameter RDA did not simply correlate with the frequency of hydrate formation. However, one would expect that the ratio of donors to acceptors will have an effect on the water environment pattern. We examined several water patterns for their frequency as we change the RDA value in ranges, see Fig. 11, where we can observe the expected effect. For example, water participating in only one or two hydrogen bond donors is favoured when the RDA value decreases, which is logical since we have fewer donor atoms and more acceptor atoms. Also the pattern DAA, (one donation and two acceptor-bonds) is more frequently observed for higher RDA values, where we have more donors available in the structure trying to find fewer acceptor atoms.
 | ||
| Fig. 11 Diagram showing the frequency of occurrences of several water environment patterns, varying for ranges of the ratio-parameter RDA. A subset of structures is selected for each range of RDA, and the percentage of each pattern recorded. Separate graphs are given for each water environment pattern using the D, A notation as in the text, Scheme 1 and Fig. 10. | ||
4. Conclusions
The survey of several donor/acceptor properties of the sample of CSD entries has shown the following:• The ratio of donor/acceptor groups, parameter RDA, does not have any significant effect on the frequency of hydrate formation
• The sum parameter SDA shows a positive correlation with the frequency of hydrate formation, and also with the number of independent water molecules included in the crystal. SDA is defined as the sum over all groups per compound of average donor contacts and average acceptor contacts.
• The difference parameter DAdiff also shows a positive correlation with the hydrate frequency. This is defined as the absolute difference of the sum of average donor contacts and the sum of the average acceptor contacts.
• As the total polar surface of molecules increases we find an increase in the frequency of hydrate formation.
• The hydrogen bond environment pattern of water in hydrates is affected by the ratio of donors/acceptors. Low values of RDA (fewer donors) favour the patterns where water donates hydrogen bonds e.g. D, DDA, and high values of RDA (more donors) favours patterns where water accepts hydrogen bonds, e.g. A, DAA.
Acknowledgements
László Fábián thanks Pfizer Inc for funding as part of the Pfizer Institute for Pharmaceutical Materials Science.References
- C. H. Gorbitz and H. P. Hersleth, On the inclusion of solvent molecules in the crystal structures of organic compounds, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 526–534 CrossRef.
- R. G. Desiraju, Hydration in Organic Crystals: Prediction from Molecular Structure, J. Chem. Soc., Chem. Commun., 1991, 426–428 RSC.
- F. H. Allen, The Cambridge structural database: a quarter of a million crystal structures and rising, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef; F. H. Allen and W. D. S. Motherwell, Applications of the Cambridge Structural Database in organic chemistry and crystal chemistry, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 407–422 CrossRef.
- L. Infantes, J. Chisholm and S. Motherwell, Extended motifs from water clusters in organic molecular crystals, CrysEngComm, 2003, 5(85), 480–486 Search PubMed.
- J. van de Streek, private communication.
- RPluto, A program for crystal structure visualisation, Cambridge Crystallographic Data Centre, Cambridge, 2002 (http://www.ccdc.cam.ac.uk, freely downloadable for non-commercial use).
- L. Infantes and S. Motherwell, The probable number of hydrogen-bonded contacts for chemical groups in organic crystal structures, Chem. Commun., 2004, 1166–1167 RSC.
- L. Infantes and W. D. S. Motherwell, Hydrogen bond competition between chemical groups: new methodology and the Cambridge Structural Database, Z. Kristallogr., 2005, 220, 333–339 CrossRef CAS.
- SYBYL 7.0, Tripos Inc., 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA.
- J. Gasteiger and M. Marsili, Prediction of proton magnetic resonance shifts: The dependence on hydrogen charges obtained by iterative partial equalization of orbital electronegativity, Org. Magn. Reson., 1981, 15, 353–360 CrossRef CAS.
- A. L. Gillon, N. Feeder, R. J. Davey and R. Storey, Hydration in Molecular Crystals – A Cambridge Structural Database Analysis, Cryst. Growth Des., 2003, 3, 663–673 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details. See DOI: 10.1039/b612529h |
| This journal is © The Royal Society of Chemistry 2007 |