Anion hopping of (and on) functional oligoarginines: from chloroform to cells
Naomi Sakai*a, Shiroh Futaki*bc and Stefan Matile*a
aDepartment of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: naomi.sakai@chiorg.unige.ch; stefan.matile@chiorg.unige.ch; Fax: +41 22 379 5123; Tel: +41 22 379 6523
bInstitute for Chemical Research, Kyoto University, Kyoto, Japan
cPRESTO, JST, Kyoto, Japan. E-mail: futaki@scl.kyoto-u.ac.jp
First published on 3rd July 2006
Abstract
Rapid progress in the understanding of counteranions as either activators or inactivators of guanidinium-rich macromolecules is summarized. From phase transfer of polyarginine into chloroform to the cytosolic delivery of green-fluorescent proteins or apoptosis-inducing peptides with cell-penetrating peptides, counteranions emerge consistently as the origin of function. A unified view of this “demystified arginine magic” is proposed, focusing on the manipulation of the underlying anion hopping of and on these sticky molecular chameleons according to the principle of Le Chatelier.
 Naomi Sakai | Naomi Sakai received her BS from Keio University (1987) and her PhD from Tokushima Bunri University (1994). After a postdoctoral stay in the group of Professor Koji Nakanishi at Columbia University (1994–1996), she focused on bioorganic biomembrane chemistry (Georgetown University 1996–1999, University of Geneva 1999-present). |
 Shiroh Futaki | Shiroh Futaki received his BS (1983) and PhD (1989) from Kyoto University. He has been involved in the design and creation of functional peptides, especially those to function with membranes and in cells (University of Tokushima 1987–1997 and Kyoto University 1997–present). |
 Stefan Matile | Stefan Matile received a Diploma (1989) and PhD from the University of Zurich (1994) for research in bioorganic porphyrin chemistry in the group of Wolf Woggon. After a postdoc with Koji Nakanishi at Columbia University (1994–1996), he developed particular interest in multifunctional supramolecular architecture in lipid bilayer membranes, first at Georgetown University (1996–1999) and then in Geneva. |
1 Introduction
Three years ago, we reported that polyarginine is soluble in chloroform.1 Even more surprisingly we discovered that polyarginine prefers chloroform to neutral water under certain conditions. How can polyarginine be lipophilic? After all, arginine is an amino acid with an alkylguanidinium cation as a side chain; polyarginine, therefore, is a polycation (Fig. 1A). Now, however, we find that, depending on the nature of their counteranions, these polycations are either clearly lipophilic or clearly hydrophilic. Similar occasional lipophilicity was not observed for other polycations such as polylysine (Fig. 1B).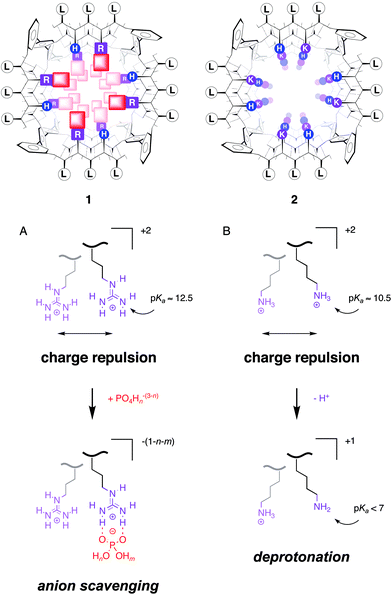 | ||
| Fig. 1 Synthetic multifunctional pore 1 with internal arginines (R, purple) scavenges internal counteranions like phosphates (red, shaded squares) to minimize charge repulsion between proximal guanidinium cations (A). Synthetic multifunctional pore 2 with internal lysines (K, magenta) does not scavenge internal counteranions because charge repulsion between proximal ammonium cations is minimized by partial deprotonation (B). As a result, pore 1 is unusually low-conducting and cation-selective, whereas pore 2 behaves as expected (high-conducting, anion-selective). Rigid-rod p-octiphenyl β-barrels 1 and 2 are shown schematically in axial view with β-sheets as solid (backbone) and dotted lines (hydrogen bonds; single-letter abbreviations. See refs 2 and 3 for specific suprastructures and general introductions to this topic. Proximal charge repulsion, but also other effects from the local environment, make the pKa's of the individual ammonium or guanidinium cations in one oligomer spread out over several units around as well as below their intrinsic pKa's. For details and specific references, also to theoretical simulations, see ref. 9). | ||
