Colloid science of mixed ingredients†
Eric
Dickinson
Procter Department of Food Science, University of Leeds, Leeds, UK LS2 9JT
First published on 14th June 2006
Abstract
Some recent advances in the colloid science of heterogeneous systems containing food biopolymer ingredients are reviewed. Understanding the instability processes controlling the shelf-life and rheology of food colloids requires a detailed knowledge of the factors affecting the nature of the interactions in emulsions and gels containing mixtures of protein + protein, protein + surfactant and protein + polysaccharide. Against this background, theoretical modelling and computer simulation are useful tools for predicting effects of system composition on stability mechanisms. Confocal microscopy combined with image analysis is providing new experimental insight into the microstructural origins of changes in macroscopic properties during processing and storage.
1. Introduction
Food colloids are complex materials composed of soft condensed matter.1 The complexity is compositional, spatial and temporal. That is, there are many different chemical components which can change their interactions and distributions during formulation, processing and storage. To try to draw up rules to make sense of this complexity, one is compelled by necessity to infer that many of the overall system attributes relating to food texture and shelf-life can be described in terms of simple mixtures of the major structure-forming ingredients.The bulk properties of colloidal systems are ultimately determined by the nature and strength of the interactions amongst the various kinds of constituent particles and polymers. The particles in food systems are entities such as emulsion droplets, gas bubbles, fat crystals and protein aggregates, whose interactions are sensitively influenced by the structure and composition of their surfaces. The polymers are proteins and polysaccharides, whose interactions with each other, and with the surfaces of the particles, are sensitively moderated by the presence of much smaller molecular species such as salts, sugars, lipids and surfactants.2–6
This article reviews recent progress in the colloid science approach to emulsions and gels based on milk proteins. The interactions between the mixed ingredients controlling the microstructure and stability properties are considered under three broad headings: (i) protein + protein, (ii) protein + surfactant and (ii) protein + polysaccharide. An underlying objective is to understand how interactions at fluid interfaces affect the spatial distributions of these ingredients, and in turn how these distributions affect the stability properties of the colloidal systems. The value of theory and computer simulation in contributing towards this understanding will be highlighted.
2. Casein: the polymer scientist's favourite protein
According to the textbooks, proteins are characterized by highly organized and specific three-dimensional structures. But most rules have exceptions. And the main exception is casein—the main protein component of mammalian milk. The individual casein molecules are flexible and disordered. They behave like heterogeneous copolymers, with a strong tendency for self-assembly7 and for sticking strongly to hydrophobic surfaces.8 Also possessing the macromolecular characteristics of polypeptides, the caseins should not be regarded, of course, as purely random coil polymers.9–11 But, compared with typical globular proteins, they have relatively little ordered secondary structure and no characteristic denaturation temperature.The casein polymers tend to aggregate in aqueous media.12 In milk, they are intimately associated with calcium phosphate in the form of colloidal particles called ‘casein micelles’ (average size ∼300 nm). Despite extensive research for many years, the detailed internal structure of the native casein micelle remains a source of active speculation and continuing controversy.13–15 But what is beyond doubt is that casein micelles are highly stable colloidal particles under the near-neutral pH conditions of fresh milk. On the other hand, after acidification and removal of the calcium phosphate, the casein polymers can be separated and individually investigated with respect to their functional properties. For emulsion stabilization, the two most important polymers in cow's milk are β-casein (209 residues, net charge −15e at pH = 7) and αs1-casein (199 residues, net charge −22e). Both these molecules are substantially phosphorylated, and hence they are ‘calcium sensitive’. And both have no internal covalent crosslinks and no ability to polymerize through intermolecular disulfide bonds. Driven mainly by hydrophobic interactions, the β-casein molecules self-assemble into surfactant-like micelles above a certain ‘critical’ concentration,12 the extent of aggregation increasing with temperature and ionic strength. In contrast, driven by a combination of specific electrostatic and hydrophobic interactions, αs1-casein molecules associate into long chain-like aggregates through a series of consecutive association steps.12
What is the simplest model capable of describing the excellent stabilizing properties of αs1-casein and β-casein? To answer this question, one might start by trying to represent them as copolymers composed of just two monomer types: hydrophobic adsorbing segments (non-polar residues) and hydrophilic non-adsorbing segments (polar and charged residues). Fig. 1 shows two such linear sequences, called polymer I and polymer II, which have been constructed respectively from the known primary sequences of αs1-casein and β-casein.16 For a copolymer composed of two segment types (A and B) the degree of deviation from a purely random sequence (f = ½) can be described by the equation17
