Soft-to-hard transformation of the mechanical properties of dynamic covalent polymers through component incorporation†
Takashi
Ono
ab,
Shunsuke
Fujii
ab,
Tadahito
Nobori
ab and
Jean-Marie
Lehn
*a
aISIS, Université Louis Pasteur, 8 allée Gaspard Monge, F-67083 Strasbourg cedex, France. E-mail: lehn@isis.u-strasbg.fr
bR&D Center, Mitsui Chemicals, Inc., 580-32 Nagaura, Sodegaura, Chiba 299-0265, Japan
First published on 14th November 2006
Abstract
The mechanical properties of acylhydrazone dynamic polymers may be converted from soft to hard by the incorporation of rigid monomeric components into the original soft polymer backbone, taking advantage of acylhydrazone bond exchange.
Dynamic polymers, dynamers,1,2 are polymeric entities of either molecular or supramolecular nature that are capable of undergoing exchange, incorporation or decorporation of their monomeric subunits, linked together by respectively reversible covalent bonds or labile non-covalent interactions. Dynamers implement constitutional dynamic chemistry,1,3 the chemistry of molecular and supramolecular entities capable of dynamically modifying their constitution by exchanging/incorporating molecular components.
On the molecular level, dynamic covalent chemistry1,3–7 has recently attracted considerable attention as a powerful methodology for discovering biologically active substances,5,7 as well as for developing new functional materials3,5,8 such as dynamic covalent polymers.1,8–13 In addition to conventional polymeric properties such as stability and strength, these dynamic polymers present the distinctive ability to exchange, incorporate and/or decorporate monomeric components linked by reversible covalent bonds, for example acylhydrazone functions,6,7,10,11,14 alkoxyamine units12 or imine metathesis ,13 depending on the external conditions. This feature offers to polymer science perspectives towards a functional plasticity that involves modification and control of the intrinsic properties of polymeric entities from within.1,10–12 Because the mechanical behavior of polymers plays a most important role in materials science, the ability to mutate the mechanical properties of polymers would be of much interest, giving access to a new class of functional polymeric materials. We now report that dynamic covalent polymers based on an acylhydrazone backbone can undergo a progressive change in their mechanical properties through the introduction of other monomeric components into their main chain.
The bis-hydrazide monomers M1 and M2 were formed by treatment of the corresponding methyl or ethylesters with hydrazine monohydrate in alcohol. The dialdehydes M3 and M4 were obtained, respectively, from 4-allyloxybenzaldehyde and hexamethyltrisiloxane using Karstedt catalyst, and from 4-hydroxy-2-methoxybenzaldehyde and 2-chloroethyl ether in presence of K2CO3 (Fig. 1).
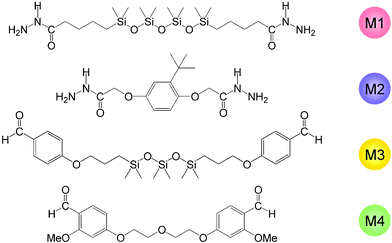 | ||
| Fig. 1 Structures of the bis-hydrazides M1 and M2, and of the dialdehydes M3 and M4. | ||
Polycondensation
‡
in CHCl3 between the monomers M1 and M3, both containing a highly flexible siloxane-derived spacer unit as soft components, yielded the alternating homopolymer P1 as a very soft and quite stretchy film F1 (of about 0.057 mm thickness) after solvent evaporation (Fig. 2). Gel permeation chromatography (GPC) gave a molecular weight Mn = 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and a distribution Mw/Mn = 1.6 (with polystyrene calibration). Differential scanning calorimetry (DSC) yielded a glass transition temperature Tg = 1 °C, respectively. Viscoelasticity measurements revealed that the storage elastic modulus E′ of F1 was 0.015 GPa at 25 °C, indicating that it possessed rubber elasticity at room temperature (Fig. 3). The profile of the loss elastic modulus, E″, gave Tg = 10 °C. These results are in line with the soft and stretchy behavior of F1, as shown in Fig. 2. The small decrease of E″ between –80 °C and –40 °C must arise from the siloxane units of the polymer P1.