To explain this puzzling phenomenon of the “occasional lipophilicity” or, maybe more precisely, the “dynamic biphilicity” of oligo/polyarginines, we considered that guanidinium cations are well known to bind strongly to certain anions (e.g., phosphates and carboxylates) by not only Coulombic but also hydrogen bonding interactions. We further noted that, because of the delocalized positive charge, guanidinium cations are less hydrophilic than ammonium cations. But in addition, we felt that the poor acidity of the guanidium cation may chiefly account for the “occasional lipophilicity” of polyarginine. In neutral water, the more acidic polylysine (intrinsic pKa = 10.5) can release protons to minimize the charge repulsion between proximal cations in the polymer (Fig. 1B). Similar reduction of individual pKa's to minimize the charge repulsion is impossible for polyarginine because of the insufficient acidity of the guanidinium cation (intrinsic pKa = 12.5). To minimize intramolecular charge repulsion, proximal guanidinium cations in polyarginine have to neutralize (or even invert) their charge not by proton release but by counteranion scavenging (Fig. 1A). Instead of an unrealistic pKa reduction by ∼six orders of magnitude, these electrostatic proximity effects in polyarginine result, therefore, in a remarkable increase of the overall stability of polyarginine–counteranion complexes. Polyarginine never exists alone, tightly bound counteranions will always be there and matter. In the company of hydrophobic counteranions, polyarginine naturally prefers chloroform to neutral water.
However, polyarginine is, of course, not only lipophilic but also hydrophilic. Exchange of hydrophobic counteranions by hydrophilic ones will transform the lipophilicity of polyarginine into hydrophilicity. The velocity of counteranion exchange is unrelated to intramolecular charge repulsion and therefore unaffected by the proximity effects with polyarginine described above. As a result, polyarginine–counteranion complexes have exceptional thermodynamic stability but poor kinetic stability. They are stable and labile. This unrestricted anion hopping on the sticky polymer is the reason why oligo/polyarginines can exhibit this puzzling “dynamic biphilicity,” i.e., can adapt—like molecular chameleons—to every environment provided the right counteranions are within reach.
Lipid bilayer membranes offer an ideal environmental complexity for adaptable molecular chameleons like oligo/polyarginines. Their hydrophobic core—either fluid or solid or both—separates the water on both sides of the membrane. Interfacial domains of variable size and composition connect the hydrophobic and hydrophilic phases. The extraordinary thermodynamic but poor kinetic stability of oligoguanidinium–counteranion complexes, however, makes their behavior in lipid bilayers quite unpredictable and, obviously, very difficult to analyze. Imagine all the possibilities of counteranion hopping not only during the function but also during the purification of, for example, an arginine-rich membrane protein.