| PAPA→B = (PAPB)½f | (1) |
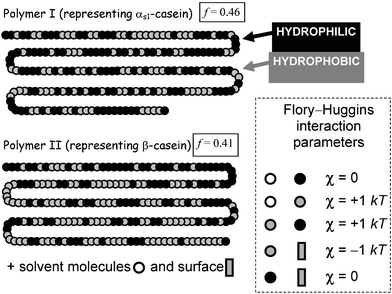 | ||
| Fig. 1 Representation of the primary sequences of αs1-casein (polymer I) and β-casein (polymer II) as copolymers composed of just two kinds of segments (hydrophobic and hydrophilic). Flory–Huggins interaction parameters determine the relative energies of the unlike pair interactions (segment–segment, segment–solvent, segment–surface). The lower is the value of the statistical parameter f, defined by eqn (1), the greater is the deviation from the segment distribution of the random copolymer (f = ½). | ||
The substantially greater proportion of charged residues near the N-terminus end of β-casein (including all 5 phosphoserines), together with its amphiphilic self-assembly behaviour in solution, leads to the simplest of all models for β-casein: the diblock copolymer.7 Even though it naïvely implies complete spatial separation of the two types of segments along the chain (PA→B → 0, f → 0), the diblock copolymer model has been successfully used to describe adsorbed β-casein at the nanoscale level.8,19,20 That is, it is supposed that β-casein adsorbs at hydrophobic surfaces in a train-loop configuration, with its hydrophilic tail (40–50 segments) dangling into the aqueous phase and the rest of the molecule strongly bound to the interface. The diblock copolymer model of adsorbed β-casein is consistent with neutron reflectivity experiments,21–24 and the representation is strongly supported by self-consistent-field (SCF) calculations.25,26
Colloidal stability depends on the establishment of repulsive forces between polymer-coated layers. The Scheutjens–Fleer SCF theory27 has been used recently16 to calculate the steric forces mediated between a pair of colloidal particles covered separately with layers of (αs1-casein-like) polymer I and (β-casein-like) polymer II. For illustrative purposes, we choose the basic set of Flory–Huggins interaction parameters given in Fig. 1, with surface coverages adjusted to conform with the equilibrium adsorption for each polymer. Fig. 2 shows the calculated force as a function of surface-to-surface separation assuming that the coverage remains constant as particles approach. We note that the β-casein-like polymer produces an effective long-range steric repulsion, whereas the αs1-casein-like polymer causes interlayer attraction at intermediate separations. This qualitative difference is consistent with the fact that, whereas the primary structure of β-casein causes it to behave like a diblock copolymer, the behaviour of αs1-casein more closely resembles that of a triblock copolymer.7 Attractive interlayer forces in systems containing triblock copolymers can be attributed to the polymer bridging effect.28
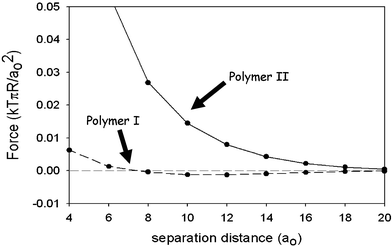 | ||
| Fig. 2 Force between two particles of radius R covered with polymer I (dashed line) and polymer II (solid line) as calculated16 from self-consistent-field theory for the model copolymers specified in Fig. 1. The surface–surface separation is measured in units of the monomer segment size a0. The force is measured in units of kTπR/a0, where kT is the thermal energy. (Adapted from ref. 16.) | ||
Experimentally, we find that oil-in-water emulsions prepared at neutral pH with αs1-casein as the sole emulsifier are, in fact, very stable under conditions of low ionic strength. This stabilization is due to the high surface charge density of the adsorbed αs1-casein layer,29 which is not properly allowed for in the crude model of Fig. 1. On the addition of an electrolyte (0.2 M NaCl), however, electrostatic screening leads to flocculation, in contrast to β-casein-coated oil droplets which are stable to salt.20,29 The significant differences in the adsorbed layer structures of αs1-casein and β-casein, and in the corresponding interparticle potentials as a function of pH and ionic strength, can be explained via SCF theory once the distributions of charged groups are included.8,26,30Fig. 3 gives a more refined version of the copolymer model of Fig. 1 involving a linear sequence of β-casein consisting of five different kinds of segments.16Fig. 4 shows the interaction potential A(d) between surfaces coated with this β-casein-like copolymer at neutral pH, as well as the equivalent interlayer energy curve for αs1-casein. The main noteworthy feature is the longer ranged steric repulsion for the ‘tail’ of β-casein than for the ‘loop’ of αs1-casein.
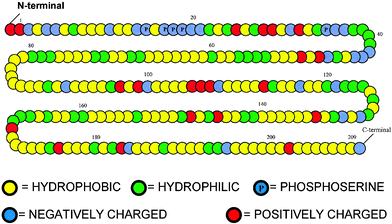 | ||
| Fig. 3 Representation of the primary sequence of β-casein as a copolymer composed of five kinds of segments (hydrophobic, uncharged hydrophilic, negatively charged, positively charged, and phosphoserine). | ||
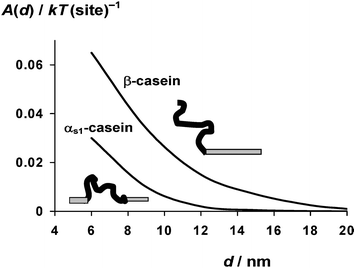 | ||
| Fig. 4 Interaction energy A(d) as a function of surface–surface separation d for the β-casein copolymer model illustrated in Fig. 3, together with that for the equivalent αs1-casein copolymer model. See ref. 16 for further details of the self-consistent-field calculations, including the assumed values of the Flory–Huggins parameters. | ||
These two biopolymers, αs1-casein and β-casein, are present together in roughly equal amounts in the commercial dairy ingredient known as sodium caseinate. Emulsions made with sodium caseinate, or with well-defined mixtures of αs1-casein and β-casein, have good stability towards added salt,31 thereby confirming the effective stabilizing ability of the latter under conditions where the former alone would lead to flocculation.32 One might wonder whether casein's excellent steric stabilizing character can demonstrate itself at low concentrations in mixtures with other food proteins.