000 and a distribution Mw/Mn = 1.6 (with polystyrene calibration). Differential scanning calorimetry (DSC) yielded a glass transition temperature Tg = 1 °C, respectively. Viscoelasticity measurements revealed that the storage elastic modulus E′ of F1 was 0.015 GPa at 25 °C, indicating that it possessed rubber elasticity at room temperature (Fig. 3). The profile of the loss elastic modulus, E″, gave Tg = 10 °C. These results are in line with the soft and stretchy behavior of F1, as shown in Fig. 2. The small decrease of E″ between –80 °C and –40 °C must arise from the siloxane units of the polymer P1.
 | ||
| Fig. 2 The soft and stretchy polymer film F1. | ||
 | ||
| Fig. 3 Temperature dependence of the storage elastic modulus E′ of the films (a) F1, (b) F8, (c) F6, (d) F7 and (e) F9, and the loss elastic modulus E″ of the films (f) F1, (g) F8, (h) F6, (i) F7, and (j) F9. | ||
On the other hand, polycondensation between the monomers M2 and M4, containing rigid spacer groups as hard components, gave the alternating homopolymer P2 as a hard but very fragile film F2 with Mn = 14000, Mw/Mn = 1.8 and Tg = 100 °C (determined by GPC and DSC, respectively). It was impossible to further study the mechanical properties of F2 as it was not strong enough for such experiments.
The homopolymers P3 and P4, and the random copolymer P5 were also prepared as reference materials, respectively, from monomers M1 and M4, M2 and M3, and all four kinds of monomer in equal molar ratio in the same manner. 1H NMR analyses in 1 : 1 DMSO-d6/CD2Cl2 solution (ca. 8 mg ml–1) allowed identification of the four different connections between hydrazides and aldehydes by the signals of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H protons of the acylhydrazone functions between 7.8–8.7 ppm (Fig. 4).
C–H protons of the acylhydrazone functions between 7.8–8.7 ppm (Fig. 4).
 | ||
| Fig. 4 Part of the 1H NMR spectra of the homopolymers (a) P1, (b) P2, (c) P3 and (d) P4, and (e) the random copolymer P5 in a 1 : 1 mixture of DMSO-d6/CD2Cl2 (ca. 8 mg ml–1) showing the C–H proton signals of the acylhydrazone functions (signal of CHCl3 remaining in the polymers is at around 8.1 ppm). | ||
Changes to the mechanical properties of acylhydrazone polymers induced by bond interchange and component incorporation was demonstrated by introducing hard components, that is M2 and M4, into the soft homopolymer structure P1. Equimolar amounts of P1, on the basis of its repeating unit, M2 and M4 were dissolved in CHCl3 in the presence of acid, followed by heating the solution to accelerate the exchange reaction. Solvent evaporation then gave the transparent film F6.§ The film F6 was distinctly harder than the parent film F1, quite enough to stand by itself (Fig. 5). The 1H NMR spectrum of F6 (in 1 : 1 DMSO-d6/CD2Cl2) showed that there were four kinds of signals of nearly equal area, which were assigned to the C–H protons of the acylhydrazone connections between M1 and M3, M1 and M4, M2 and M3, and M2 and M4 (Fig. 6). This observation indicated that monomers M2 and M4 were introduced into the main chain of the homopolymer P1, generating a random copolymer P6, obtained as the film F6, which should be similar to the random copolymer P5, obtained from the mixture of the four components M1–M4. GPC gave Mn = 59000 and Mw/Mn = 1.5 (with polystyrene calibration) for P6, of the same order as the parent homopolymer P1. However, determining how random the sequences are is not possible from the NMR spectra.
 | ||
| Fig. 5 Evolution in appearance of the mechanical properties of the polymer films (a) F1, (b) F8, (c) F6 and (d) F7. | ||
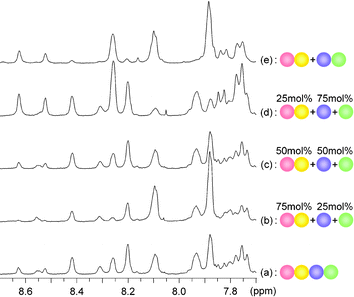 | ||
| Fig. 6 Part of the 1H NMR spectra of (a) the random copolymer P5, the modified polymers (b) P8, (c) P6 and (d) P7, and (e) the polymer blend P9 in a 1 : 1 mixture of DMSO-d6/CD2Cl2 (ca. 8 mg ml–1) showing the C–H proton signals of the acylhydrazone functions (the signal of CHCl3 remaining in the polymers at around 8.1 ppm). | ||
Introduction of M2 and M4 into homopolymer P1 led to a striking evolution in its mechanical properties. The glass transition temperature, Tg, of P6 increased to 56 °C, beyond room temperature, compared to 10 °C for P1 (based on the loss elastic modulus E″). This increment nicely reflected the difference in film hardness at room temperature between F1 and F6, illustrated in Fig. 5. The E′ profile of F6 indicated that it had a typical glass state up to around 50 °C and rubber elasticity around 70 °C.
The degree of evolution in mechanical properties was correlated with the ratio of M2 and M4 incorporated into the resulting polymers (Table 1). Three molar equivalents of M2 and M4, on the basis of P1 units, led to a film F7 (harder than F6), whereas one third of both M2 and M4 gave film F8 (softer than F6) (Fig. 5). The 1H NMR spectra of F7 and F8 showed that both were also random copolymers P7 and P8, respectively, consisting of M1, M2, M3 and M4 in approximately the expected ratios. In the case of P7, for instance, the ratio of the peak areas assigned to the connections between M1 and M3, M1 and M4, M2 and M3, and M2 and M4 was about 1 : 3 : 3 : 9, whereas P7 was a random copolymer made of M1, M2, M3 and M4 in about a 1 : 3 : 1 : 3 molar ratio, as expected. Tg and viscoelasticity values were in accordance with the proportion of M2 and M4 in the polymers. The mechanical properties of these resulting polymers were thus markedly different from those of the parent homopolymer P1 or P2, implying that the mechanical properties of the acylhydrazone polymers are adjustable via the nature and/or the proportion of the different components incorporated.
| Film | Ratio of M2 and M4 to P1 (mol%)b | Thickness/mm | M n c | M w/Mnc | T g (DSC)/°Cd | T g (E″)/°Ce | E′/GPaf |
|---|---|---|---|---|---|---|---|
| a In presence of 2.5 mol% acid per acylhydrazone unit for F6–F8; no acid added for F9. b Total molar ratio of M2 and M4 in the resulting polymers on the basis of monomer units. c Molecular weight Mn and distribution Mw/Mn determined by GPC with polystyrene calibration. d Glass transition temperature determined by DSC. e Glass transition temperature determined by the profile of the loss elastic modulus E″. f Storage elastic modulus E′ at 25 °C. g Not determined. | |||||||
| F1 | 0 | 0.057 | 41000 |
1.6 | 1 | 10 | 0.015 |
| F2 | 100 | —g | 14000 |
1.8 | 100 | —g | —g |
| F8 | 25 | 0.049 | 47000 |
1.5 | —g | 32 | 0.48 |
| F6 | 50 | 0.049 | 59000 |
1.5 | 46 | 56 | 1.1 |
| F7 | 75 | 0.055 | 75000 |
1.5 | —g | 88 | 1.3 |
| F9 | 50 | 0.039 | 34000 |
2.1 | 19 | 30 | 0.59 |
In contrast, lack of incorporation did not bring evolution in mechanical properties, leading only to the behavior expected for a blend of the parent polymers P1 and P2. Thus, a slightly cloudy film, F9, was obtained from an equimolar ratio mixture of P1 and P2 heated in CHCl3 solution for 24 h in the absence of acid, followed by solvent evaporation. The 1H NMR spectrum of F9 showed that it was just a polymer blend, P9, of P1 and P2 with only a very small amount of exchange (<5%). The film F9 seemed to be softer than F6, although both F9 and F6 consisted originally of the same segments. Single glass transition temperatures for P9 were observed based on both DSC and the loss elastic modulus E″, respectively, and were about 20 °C higher than those of P1. The profile of the storage elastic modulus, E′, showed that P9 possessed a glass state up to around 30 °C, and that E′ itself decreased with relative moderation above Tg as the temperature increased. These observations implied that P2 was microdispersed in a P1 matrix at a nanometer scale in the polymer blend P9.
In conclusion, the present results show that the mechanical properties of polyacylhydrazone dynamers can be varied through incorporation of other components into the main chain of the original polymer, making use of the reversible nature of the acylhydrazone bond. This feature provides a very useful methodology for modifying the mechanical properties of polymers, giving access to smart and adaptive dynamic materials1,8 such as self-strengthening polymeric materials controlled-by and responding-to external stimuli. Such behavior by dynamers is thus a result of the application of the principles of constitutional dynamic chemistry1,3 to polymer science.
We thank Mr. Sokei Sekine, Mr. Takumi Yamanoue and Dr. Tomoyoshi Sasakawa for viscoelasticity measurements and DSC analyses, and Dr. Masaki Okazaki for GPC measurements.
Notes and references
- J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814 CrossRef CAS.
- J.-M. Lehn, Polym. Int., 2002, 51, 825 CrossRef CAS.
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763 CrossRef CAS; J.-M. Lehn, Science, 2002, 295, 2400 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898 CrossRef.
- J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455 CrossRef CAS.
- G. R. L. Cousins, S.-A. Poulsen and J. K. M. Sanders, Curr. Opin. Chem. Biol., 2000, 4, 270 CrossRef CAS.
- O. Ramström and J.-M. Lehn, Nat. Rev. Drug Discovery, 2001, 1, 26.
- J.-M. Lehn, in Supramolecular Science: Where It Is and Where It Is Going, ed. R. Ungaro and E. Dalcanale, Kluwer, Dordrecht, The Netherlands, 1999, pp. 287 Search PubMed.
- J.-M. Lehn, in Supramolecular Polymers, ed. A. Cifrri, Taylor Francis, New York, 2nd edn, 2005, ch. 1, pp. 3 Search PubMed.
- W. G. Skene and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8270 CrossRef CAS.
- T. Ono, T. Nobori and J.-M. Lehn, Chem. Commun., 2005, 1522 RSC.
- H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, Chem. Commun., 2002, 2838 RSC; H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, J. Am. Chem. Soc., 2003, 125, 4064 CrossRef CAS; G. Yamaguchi, Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2005, 38, 6316 CrossRef CAS.
- T. Nishinaga, A. Tanatani, K. Oh and J. S. Moore, J. Am. Chem. Soc., 2002, 124, 5934 CrossRef CAS.
- R. Nguyen and I. Huc, Chem. Commun., 2003, 942 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterisation data for the monomers. See DOI: 10.1039/b612035k |
| ‡ The bis-hydrazide(s) and dialdehyde(s) at concentrations of around 0.1 M were each dissolved in a 1 : 1 stoichiometry in CHCl3 and heated to 60 °C for 3 h. The solution was poured into a 50 mm diameter Petri dish made of fluoroplastic, followed by evaporation at 60 °C at normal pressure until most of the solvent had disappeared, and then kept at 60 °C in vacuo for 24 h. About 200 mg of the total amounts of the monomers were used to obtain the polymer film of around 0.04–0.06 mm thickness. The films thus obtained were used as such for the study of their mechanical properties and usually contained trace amounts of CHCl3, as determined by 1H NMR. |
| § Equimolar amounts of polymer P1, on the basis of its repeating unit, bis-hydrazide M2 and dialdehyde M4 were dissolved in CHCl3 (the concentration was around 0.05 M), followed by the addition of pentadecafluorooctanoic acid in 0.025 molar ratio with respect to the resulting total acylhydrazone bonds. The solution was heated to 60 °C for 24 h and then poured into a Petri dish, followed by evaporation at 60 °C at normal pressure until most of the solvent had disappeared, and then in vacuo for 24 h at the same temperature. |
| This journal is © The Royal Society of Chemistry 2007 |