We tended to ignore the functional importance of counterions and literally stumbled into the study of counterion-mediated function while working on functional oligoarginines in lipid bilayer membranes. Pondering over “mysterious” behavior of arginine-rich synthetic multifunctional pores in Geneva2,3 and cell-penetrating peptides (CPPs) in Kyoto,4 we met in the summer of 2003 in Boston at the arginine-rich PEM-III (the 3rd Peptide Engineering Meeting)5 and decided to investigate together the importance of the elusive counteranions for what is often referred to as “arginine magic.” Highlights from the resulting collaboration are briefly recalled in the following. First, we describe the evidence for why contributions of counteranions to the function of oligoarginines in lipid bilayer membranes cannot be ignored. Then, we move on to spotlight how counteranions can be used to change the function of oligoarginines in lipid bilayer membranes, ending up with a very recent breakthrough on the rapid and efficient cytosolic delivery of CPPs with counteranions.6
2 Counterions and synthetic multifunctional pores
Synthetic multifunctional pores are pores made from abiotic scaffolds that can not only mediate molecular translocation across bilayer membranes but are also capable of molecular recognition and catalysis.2,3 Classical synthetic multifunctional pores are formed by rigid-rod β-barrels. These barrel–stave supramolecules are constructed from p-oligophenyl scaffolds. These staves are brought together with β-sheets formed between interdigitating peptide strands from neighboring rigid rods. Because of the opposite orientation of proximal amino-acid side chains in β-sheet conformation, the functionalization of both inner and outer surface of rigid-rod β-barrels is very straightforward. Fig. 1 shows the schematic axial view of two synthetic multifunctional pores.The precise pore structure and multifunctionality is not important in the context of this article; interested readers are referred to recent reviews on this topic.2,3 Important, however, are the differences between pore 1 and 2. The inner surface of pore 1 is decorated with arginines, whereas that of pore 2 is rich in lysines. In routine characterization of both pores in planar bilayer conductance experiments, pore 2 and related lysine-rich pores behaved completely normally.7,8 Pore 1, however, had a much lower conductance than expected and was cation selective.9 In particular, the finding that a polycationic pore selects for cations did not make sense, and precedence for cation-selective arginine-rich pores10 did not help to find an explanation for this behavior either.
The explanation came from the mass spectra of pores 1 and 2.7,9 Both were prepared as TFA salts, pore 1 appeared together with multiple phosphate counterions. This finding revealed that the arginine-rich pore 1 (but not the lysine-rich pore 2) can scavenge trace amounts of phosphate in the environment. The low conductance of pore 1 suggested that these counteranions are so strongly immobilized that they are felt as part of the pore by electrolytes passing by. This was neither the case with internal counteranions in the high-conducting, lysine-rich pore 2 nor with internal magnesium countercations in the corresponding, equally high-conducting rigid-rod pores with internal aspartates.7,9 The cation selectivity of pore 1 demonstrated that internal phosphates overcompensate the charge of the guanidinium cation. The observed, intriguing inversion of anion/cation selectivity with decreasing pH was consistent with either protonation of the internal phosphate counteranions, the neighboring histidines, or both.
Aware of ongoing discussions in the literature,1 it was this initial evidence for the functional relevance of counteranions that prompted us to study their possible role for the function of oligoarginines in bilayer membranes in a more general context.
3 Counterions and oligo/polyarginine carriers in vesicles
Phase transfer of octa- and polyarginine (Fig. 2, 3) from neutral water into non-polar solvents, the simplest, most fundamental and most convincing finding in support of the functional importance of counteranions, has been described and interpreted in the introduction.1,11 This process was enabled by hydrophobic anions like phosphatidylglycerol (PG), sodium dodecylsulfate (SDS, 4) below the critical micelle concentration, stearate, laurate (5), cholesterol sulfate, and so on (Fig. 2). It was inhibited by hydrophilic anions like sulfate, heparin (6), ATP, SDS micelles, and so on.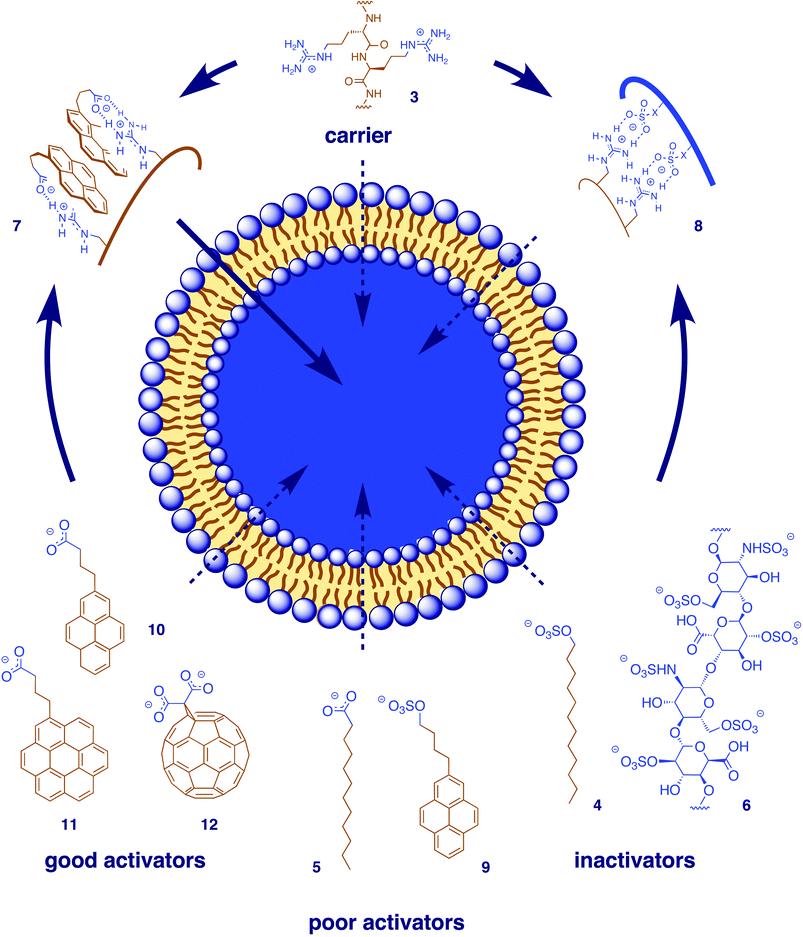 | ||
| Fig. 2 Counteranion–oligo/polyarginine complexes 7 as symbiotic anion carriers in vesicles. Aromatic carboxylates (like 10–12) are better counteranion activators than alkyl carboxylates (like 5) or alkyl and aryl sulfates (like 4 or 9), multivalent counteranions like heparin (6) or micellar SDS (4) act as inactivators because they “irreversibly” form hydrophilic counteranion–oligo/polyarginine complexes 8. | ||
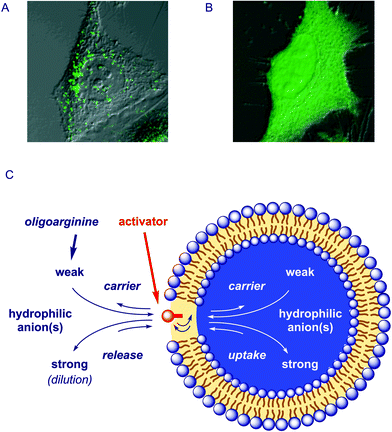 | ||
| Fig. 3 A unified view of the functions of oligoarginine–counteranion complexes (C) emerges from the comparison of results from uptake of fluorescently-labeled octaarginine into HeLa cells (A) in the absence and (B) in the presence of pyrenebutyrate with results on anion carrier activity in bulk membranes and vesicles (Fig. 2): According to the principle of Le Chatelier, anion hopping of the sticky polycation is directed toward uptake in the presence of “strong” internal hydrophilic anions, toward release in the presence of “strong” external hydrophilic anions (or repeated external dilution), and toward carrier activity in the presence of a transmembrane gradient of “weak” hydrophilic anions (and the absence of “strong” hydrophilic anions), with the counteranion activator acting in the membrane as a “translocation catalyst.” A and B are adapted with permission from ref. 6, © 2006 American Chemical Society | ||
Unproblematic co-transfer of reporter anions like 5(6)-carboxyfluorescein (CF) into non-polar solvents provided the first indication that lipophilic polyarginine–counteranion complexes like 7 (but not hydrophilic complexes like 8) might act as symbiotic anion carriers in bulk and lipid bilayer membranes. Studies in the U-tube confirmed octa- and polyarginine 3 as anion carriers in the presence of the hydrophobic anion activators introduced above. Similarly, octa- and polyarginine 3 were unable to mediate the release of CF from large unilamellar vesicles (LUVs) composed of the neutral phosphatidylcholine (PC-LUVs⊃CF). This situation changed in the presence of anionic PG in mixed PG–PC-LUVs⊃CF. Increasing CF efflux was mediated by octa- and polyarginine with increasing PG content in the PG–PC-LUVs⊃CF. This activation of octa- and polyarginine is contrary to the inactivation of lysine-rich pores like melittin in anionic vesicles; no activity was observed for polylysine under any conditions.