3. Mixed proteins: protecting emulsions against heat-induced flocculation
The other main class of proteins in milk are called whey proteins. These are typical globular proteins, and they adsorb at liquid interfaces to produce close-packed viscoelastic monolayers.18–20 Being thinner, the whey protein monolayers have distinctly less steric stabilizing capacity than the casein monolayers. Another important difference between these two classes of proteins is that, whereas the disordered caseins are extremely stable to heat, the globular whey proteins—especially the main component, β-lactoglobulin—are readily denatured at temperatures above ∼75 °C. The denatured protein is highly susceptible to aggregation and cross-linking. This makes whey protein-stabilized emulsions rather sensitive to heat-induced flocculation and ‘thickening’.33,34Fig. 5(a) shows the heterogeneous flocculated state of a concentrated β-lactoglobulin emulsion (mean droplet size <1 µm), as observed by confocal microscopy after heat treatment at 90 °C for a few minutes.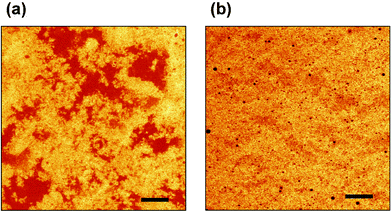 | ||
| Fig. 5 Effect of replacing a small fraction of β-lactoglobulin by sodium caseinate on the state of aggregation of a concentrated oil-in-water emulsion (45 vol% oil, 3 wt% total protein, pH 6.8, ionic strength 0.03 M) heated for 6 minutes at 90 °C. Confocal micrographs were obtained with Rhodamine B as the fluorescent protein stain: (a) emulsion containing 3 wt% β-lactoglobulin; (b) emulsion containing 2.85 wt% β-lactoglobulin + 0.15 wt% sodium caseinate. Scale bar = 20 µm.(Reproduced with permission from ref. 36.) | ||
The impressive steric stabilizing capacity of the casein polymer, when present as a minor component of a protein ingredient mixture, has recently been demonstrated35–37 in experiments on thermally processed whey protein emulsions. Replacement of just a small proportion (a few percent) of whey protein isolate35 or pure β-lactoglobulin36 by casein prevents droplet flocculation and the associated emulsion viscosity increase typically observed on heating. For instance, Fig. 5(b) shows the same emulsion system as in Fig. 5(a), except that 5% of the β-lactoglobulin used as the emulsifying agent to make the emulsion (45 vol% oil, 3 wt% total protein) was replaced by sodium caseinate. This level of casein incorporation (0.15 wt% protein in absolute terms) would be far too low to prepare such a fine stable emulsion when used on its own; but when present together with the whey protein it produces a high degree of synergistic stabilization. Furthermore, as we might expect from Fig. 4, it appears that β-casein is a better protective agent than αs1-casein. Indeed, in model emulsions heated to 90 °C for 3 minutes, it was found36 that the replacement of less than 1% of the β-lactoglobulin present by β-casein could eliminate completely the heat-induced viscosity increase. This corresponds to just a single β-casein molecule for every 50–100 whey protein molecules in the monolayer around the surface of these emulsified oil droplets!
To test whether SCF theory is able to explain this striking phenomenon, we have recently modelled16 the saturated whey protein monolayer as a dense brush-like layer of short tethered linear chains, each consisting of 25 monomers of alternating hydrophobic and hydrophilic segments (f → 1). With this representation, we have calculated the effect on the adsorbed layer structure and the surface–surface interactions of replacing a small proportion of the short chains in this hypothetical whey protein monolayer by β-casein-like copolymers of the type shown in Fig. 3. Calculated interaction potentials of the mixed layers as a function of interlayer separation are shown in Fig. 6. For surfaces coated with β-lactoglobulin (no casein present), the theory predicts an attraction of range d ≈ 2 nm (curve A). Similarly, for only a low density of grafted β-casein chains (2.5% replacement), in the absence of whey protein, theory also predicts an attraction, but now of longer range (3–4 nm), due to casein polymer bridging (curve D). But when the whey protein and β-casein (2.5% replacement) are present together, the theory predicts a stabilizing interlayer repulsion and a maximum in A(d) at d ≈ 1–2 nm (curve B). The height of the predicted ‘primary’ maximum increases from Amax ≈ 1 kT for 2.5% β-lactoglobulin substitution (curve B) to Amax > 15 kT for 5% substitution (curve C). So the simple SCF theory does indeed demonstrate that the replacement of just a few per cent of whey protein by β-casein tails can produce enough steric repulsion to transform the system from ‘flocculated’ to ‘stable’. The theory also predicts enhanced steric stabilization on replacement of whey protein by αs1-casein, but to a lesser extent.16
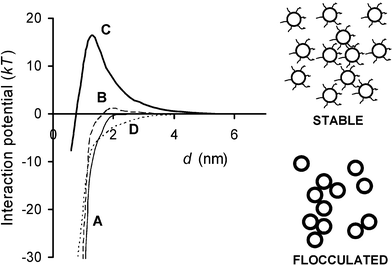 | ||
| Fig. 6 Interaction potential for model whey protein layer consisting of densely packed brush-like tethered chains with small fraction of whey protein replaced by β-casein as represented by the copolymer model of Fig. 3. The energy A(d) is plotted as a function of surface-surface separation d: A, no β-casein; B, 2.5% β-casein; C, 5% β-casein; D, 5% β-casein alone (without whey protein layer). Potentials A, B and D imply flocculated emulsion systems; potential C implies a stable emulsion state. | ||
These polymer-type calculations explain how a very low surface coverage of casein has the capability to convert a heat-sensitive globular protein-stabilized emulsion into one that is stable to thermal treatment. The entropic driving force is the excluded volume interaction between the spaced-out casein chains and the dense brush-like layer that we use to represent the β-lactoglobulin adsorbed monolayer. In what we rather fancifully describe as an ‘overgrown garden’ model,16 repulsion between the ‘grass-like’ whey protein layer and the ‘weed-like’ casein molecules increases the extension of the latter from the surface, thereby greatly increasing the steric stabilizing capacity of the mixed ingredient interface.
4. Protein + surfactant mixtures: heterogeneity from competitive adsorption
While mixtures of caseins and whey proteins can produce interfacial interactions that are synergistic,17 the surface interactions in mixtures of proteins and small-molecule surfactants are typically antagonistic. This is because of the ability of surfactants to disrupt the structure of a protein adsorbed layer, and, at high enough concentrations, to completely displace the protein from an emulsion droplet surface.38–41 Enhancement of fat droplet clumping by surfactant-induced disruption of interfacial protein layers is of considerable practical importance in the manufacture of ice-cream.42,43The balance of protein–protein and protein–surfactant interactions may change dramatically when emulsions containing globular proteins are heated beyond the protein denaturation temperature. In turn, this may lead to large changes in emulsion stability and microstructure. This is illustrated in Fig. 7 for the case of a β-lactoglobulin-stabilized emulsion (20 vol% oil) of high protein content (13.2 wt%) with non-ionic surfactant (Tween 20) present at a surfactant/protein molar ratio of R = 2.44 The predominantly yellow appearance of the emulsion before heating (Fig. 7(a)) indicates that the protein-coated droplets (mean size <0.5 µm) and the excess protein present in the aqueous phase are closely associated. The microscopic heterogeneity in Fig. 7(a) is due to weak reversible flocculation induced by precipitation of the excess protein onto the surface of the emulsion droplets in the relatively ‘poor solvent’ environment (low ionic strength).44 After heating the sample to 85 °C for 30 minutes, and then cooling it down again, the original liquid-like emulsion has been transformed into a solid-like emulsion gel. In the micrograph (Fig. 7(b)), we see that the oil droplets (green colour) are still flocculated, but the droplet-rich areas have become depleted of protein (red colour). This implies that the Tween 20 has displaced the protein from the oil–water interface during the thermal treatment. That is, the system has been transformed from a protein-stabilized emulsion into a surfactant-stabilized emulsion. In contrast to the original protein-coated droplets, the resulting surfactant-coated droplets are thermodynamically incompatible with the high content of protein in the aqueous phase, now thermally denatured and transformed into an aggregated network. In material science terms, we might say that the surfactant-coated droplets act as ‘inactive’ filler particles, lowering the elastic modulus of the final emulsion gel compared with the surfactant-free system.45–47 Another effect of heat treatment is a distinct coarsening of the emulsion, as indicated by the sight of some large oil droplets in Fig. 7(b). This would seem to imply that protein denaturation–aggregation, and the consequent competitive displacement of protein from the oil–water interface, is accompanied by loss of integrity of the thin films between adjacent oil droplets, i.e., inducing heat-induced droplet coalescence.48
 | ||
| Fig. 7 Confocal micrographs captured at 25 °C for an oil-in-water emulsion (20 vol% oil, 13.2 wt% β-lactoglobulin in aqueous phase) containing Tween 20 at surfactant/protein molar ratio R = 2: (a) before heating; (b) after heating (85 °C for 30 min).44 The sample was stained with Nile Blue for protein (appears as red colour) and Nile Red for oil (appears as green colour). The yellow appearance of image (a) implies that the protein and oil droplets are in a closely associated state. Some large individual oil droplets in image (b) are clearly evident. Scale bar = 100 µm. (Reproduced with permission from ref. 44.) | ||
The kinetic mechanism whereby small surface-active molecules disrupt and displace adsorbed protein from an oil–water interface has been investigated by a Brownian dynamics computer simulation.49–51 The model consists of monodisperse spherical particles49 that adsorb at a hypothetical planar fluid interface by virtue of an external square-well shaped potential. To account for the intermolecular cross-linking behaviour of partly unfolded globular proteins, the adsorbed particles are allowed to interact with each other through flexible bonds. Depending on the parameter values chosen, these bonds may be transient or effectively permanent over the time-scale of the simulation, thereby reproducing the range of interfacial structural and rheological properties found with adsorbed globular proteins.52,53 The surfactant molecules are represented as small non-bonding species with a higher surface binding energy per unit area than the protein particles.
The structure of a simulated protein + surfactant layer with breakable protein–protein bonds is illustrated in Fig. 8. The assumed diameter ratio of protein film particles and surfactant displacer particles is 2 ∶ 1. Full details of the model and the simulation procedure are described elsewhere.51 Suffice it to say here that, after adsorption of the protein particles to form a nearly close-packed monolayer and subsequent cross-linking at the interface to form a gel-like film, the smaller surfactant species are then introduced beneath the interface. Initially, individual surfactant molecules adsorb at gaps and defects in the dense protein monolayer (Fig. 8(a)). Then, with increasing number density of adsorbed surfactant molecules, these holes grow in size, and the protein-rich regions become increasingly compressed by the growing surfactant domains. Because the protein–protein bonds are not permanent in this particular simulation, the compression of the protein-rich regions is accompanied by stress relaxation in the protein film through bond breakage and the accompanying structural rearrangements. In the later stages of the displacement process, the two-phase structure of the simulated mixed film quite closely resembles the high-resolution images of mixed layers of β-lactoglobulin + Tween 20 derived from atomic force microscopy.51,54,55
 | ||
| Fig. 8 Effect of a competing surfactant on the interfacial structure of a simulated globular protein film composed of bond-forming particles: (a) at a very low surfactant surface coverage; (b) at a moderately high surfactant coverage. Protein-like particles and surfactant displacer particles are coloured white and red, respectively. | ||
The relative contributions of short-range forces and cross-links to the surface rheology of protein layers depends on many factors, including the surface coverage, the age of the adsorbed film, and the susceptibility of the protein molecules towards unfolding at the interface.56–61 Small-deformation surface rheology experiments on freshly adsorbed layers can be interpreted entirely in terms of the effect of the short-range repulsive forces between closely packed non-bonded spheres.52,62,63 On the other hand, the ‘wrinkling’ and fracture of protein films when subjected to large deformations in compression or extension seems to require a model with transient or permanent cross-links.53,64,65
Under certain conditions, an adsorbed globular protein layer behaves like a nanoparticle monolayer. Such a monolayer is illustrated by the images in Fig. 9 for an interface subjected to uniaxial compression. The simulated monolayer of identical spherical particles with attractive short-range forces adsorbed at a planar interface66 is compared with an experimental monolayer of monodisperse gold nanoparticles (<10 nm diameter) visualized by scanning electron microscopy.67 Initially, following adsorption, the ‘sticky’ particles aggregate leaving gaps in the monolayer (images A and D). Upon compression, some ordered (crystalline) domains are formed, separated by defect boundaries (images B and E). Finally, the collapse of the original interfacial monolayer leads to the appearance of heterogeneous anisotropic secondary layers; these take the form of widening strips perpendicular to the direction of compression (images C and F). Replacing the ‘sticky’ particle interactions in the simulation by transient (reversible) bond formation leads to no significant change in these qualitative features. But when bonds are allowed to become permanent (irreversible), the monolayer crumples after collapse, and the density of the wrinkles diminishes with decreasing bond breakability.65
 | ||
| Fig. 9 Structure of monolayers of monodisperse spherical particles subjected to uniaxial compression: A–C, scanning electron micrographs of gold nanoparticles compressed on a Langmuir trough;67 D–F, simulated monolayers from Brownian dynamics.66 Each set of images shows gaps, defects, and displaced secondary layers, depending on whether the monolayer is in the uncompressed state (A and D), is compressed to an interfacial stress just below the collapse point (B and E), or is compressed to a stress beyond the collapse point (C and F). Image dimensions: A, B, 350 × 350 nm; C, 3 × 3 µm. | ||
As an interfacial mixture bond-forming species (protein) and non-bond-forming species (surfactant) is compressed beyond the collapse point, there are two different scenarios for the structural development, depending on the bond strength.65 In the case of transient bonds, the aggregates from the bond-forming species tend to remain adsorbed under compression, whilst the non-bond-forming species get ‘squeezed out’ of the interface. Conversely, when protein–protein bonds are permanent, the bond-forming species develop a cross-linked network at the interface, which then buckles and desorbs on compression, with the non-bond-forming species remaining at the interface.
5. Proteins + polysaccharide mixtures in emulsions: shelf-life issues
A commonly encountered type of instability in emulsions is depletion flocculation due to non-adsorbed nanoparticles or polymers. This type of weak reversible flocculation has its entropic origin in the exclusion of non-adsorbed species from the narrow gap between closely approaching droplet surfaces. Three main classes of species can cause depletion flocculation of casein-stabilized emulsions—surfactant micelles,68 caseinate nanoparticles,69–71 and polysaccharides.72,73 While the destabilization of caseinate-based emulsions containing excess unadsorbed protein is thermodynamically attributable71 to the presence of sub-micelles (nanoparticles of size 10–20 nm), the microlayering of these sub-micelles can provide a kinetic barrier to flocculation in concentrated systems due to the induced oscillatory potential of mean force.74 In addition to the influence of polysaccharides, the strength of the depletion attraction for caseinate-stabilized emulsion droplets is affected by other ingredients such as ethanol,75,76 surfactants68,77 and calcium salts,76–78 as well as by the pH79,80 and the overall ionic strength.81Hydrophilic polymers (‘hydrocolloids’) like xanthan gum are routinely used in the food industry to improve the texture and physical shelf-life of pourable oil-in-water emulsions such as salad dressings. The purpose of this added ‘stabilizer’ is to inhibit gravity-induced creaming and/or serum separation during long-term storage. The traditional explanation2,5,82 for the stabilizing effect is through the hydrocolloid's ability to control the rheology of the aqueous continuous phase. This is undoubtedly correct at low oil volume fractions, where well-dispersed droplets can be separately immobilized in an entangled polysaccharide network, and the small buoyancy force acting on an individual droplet is insufficient to overcome the effective yield stress of the surrounding weak gel-like matrix.82 On the other hand, for concentrated emulsions (say ≥30 vol% oil), the explanation is not so straightforward, because the evolving microstructure of the system becomes highly heterogeneous due to local phase-separation induced by the non-adsorbing polysaccharide.3
Whereas a very low concentration of xanthan (≪0.1 wt%) causes depletion flocculation, gradually increasing the xanthan content towards ∼0.1 wt% leads to a much reduced creaming rate, and also a considerable delay in the onset of visible serum separation. Fig. 10 shows data for the rheology, stability and microstructure of a 30 vol% caseinate-stabilized emulsion made with 1-bromohexadecane as the oil phase and with various concentrations of xanthan incorporated. The emulsion without added hydrocolloid is a low-viscosity Newtonian liquid. With increasing xanthan content (up to 0.07 wt% in the aqueous phase), it becomes increasingly shear-thinning with the measured low-shear-rate viscosity reaching values more than 103 times larger than the high-shear-rate viscosity (Fig. 10(a)).83 When stored quiescently in glass tubes for several hours, the emulsions containing 0.02–0.06 wt% xanthan show visible serum layers at the top. (The ‘creaming’ here actually occurs downwards because the protein-coated droplets have a net density slightly greater than that of the aqueous continuous phase). After 10 hours storage, the 0.04 wt% xanthan sample has the thickest serum layer. There is no visible serum layer for xanthan concentrations above 0.07 wt% (Fig. 10(b).73 And at contents of around 0.2–0.3 wt% xanthan, in the presence of an anti-microbial agent, the emulsions remain stable to visible creaming for weeks or even months.
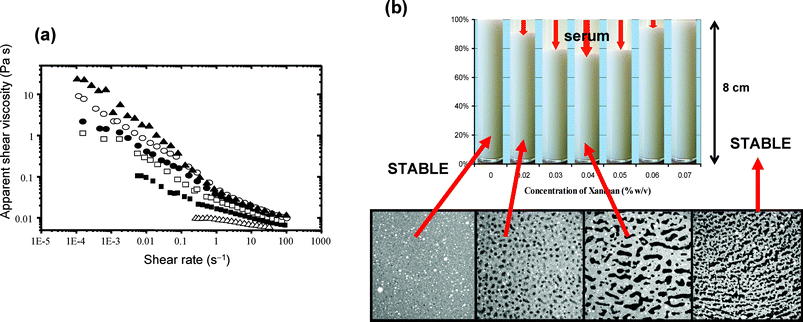 | ||
| Fig. 10 Influence of xanthan gum on the physical properties of oil-in-water emulsions (30 vol% 1-bromohexadecane, 1.4 wt% sodium caseinate, pH = 6.8, mean droplet size ∼0.5 µm). (a) Apparent viscosity against shear-rate at 20 °C for oil-in-water emulsions containing different concentrations of polysaccharide in the aqueous phase: △, 0 wt%; ■, 0.03 wt%; □, 0.04 wt%; ●, 0.05 wt%; ○, 0.06 wt%; ▲, 0.07 wt%.83 (b) Visual appearance of samples stored quiescently at ambient temperature for 10 hours, showing maximum serum separation occurring at 0.04 wt% xanthan. The four confocal micrographs refer to emulsions with xanthan contents of 0.01, 0.02, 0.04 and 0.1 wt% in images (250 × 250 µm) captured 10 minutes after sample stirring has stopped.73 (Reproduced with permission from refs 73 and 83.) | ||
It has long been recognized84–86 that this type of emulsion (in)stability, caused by the addition of non-adsorbing polysaccharide, tends to correlate more with the rheology of the whole emulsion than with that of the equivalent aqueous solution of the polysaccharide. This would seem to imply that the emulsion is prevented from creaming by the existence of some kind of particle gel network.1 In the context of pourable salad dressings, it was persuasively argued87 that this transient (but potentially long-lasting) network is formed from droplets that have become flocculated under the influence of attractive depletion interactions induced by the non-adsorbed polysaccharide.
In a bid to more clearly establish the underlying mechanism, we have recently been using confocal microscopy73,83 to explore the evolving microstructure of caseinate-based emulsions with added xanthan. As illustrated in Fig. 10(b), there is microscopic separation into oil-rich and oil-depleted regions over the polysaccharide concentration range 0.02–0.1 wt%. There was no such phase separation in an emulsion without added xanthan or one containing just 0.01 wt%. In the range 0.02–0.05 wt% polysaccharide, the microphase regions were observed to exhibit shape relaxation, coarsening and coalescence phenomena.73Fig. 11 shows a confocal micrograph of an emulsion sample (0.05 wt% xanthan) where protein, oil and xanthan have each been separately labelled.83 Once stirring stops, the interfacial tension drives the elongated shapes of phase-separating xanthan-rich domains into more circular shapes. With increasing xanthan concentration, however, the relaxation rate is increasingly retarded, and for ≥0.06 wt% there was found to be no microstructural evolution over the experimental time-scale (a few hours). While the characteristic domain relaxation time derived from image analysis could be roughly correlated with the polysaccharide solution rheology,73 one key question still remained: are the observed kinetics of global phase separation (i.e., macroscopic serum separation) mainly dependent on the rheology of the xanthan-rich regions or on the rheology of the concentrated oil droplet regions?
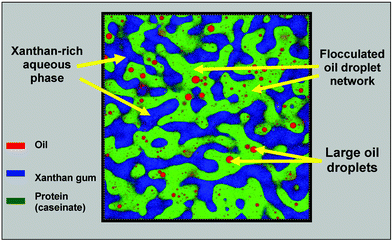 | ||
| Fig. 11 Confocal image of protein-stabilized emulsion (30 vol% oil, 1.4 wt% sodium caseinate) containing 0.05 wt% xanthan. Triple fluorescent labelling indicates locations of oil droplets (Nile Red), protein (Nile Blue) and xanthan (fluorescein-5-isothiocyanate).83 The sample (with dyes present) was stirred for 5 min in a shear cell at 1 rad s−1, and the image was captured ∼10 min after stirring ceased. Note that only the very largest of the emulsion droplets are visible, most of them being smaller than the optical resolution (∼0.5 µm). Image dimensions: 100 μm × 100 μm.. | ||
To answer this question, we have used confocal microscopy to monitor the mobility of colloidal probe particles within these phase-separating regions. The technique of particle tracking has been used for many years by biologists and material scientists to probe the microrheology of non-uniform systems.88–92 But the potential of particle tracking in the field of food colloids remains so far largely unexplored.
Fig. 12 shows individual trajectories of carboxylate-modified polystyrene microspheres (diameter 0.21 µm, ∼4 charged groups per nm2 of surface) monitored over a constant time period in an emulsion sample containing 0.05 wt% xanthan. We can see that the probe particles are distributed fairly evenly between the microphase regions; and some particles (yellow coloured trajectories) move freely across the ‘boundary’ between the different microphases during the observational time period. We can also see from Fig. 12 that the diffusive motion of the probe particles in the emulsion droplet regions (white coloured trajectories) is much more restricted, on average, than that of particles in the xanthan-containing regions (green coloured trajectories).
 | ||
| Fig. 12 Trajectories of fluorescent colloidal particles in the phase-separating emulsion (0.05 wt% xanthan) illustrated in Fig. 11. The axis units are expressed in pixels (1 pixel = 0.183 µm). Three different types of trajectories are distinguished by colour: green, in the xanthan-rich region; white, in the oil-droplet-rich region; yellow, traversing the boundary between the different regions.83 | ||
Fig. 13 compares the mean-square displacement (MSD) of particles in (a) xanthan-rich regions and (b) oil-droplet-rich regions for emulsions containing different xanthan concentrations (in the emulsion aqueous phase) in the range 0.03–0.07 wt%. The time-averaged MSD in the image plane is defined by
| <Δr2(τ)> = <[x(t + τ) − x(t)]2 + [y(t + τ) − y(t)]2> | (2) |
| <Δr2(τ)> = 4Dτ | (3) |
| D = kT/6πηa | (4) |
| <Δr2(τ)> ∼τα | (5) |
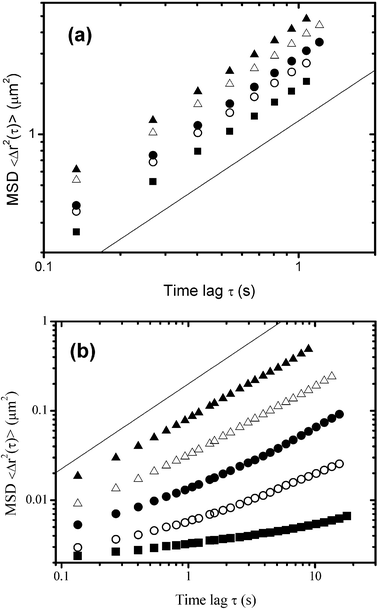 | ||
| Fig. 13 Effect of the xanthan concentration on the diffusion of fluorescent microspheres as determined by individual particle tracking in different regions of phase-separating emulsions (30 vol% oil, 1.4 wt% sodium caseinate).83 The ensemble-average mean-square displacement (MSD), <Δr2(τ)>, is plotted on a log–log scale against the time lag τ for (a) xanthan-rich aqueous regions and (b) floccculated oil-droplet rich regions: ▲, 0.03 wt%; △, 0.04 wt%; ●, 0.05 wt%; ○, 0.06 wt%; ■, 0.07 wt%. The straight lines of slope unity correspond to purely diffusive behaviour. | ||
Fig. 14 shows inferred viscosities based on the complete set of MSD data in Fig. 13. We note that the viscosity values calculated for the oil-droplet-rich regions are 102–103 times larger than those for the xanthan-rich regions. Moreover, the oil-droplet-rich microphase viscosity is seen to increase dramatically with xanthan concentration. Therefore, it is clear that, although the xanthan-containing phase does become more viscous with more xanthan present in the system, the main influence of the hydrocolloid stabilizer on the overall rheology of the emulsion is through its effect on the oil droplet network. It would appear that the kinetics of phase separation, leading eventually to enhanced gravity creaming and macroscopic serum separation, is predominantly controlled by the rheological behaviour of the interconnected oil droplet regions. As the microstructure coarsens with time, the restructuring and reorganization of the flocculated droplets leads to a loss of connectivity in the aggregated microstructure, and finally the transient network collapses.93 The time of onset of network collapse depends on the effective yield stress of the oil-droplet network and on the densities and relative proportions of the microphases. This type of system has been observed to exhibit a characteristic delay time before the structure collapses completely.87
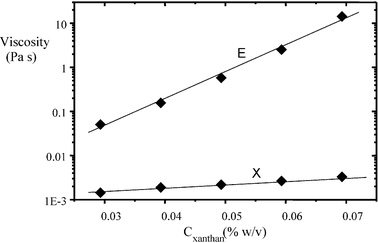 | ||
| Fig. 14 Apparent shear viscosities from particle tracking in phase-separating regions of caseinate-stabilized emulsions (30 vol% oil, 1.4 wt% protein, pH = 6.8) as a function of the xanthan concentration Cxanthan: X, xanthan-rich aqueous regions; E, flocculated emulsion droplet-rich regions.83 At Cxanthan = 0.07 wt%, the microviscosity in the oil-droplet-rich phase is more than 103 times that in the xanthan-rich phase. | ||
What are the implications of these combined results (Fig. 10–14) for the role of added hydrocolloid in a pourable salad-dressing-type emulsion (or some other emulsion product)? It is clear that the answer does not lie simply in bulk rheological control by the aqueous polymer solution. Rather, it appears that the essential stabilizing mechanism involves a thermodynamically driven reorganization of the emulsion system into a locally concentrated, depletion-flocculated microstructure. We could say that the gravitationally unstable ‘thin’ liquid-like emulsion is transformed—through changes in microscopic morphology and microrheology—into a more stable ‘thick’ (mayonnaise-like) emulsion containing trapped ‘blobs’ of polysaccharide-structured water.
There is an alternative strategy for protecting a protein-stabilized emulsion against gravity-induced destabilization in the presence of added polysaccharide. This involves manipulating the aqueous solution conditions so that the protein–polysaccharide interaction switches from net repulsive to net attractive. Such a change in ingredient interactions can be achieved during the acidification of a caseinate-stabilized emulsion containing added pectin.80,94–96
Fig. 15 shows photographs of sets of emulsion samples (30 vol% sunflower oil, 1.4 wt% protein, 45 wt% sucrose) containing different concentrations of low-methoxyl amidated pectin stored under quiescent conditions for 7 days: (a) at neutral pH, and (b) after acidification with glucono-δ-lactone.95 As the pH of the caseinate-stabilized emulsion is lowered towards the protein's isoelectric point, the steric stabilizing droplet–droplet repulsion is converted into a net attraction, and the liquid-like emulsion is transformed into a soft solid-like flocculated emulsion.97,98 The time-dependent rheology and microstructure of such an emulsion gel is sensitive to the presence of other ingredients like sugars,99 surfactants100 and polysaccarides.95 For the emulsion samples shown in Fig. 15, containing various additions of pectin, the pH at gelation lies in the range 5.7–5.9. At neutral pH, above a certain pectin concentration (0.05 wt%), the emulsion was found to phase separate within one day of preparation (Fig. 15(a)). With increasing pectin content, the bottom serum layer was gradually transformed from turbid to transparent and its height was reduced. This behaviour at neutral pH is consistent with the mechanism of reversible depletion flocculation due to a non-adsorbing polymer. At a higher pectin concentration, the strength of the depletion flocculation is enhanced, and the local structure is immobilized by the colloidal gel (or colloidal glass-like101,102) state formed from the flocculated droplets. After acidification, the instability caused by the presence of pectin at neutral pH becomes inhibited: each emulsion gel sample has a uniform single-phase appearance (Fig. 15(b)). The character of the protein–polysaccharide interaction has changed from net repulsive at pH ≈ 7 (which causes depletion flocculation and micro-phase separation) to net attractive at low pH (implying bridging flocculation and complexation).95
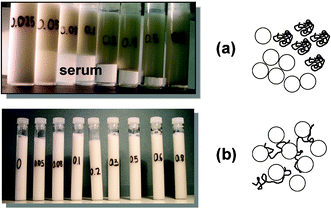 | ||
| Fig. 15 Stability of caseinate-stabilized emulsions (30 vol% oil, 45 wt% sucrose, 1.4 wt% protein) containing various concentrations of low-methoxyl pectin (0.025, 0.05, 0.08, 0.1, 0.2, 0.3, 0.5 and 0.8 wt%). Sample tubes were stored quiescently at ambient temperature for 7 days and the appearance with respect to serum separation was observed: (a) emulsions prepared and stored at neutral pH conditions (repulsive casein–pectin interactions, depletion flocculation); (b) emulsions prepared at pH = 7, acidified to pH ≈ 4 (over several hours) with glucono-δ-lactone, and then stored at low pH (favouring attractive casein–pectin interactions and emulsion bridging flocculation). The schematic diagrams illustrate the two flocculation mechanisms. (Adapted from ref. 95.) | ||
Mixed biopolymer systems are offering exciting opportunities in the rapidly developing areas of nanotechnology and microencapsulation. Electrostatic interactions between polysaccharides and protein-coated surfaces may be beneficial or detrimental to colloid stability, depending on factors such as the interaction strength, the electrolyte conditions, and the polymer surface coverage.103–105 The potential benefits of electrostatic deposition are being exploited in the powerful ‘layer-by-layer’ approach106,107 to the stabilization of emulsions by mixed biopolymers.
In naturally occurring gums and stabilizers, the surface-active character and/or emulsifying ability of the polysaccharide component typically arises from the presence of a covalently bound protein moiety.85,103 A simple biomimetic analogue, having excellent interfacial and emulsion stabilizing properties, can be made by dry-heating a mixture of protein + polysaccharide, e.g., a freeze-dried mixture of β-lactoglobulin (or commercial whey protein) + dextran held at 60–80 °C for few hours.108,109 The excellent steric stabilizing of such hybrid biopolymers has been demonstrated theoretically using SCF theory.110 A practical advantage of covalently linked protein–polysaccharide conjugates over analogous electrostatic complexation is the maintenance of molecular structural integrity over a wide range of pH and electrolyte conditions, without complications from complex coacervation or biopolymer precipitation.105,111 Hence, for instance, a casein–maltodextrin conjugate can be made112 that remains soluble, and is a highly effective emulsifying agent, under the same acidic solutions conditions where the normal pure casein ingredient would become aggregated and functionally ineffective.
References
- R. Mezzenga, P. Schurtenberger, A. Burbidge and M. Michel, Nat. Mater., 2005, 4, 729 CrossRef CAS.
- E. Dickinson and G. Stainsby, Colloids in Food, Applied Science, London, 1982 Search PubMed.
- E. Dickinson and D. J. McClements, Advances in Food Colloids, Blackie, Glasgow, 1995 Search PubMed.
- Food Colloids, Biopolymers and Materials, ed. E. Dickinson and T. van Vliet, Royal Society of Chemistry, Cambridge, 2003 Search PubMed.
- D. J. McClements, Food Emulsions, CRC Press, Boca Raton, FL, 2nd edn, 2005 Search PubMed.
- Food Colloids: Interactions, Microstructure and Processing, ed. E. Dickinson, Royal Society of Chemistry, Cambridge, 2005 Search PubMed.
- D. S. Horne, Int. Dairy J., 1998, 8, 171 CrossRef CAS.
- E. Dickinson, Int. Dairy J., 1999, 9, 305 CrossRef CAS.
- T. F. Kumosinski, E. M. Brown and H. M. Farrell, Jr., J. Dairy Sci., 1991, 74, 2889 CrossRef CAS.
- T. F. Kumosinski, E. M. Brown and H. M. Farrell, Jr., J. Dairy Sci., 1993, 76, 931 CrossRef CAS.
- Y. D. Livney, A. L. Schwan and D. G. Dalgleish, J. Dairy Sci., 2004, 87, 3638 CrossRef CAS.
- D. G. Schmidt, in Developments in Dairy Chemistry, ed. P. F. Fox, Applied Science, London, 1982, p. 61 Search PubMed.
- C. G. de Kruif and C. Holt, in Advanced Dairy Chemistry—Proteins, ed. P. F. Fox and P. L. H. McSweeney, Kluwer/Plenum, New York, 3rd edn, 2003, vol. 1, p. 213 Search PubMed.
- H. M. Farrell, Jr., E. L. Malin, E. M. Brown and P. X. Qi, Curr. Opin. Colloid Interface Sci., 2006 DOI:10.1016/j.cocis.2005.11.005.
- D. S. Horne, Curr. Opin. Colloid Interface Sci., 2006 DOI:10.1016/j.cocis.2005.11.004 , in press.
- E. L. Parkinson, R. Ettelaie and E. Dickinson, Biomacromolecules, 2005, 6, 3018 CrossRef CAS.
- A. C. Balazs, Acc. Chem. