Scaling out of electrolyte free electrosynthesis in a micro-gap flow cell
Ping
He
a,
Paul
Watts
a,
Frank
Marken
b and
Stephen J.
Haswell
*a
aDepartment of Chemistry, University of Hull, Hull, UK HU6 7RX
bDepartment of Chemistry, University of Bath, Bath, UK BA2 7AY. E-mail: s.j.haswell@hull.ac.uk
First published on 30th October 2006
Abstract
The electro-reductive coupling of activated olefins and benzyl bromide derivatives has been selected to compare the performance of single and multiple channel (scaled-out) micro-gap electrochemical flow reactors. Two working electrode configurations were evaluated; in the first a single set of electrodes was used in conjunction with a multiple flow manifold to give two and four separate flow channels; in the second independent electrodes were used within the same flow manifold. Problems with shunt currents and Joule heating in the first configuration meant that only the second configuration was reliable, giving results comparable to those obtained for the single flow cell. Excellent yields of the coupling products such as 2-benzyl-succinic acid dimethyl ester and derivatives were obtained. This demonstrates micro reactor scale-out for unsupported electrosyntheses.
Introduction
Electrosynthesis offers a potentially clean and versatile methodology for the generation of anion and cation radical intermediates in organic synthesis under relatively mild reaction conditions.1 At present, however, the main disadvantage of using micro reactor based methodology is the low quantities of product produced. In order to overcome this problem, whilst maintaining the practical and chemical advantages, the concept of scale-out can be employed.2 Scale-out systems have included multisectioned flow-through porous electrodes,2a coplanar platinum interdigitated microband electrodes2b and a miniaturized parallel plate electrochemical cell.2c However, all of these cited electrochemical cells have involved the use of supporting electrolytes or relatively low conversion without intentionally added supporting electrolyte.In this note, the electro-reductive coupling of activated olefins and benzyl bromide derivatives has been selected to compare the performance of single and multiple channel micro-gap flow reactors.3
Experimental
Construction of micro-gap flow electrochemical cells
The details of constructing a single channel electrochemical cell have been described previously.4 In brief, a single channel cell consisted of two glass plates (3 cm length, 2 cm width, 6 mm thickness) in which two holes are drilled in the top plate to enable PEEK tubes (id 0.24 mm) to be connected in order to allow inlet and outlet flow. Two equally sized Pt foils (4 mm width and 15 mm length, 50 µm thickness, Goodfellow Cambridge Limited, purity 99.99%) were used as the working and counter electrodes, and two PTFE spacers (120 µm thick, Bohlender GmbH, Germany) with a rectangular flow reaction zone (3 mm width and 15 mm length) were used to produce the single channel cell with a working area of 45 mm2 and inter-electrode distance of 160 µm. Scaling out systems were made with multiple channels and using two distinct cell configurations. In the first configuration, a single set of electrodes was used in conjunction with a multiple flow manifold to give two (see Fig. 1(a)) and four separate flow channels. In the second configuration, independent electrodes were used within the same flow manifold (see Fig. 1(b)). In the first configuration, the electrode width was 8 mm and 16 mm for making two channels and four channels, respectively. In the second configuration, each channel was built by using the same procedures as that for making single cells (see above).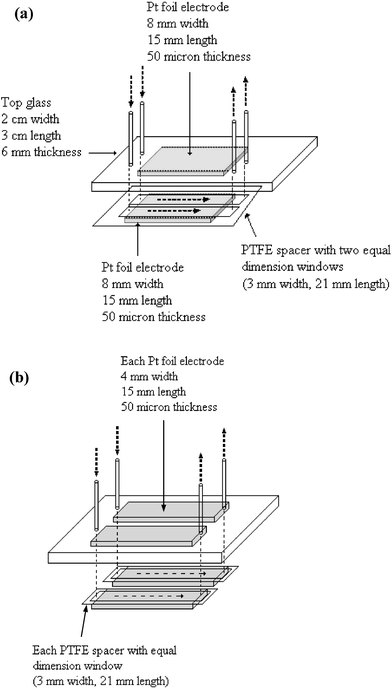 | ||
| Fig. 1 Schematic representation of scale out of micro-gap flow cell showing (a) a single pair of electrodes and (b) independent electrode configuration for a multiple double flow system. The arrows show reagent flow direction. | ||
The electrochemical coupling of an activated olefin with benzyl bromide
For electrosynthesis in single reaction channel cells, the procedure described previously4 was employed. For 2 and 4 independent reaction channel cells, each cell was connected in parallel linkage and each circuit consisted of a power supply (BPS4000, CALEX Electronics Ltd.), an ammeter and a voltmeter (TTi1906 Computing Multimeter, RS Components). A solution containing 5 mM activated olefin and 5 mM benzyl bromide in DMF (N,N-dimethylformamide, Fluka, 99%, stored over molecular sieve, H2O ≤ 0.01%, which was further dried over molecular sieve 3A (Lancaster, 1–2 mm beads) for 72 h prior to use and kept in a desiccator5) was continuously pumped (Harvard PHD 2000 syringe pump) through the reaction cell in which two platinum electrodes with a working area of 45 mm2 were positioned with an inter-electrode gap of 160 µm (see Fig. 2). During typical electrosynthesis runs, product samples were collected in a vial from each channel for 5 min in order to obtain sufficient material for subsequent GC/MS analysis. The conditions for GC/MS analysis and identification of products using 1H and 13C NMR analysis were described previously.4a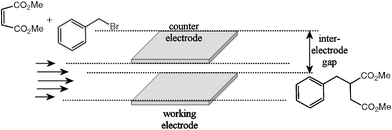 | ||
| Fig. 2 Schematic representation of the C–C coupling reaction during micro reactor electrosynthesis. A flow of reagents through a rectangular duct with working and counter electrode facing each other results in the formation of products. | ||
Resistance measurements (80 mA, 6430A Precision component analyzer, Wayne Kerr) for independent sets of electrodes were performed and indicate that each cell has resistance of typically 1.5 kΩ (for DMF/5 mM benzylbromide/5 mM dimethylfumarate) and the total resistance for 2 and 4 parallel connections is 750 Ω and 375 Ω, respectively. The total resistance for ten cells in parallel connection would be 150 Ω, compared to 170 Ω total resistance for ten cells in series connection reported for a similar system.2a However, resistance effects under no electrolyte conditions are complicated and the current distribution within individual cells may have effects on the local resistance.
Results and discussion
Initially, experiments were conducted for the coupling of an activated olefin with benzyl bromide at constant current (under conditions employed here constant current and constant potential modes give essentially the same results) in the absence of intentionally added supporting electrolyte in a single channel cell at 10 µl min−1 flow rate of reactant solution. Under these conditions voltages of typically 4–4.4 V (applied between working and counter electrodes) are applied to maintain a current of 0.6 mA. Table 1 summarizes the typical product yields. It can be seen that sufficiently high levels of product (yield >93%) can be obtained. Interestingly, it was noted that the unwanted dimerization of olefins was occurring to an extent of less than 2%. A very low amount of toluene (from debromination of benzyl bromide) was observed and no dimerization of benzyl bromide was detected. No obvious detrimental anode processes occurred at the counter electrode (i.e. the oxidation of bromide ions formed during the coupling reaction to produce bromine, was minimal4a).| Entry | Olefin | R2–Br | Cell | Flow/ µl min−1 | Yield (R1–R2) (%)b |
|---|---|---|---|---|---|
| R1 | R2 | ||||
| a Olefin is 5 mM, halide is 5 mM, solvent is DMF, electrode gap is 160 µm, voltage is 4–4.4 V to maintain constant current of 0.6 mA. b Yield was determined using GC based on the quantity of the product after reaction using n-decane as an internal standard. Side products include dimerization of olefin and debromination of benzyl bromides, no dimerization of benzyl bromides is detected. | |||||
| 1 | Dimethylfumarate | Benzyl | Single | 10 | 98 |
| 2 | Dimethylfumarate | 4-Methoxybenzyl | Single | 10 | 94 |
| 3 | Dimethylfumarate | 4-Methylbenzyl | Single | 10 | 94 |
| 4 | Dimethylfumarate | 4-Bromobenzyl | Single | 10 | 99 |
| 5 | Dimethylfumarate | 1-Phenylethyl | Single | 10 | 98 |
In order to further explore the reactant flow within the micro reactor, we estimate the average residence time and the approximate inter-diffusion time for reactants travelling between the two electrodes. At a flow rate of 10 µl min−1, the residence time and the average linear velocity are about 43 s and 0.35 mm s−1, respectively. Using the Einstein–Smoluchowski equation6 (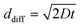 ; ddiff = distance travelled by diffusion, D = diffusion coefficient, t = time) the diffusion time across the electrode gap is estimated as typically 12–13 s. This suggests that inter-diffusion is possible. High conversion is probably due to effective inter-diffusion of the dimethylfumarate radical anion and benzyl bromide, and reactive electron transfer between the dimethylfumarate radical anion and benzyl bromide.4a
; ddiff = distance travelled by diffusion, D = diffusion coefficient, t = time) the diffusion time across the electrode gap is estimated as typically 12–13 s. This suggests that inter-diffusion is possible. High conversion is probably due to effective inter-diffusion of the dimethylfumarate radical anion and benzyl bromide, and reactive electron transfer between the dimethylfumarate radical anion and benzyl bromide.4a
To further evaluate the scale out methodology, the same coupling reactions were conducted using double and quadruple micro-gap flow cells (see Fig. 1). The individual flow rates for each of the quadruple cells were measured and found to vary by less than 5% compared to each other and the single channel device. The cells were operated in two distinct configurations as described in the experimental section. It is important to stress that the two and four parallel flow cells used allowed the geometry, flow rate, and applied potentials of the previously optimised single flow cell to be maintained. Using the double and quadruple micro-gap flow cells the coupling reactions were carried out using both electrode configurations and the results are summarised in Table 2. Comparison with Table 1 indicates that the double and quadruple flow cells show the same level of product yield as that obtained with the single cell under the same conditions. However, volumetric flow rates equivalent to 20 µl min−1 (i.e. 10 µl min−1 × 2 flow cells) and 40 µl min−1 (i.e. 10 µl min−1 × 4 flow cells) could be achieved. This demonstrates that scale-out can be achieved without loss of performance compared to that obtained for a single cell. The performance however of the quadruple flow cell configured with only one set of electrodes was poor (configuration 1, Entry 16 in Table 2). This is due presumably to shunt currents (localised resistance changes) and a pronounced Joule heating effect.2d These problems lead to the generation of bubbles and disruption to the flow. No such problems were observed in the second configuration with electrode current individually controlled.
| Entry | Olefin | R2–Br | Cellb | Electrode/reactor numberc | Flow/d µl min−1 | Product (R1–R2) yield (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | Cell-1 | Cell-2 | Cell-3 | Cell-4 | ||||
| a Olefin is 5 mM, halide is 5 mM, solvent is DMF, electrode gap is 160 µm. For the double cell (D) experiments, voltage was 3.5–4 V to maintain constant current of 0.6 mA. For multiple cell experiments (M), each cell has similar voltage value to the single cell in order to maintain a constant current of 0.6 mA. b When using the double cell geometry, a single electrode was divided into two single flow cells. In multiple cell geometry, two and four independent electrodes were used for generating 2 (M2) and 4 (M4) parallel flow cells. c The values present the number of electrode and reactor used in the cell configuration. d The flow value represents the total flow rate. e The quadruple flow cell with a single electrode (configuration 1). | |||||||||
| 1 | Dimethylfumarate | Benzyl | D | 1/2 | 20 | 98 | 97 | — | — |
| 2 | Dimethylfumarate | 4-Methoxybenzyl | D | 1/2 | 20 | 93 | 94 | — | — |
| 3 | Dimethylfumarate | 4-Methylbenzyl | D | 1/2 | 20 | 93 | 94 | — | — |
| 4 | Dimethylfumarate | 4-Bromobenzyl | D | 1/2 | 20 | 99 | 99 | — | — |
| 5 | Dimethylfumarate | 1-Phenylethyl | D | 1/2 | 20 | 98 | 97 | — | — |
| 6 | Dimethylfumarate | Benzyl | M2 | 2/2 | 20 | 98 | 98 | — | — |
| 7 | Dimethylfumarate | 4-Methoxybenzyl | M2 | 2/2 | 20 | 94 | 94 | — | — |
| 8 | Dimethylfumarate | 4-Methylbenzyl | M2 | 2/2 | 20 | 94 | 95 | — | — |
| 9 | Dimethylfumarate | 4-Bromobenzyl | M2 | 2/2 | 20 | 99 | 99 | — | — |
| 10 | Dimethylfumarate | 1-Phenylethyl | M2 | 2/2 | 20 | 98 | 98 | — | — |
| 11 | Dimethylfumarate | Benzyl | M4 | 4/4 | 40 | 98 | 98 | 97 | 98 |
| 12 | Dimethylfumarate | 4-Methoxybenzyl | M4 | 4/4 | 40 | 93 | 94 | 93 | 94 |
| 13 | Dimethylfumarate | 4-Methylbenzyl | M4 | 4/4 | 40 | 95 | 93 | 94 | 93 |
| 14 | Dimethylfumarate | 4-Bromobenzyl | M4 | 4/4 | 40 | 99 | 98 | 99 | 99 |
| 15 | Dimethylfumarate | 1-Phenylethyl | M4 | 4/4 | 40 | 98 | 97 | 97 | 98 |
| 16 | Dimethylfumarate | Benzyl | Q4e | 1/4 | 40 | 70 | 20 | 30 | 65 |
Conclusions
In this preliminary scale-out study, it has been demonstrated that electrosynthesis in a multi-channel micro-gap electrochemical flow cell is possible in the absence of intentionally added supporting electrolyte. The micro flow electrochemical reactor can be easily multiplexed to generate a number of parallel flow cells (scale-out) which offer the performance of the single cell whilst increasing the volumetric throughput of the system.References
- J. Utley, Chem. Soc. Rev., 1997, 26, 157 RSC; P. T. Anastas and T. C. Williamson, Green Chemistry, Oxford University Press, Oxford, 1998, p. 167 Search PubMed; E. Steckhan, T. Arns, W. R. Heineman, G. Hilt, D. Hoorman, J. Jorissen, L. Kroner, B. Lewall and H. Putter, Chemosphere, 2001, 43, 63 Search PubMed; D. Pletcher and N. L. Weinberg, Chem. Eng., 1992, 99, 98 CrossRef CAS; Microreaction Technology, ed. W. Ehrfeld, Springer, Berlin, 1998 Search PubMed; W. Ehrfeld, V. Hessel and H. Löwe, Microreactors: New Technology for Modern Chemistry, Weinheim, Wiley-VCH, 2000 Search PubMed; V. Hessel, S. Hardt and H. Löwe, Chemical Micro Process Engineering, Weinheim, Wiley-VCH, 2004 Search PubMed; D. Horii, M. Atobe, T. Fuchigami and F. Marken, J. Electrochem. Soc., 2006, 153, 143 Search PubMed.
- (a) C. Vallieres and M. Matlosz, J. Electrochem. Soc., 1999, 146, 2933 CrossRef CAS; (b) C. Belmont and H. H. Girault, J. Appl. Electrochem., 1994, 24, 719 CrossRef CAS; (c) S. Rode, S. Altmeyer and M. Matlosz, J. Appl. Electrochem., 2004, 34, 671 CrossRef CAS; (d) M. Küpper, V. Hessel, H. Löwe, W. Stark, J. Kinkel, M. Michel and H. Schmidt-Traub, Electrochim. Acta, 2003, 48, 2889 CrossRef CAS.
- (a) J. P. Devlin, W. D. Ollis, J. E. Thorpe, R. S. Wood, B. J. Broughton, P. J. Warren, K. R. H. Wooldbridge and D. E. Wright, J. Chem. Soc., Perkin Trans. 1, 1975, 830 RSC; (b) N. A. Porter, D. M. Scott, I. J. Rosenstein, B. Giese, A. Veit and H. G. Zeitz, J. Am. Chem. Soc., 1991, 113, 1791 CrossRef CAS; (c) D. E. Levy, F. Lapierre, W. Liang, W. Ye, C. W. Lange, X. Li, D. Grobelny, M. Casabonne, D. Tyrrell, K. Holme, A. Nadzan and R. E. Galardy, J. Med. Chem., 1998, 41, 199 CrossRef CAS; (d) W. C. Groutas, M. J. Brubaker, M. A. Stanga, J. C. Castrisos, J. P. Crowley and E. J. Schatz, J. Med. Chem., 1989, 32, 1607 CrossRef CAS; (e) R. B. Bates, R. S. Cuther and R. M. Freeman, J. Org. Chem., 1977, 42, 4162 CrossRef CAS; (f) G. Sabitha, R. Srividya and J. S. Yadav, Tetrahedron, 1999, 55, 4015 CrossRef CAS; (g) S. C. Bergmeier and K. A. Ismail, Synthesis, 2000, 1369 CrossRef CAS; (h) R. Ballini, G. Bosica, D. Fiorini and P. Righi, Synthesis, 2002, 681 CrossRef CAS; (i) M. Mella, M. Fagnoni and A. Albini, Eur. J. Org. Chem., 1999, 9, 2137 CrossRef.
- (a) P. He, P. Watts, F. Marken and S. J. Haswell, Angew. Chem., Int. Ed., 2006, 45, 4146 CrossRef CAS; (b) P. He, P. Watts, F. Marken and S. J. Haswell, Electrochem. Commun., 2005, 7, 918 CAS.
- R. D. Burfield and R. H. Smithers, J. Org. Chem., 1978, 43, 3966 CrossRef CAS.
- I. Levine, Physical Chemistry, McGraw-Hill, 2nd edn, 1983 Search PubMed.
| This journal is © The Royal Society of Chemistry 2007 |