Fluorescently-labeled oligoarginines (FL-R8 and FL-R16) were used to study the mechanism of transport in anionic vesicles in detail.12 We found that the shorter FL-R8 accumulates at the interface of anionic vesicles, whereas the longer FL-R16 locates deeper in the membrane. Both oligoarginines could be re-extracted from the membrane with heparin, but only FL-R8 exhibited biphasic kinetics. This key finding revealed the counteranion-mediated, slow translocation of FL-R8 from the internal interface across intact spherical bilayers after the initial fast removal of the octaarginine from the external interface.
These findings provided excellent evidence that the actions of oligoarginines in bilayer membranes will not be understood as long as the elusive counteranion exchanges on the sticky polymer remains ignored. With the “arginine magic” as such presumably demystified, we were eager to participate creatively and teach the oligoarginine “magicians” new tricks by giving them new counteranions up their sleeves.13,14 We first focused on CF release from the otherwise inaccessible neutral PC-LUVs⊃CF. Not all hydrophobic anions could activate polyarginine under these conditions. Arylsulfates such as pyrene 9 had modest activity, probably because the sulfate–guanidinium ion pairs are too inert. Dominance of ion pairing in these inefficient complexes was indicated by decreasing stability with increasing ionic strength.
Alkylcarboxylates such as laurate 5 were nearly inactive. The best activity was found for carboxylates of higher arenes such as pyrenes (10), coronenes (11), fullerenes (12) or various calixarenes made and studied in the Coleman group in Lyon. The preference of aryl over alkyl activators attracted our attention early on. The dominance of interactions other than ion pairing in these efficient complexes was confirmed by increasing stability with increasing ionic strength. Strong excimer emission confirmed the importance of aromatic interactions. Arene-templated guanidinium–carboxylate pairing15 as indicated in the functional pyrene-complex 7 (Fig. 2) was a particularly appealing explanation for stable yet labile arylcarboxylate complexes. In analogy to the role of tryptophans in membrane proteins, interfacal preference16,17,12 emerged as another possible contribution to the superb performance of higher arenes as activators. Different dependence on membrane fluidity and size of the transported anions including CF-dextran suggested that contributions from transient pores may be more dominant with planar activators (pyrenes, coronenes) than with spherical activators (fullerenes, calixarenes). This finding was consistent with the higher efficiency of planar compared to spherical arene activators in live cells.
4 Counterions and cell-penetrating peptides in live cells
Cell-penetrating peptides (CPPs) have attracted much interest as potential drug-delivery agents.1,4,6,11 Arginine oligomers including the HIV-1 Tat transduction domain RKKRRQRRR are among the simplest of the CPPs. With multiple guanidinium cations as only structural requirement, a rapidly growing collection of synthetic CPPs already includes branched D- and L,D-oligoarginines, oligoarginine–stearate conjugates, guanidinium-rich peptoids, β- and α/β-peptides, carbamate oligomers and peptide nucleic acids.1 However, the mode of action of CPPs is under debate. Increasing evidence is accumulating in support of conventional endocytic uptake into endosomes (Fig. 3A) rather than the earlier suspected direct entrance into the cytosol with a controversial diffusion across cell membranes as if these barriers would not exist.In contrast to the inefficient spherical fullerene (12) and calixarenes,14 the planar pyrenebutyrate 10 had a dramatic impact on the delivery of various CPPs into live cells (Fig. 3B).6 First, we observed that CPP uptake by HeLa cells increased up to five times in the presence of pyrenebutyrate. Second, the strong dependence of presumably endocytic CPP uptake on temperature disappeared in the presence of pyrenebutyrate, leading to an up to 25 times increase in uptake at 4 °C. Third, CPP uptake in the presence of pyrenebutyrate was accelerated by inside-negative membrane potentials; little uptake was observed in depolarized HeLa cells. Increased efficiency and dependence on polarization as well as reduced dependence on temperature were all in support of direct, counteranion-mediated, polarization-supported diffusion through cell membranes.
The impact of counteranions on the delivery mechanism of CPPs had direct and spectacular consequences on their intracellular distribution. Confocal fluorescence microscopy revealed that in the presence of pyrenebutyrate, CPPs do not end up in endosomes (Fig. 3A) but directly and rapidly reach cytosol as well as the nucleus of HeLa cells (Fig. 3B). With counteranion activators, presumably non-endocytic CPP delivery not only into HeLa cells but also into otherwise challenging cells such as rat hippocampal primary cultured neurons became possible.
Rapid cytosolic delivery of proteins as large as enhanced green fluorescent protein (EGFP) attached to CPPs was possible in the presence of pyrenebutyrate. This finding was consistent with the formation of transient pores by counteranion–CPP complexes in the presence of arene activators such as pyrenebutyrate implied from size-exclusion experiments in vesicles. The same counteranion–CPP synergism allowed for the cytosolic delivery of peptides that act in the cytosol. Pyrenebutyrate-mediated delivery of the apoptosis-inducing peptide PAD attached to octaarginine did cause programmed cell death by the expected mitochondrial membrane disruption.
5 Conclusions
Upon close inspection of oligoarginine function in membranes, counteranions emerge consistently as the key players. They can phase transfer polyarginine into chloroform,1,12 determine the characteristics of arginine-rich synthetic multifunctional pores,2,3,9 transform oligo/polyarginines into anion carriers in bulk and lipid bilayer membranes,1,12–14 and account for direct and fast cytosolic delivery of oligoarginines.6 Cytosolic delivery of large proteins implies that phase transfer mediated by oligoarginine–counterion complexes can, at least in some cases, include the formation of transient, so far not verified pores.The attractive reversibility of transmembrane CPP translocation by counteranion activators12 provides yet another fine illustration of the multiple equilibrium processes that account for diversity and complexity of counteranion-mediated functions. Without control, this elusive hopping of oligoarginines from one anion to another as elaborated in the introduction may appear as “mysterious” behavior. However, the multi-equilibrium processes underlying both function and destination of oligo/polyarginines in bulk membranes, vesicles and live cells may all ultimately be explicable with the principle of Le Chatelier.18 Following Le Chatelier's principle, CPPs may choose to move from the counteranion activator in the membrane to cytosol and nucleus because they are attracted by “strong” hydrophilic polyanions such as RNA, DNA or ATP (Fig. 3C). Reversibility of CPP uptake by dilution is obvious in the context of multiple equilibrium processes, and so is the extractability of octaarginine from vesicles by addition of external “strong” polyanions like heparin. With pyrenebutyrates confirmed to accumulate in the membrane, the counteranion activators emerge as “translocation catalysts,” accelerating CPP movement.
Le Chatelier's principle further suggests that “strong” hydrophilic anions disable oligo/polyarginines as anion carriers by removing them from their place to act, i.e., the bulk or lipid bilayer membrane (Fig. 3C). Bound to the “translocation catalysts,” polyarginine carriers shuttle “weak” hydrophilic anions across the membrane, driven, again, by Le Chatelier, here imposed in the form of a concentration gradient of weak hydrophilic anions.
So in short, many mysterious functions of oligoarginines in membranes can be explained by the unified view of (a) exchangeable counteranions and (b) multiple equilibrium processes controlled by Le Chatelier's principle. Immobilization and/or compartmentalization of oligoarginine, counteranion, or both emerges as key variable to modulate the synergistic action of oligoarginine–counteranion complexes. In cytosolic delivery, the counteranion is immobilized in the membrane as “translocation catalyst” and the oligoarginine moves freely to the best-binding polyanion. In synthetic multifunctional pores, the arginines are immobilized at the inner pore surface, and as synergistic anion carriers, both oligoarginine and counteranions accumulate in the membrane. As far as application to biological problems beyond CPPs are concerned, we note that, for example, exchangeable counteranions may constructively contribute to the still ongoing debate on the molecular basis of the voltage gating of biological ion channels with arginine-rich paddles.19 Our own current interests reach from intra- as well as intercellular targeting to biphasic synthetic organic chemistry and the development of biochemical probes and sensors.
Acknowledgements
We warmly thank all coworkers and collaborators who contributed to this research, and the Swiss NSF and Japan Science and Technology Agency (JST) for financial support.References
- N. Sakai and S. Matile, J. Am. Chem. Soc., 2003, 125, 14348–14356 CrossRef CAS.
- N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2005, 38, 79–87 CrossRef CAS.
- N. Sakai and S. Matile, Chem. Commun., 2003, 38, 2514–2523 RSC.
- S. Futaki, Ed. Arginine-rich Peptides. Curr. Protein Pept. Sci., 2003, 4, 87–157 Search PubMed.
- C. Deber, Biopolymers, 2004, 76, 3 CrossRef CAS.
- T. Takeuchi, M. Kosuge, A. Tadokoro, Y. Sugiura, M. Nishi, M. Kawata, N. Sakai, S. Matile and S. Futaki, ACS Chem. Biol., 2006, 1, 299–303 Search PubMed.
- S. Litvinchuk, G. Bollot, J. Mareda, A. Som, D. Ronan, M. R. Shah, P. Perrottet, N. Sakai and S. Matile, J. Am. Chem. Soc., 2004, 126, 10067–10075 CrossRef CAS.
- N. Sakai, D. Houdebert and S. Matile, Chem.—Eur. J., 2003, 9, 223–232 CrossRef CAS.
- N. Sakai, N. Sordé, G. Das, P. Perrottet, D. Gerard and S. Matile, Org. Biomol. Chem., 2003, 1, 1226–1231 RSC.
- T. Iwata, S. Lee, O. Oishi, H. Aoyagi, M. Ohno, K. Anzai, Y. Kirino and G. Sugihara, J. Biol. Chem., 1994, 269, 4928–4933 CAS.
- J. B. Rothbard, T. C. Jessop, R. S. Lewis, B. A. Murray and P. A. Wender, J. Am. Chem. Soc., 2004, 126, 9506–9507 CrossRef CAS.
- N. Sakai, T. Takeuchi, S. Futaki and S. Matile, ChemBioChem, 2005, 6, 114–122 CrossRef CAS.
- F. Perret, M. Nishihara, T. Takeuchi, S. Futaki, A. N. Lazar, A. W. Coleman, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 1114–1115 CrossRef CAS.
- M. Nishihara, F. Perret, T. Takeuchi, S. Futaki, A. N. Lazar, A. W. Coleman, N. Sakai and S. Matile, Org. Biomol. Chem., 2005, 3, 1659–1669 RSC.
- S. E. Thompson and D. B. Smithrud, J. Am. Chem. Soc., 2002, 124, 442–449 CrossRef CAS.
- W. M. Yau, W. C. Wimley, K. Gawrisch and S. H. White, Biochemistry, 1998, 37, 14713–14718 CrossRef CAS.
- M. Schiffer, C. H. Chang and F. J. Stevens, Protein Eng., 1992, 5, 213–214 CrossRef CAS.
- The Le Chatelier principle, after the French chemist Henri Louis Le Chatelier (1850–1936), suggests that “if there is a change in the condition of a system in equilibrium, the system will adjust itself in such a way as to counteract, as far as possible, the effect of that change.” K. J. Laidler and J. H. MeiserPhysical Chemistry, 3rd edition, Houghton Mifflin, Boston, MA, 1999, 169 Search PubMed.
- R. B. Long, E. B. Campbell and R. MacKinnon, Science, 2005, 309, 903–908 CrossRef.
| This journal is © The Royal Society of Chemistry 2006 |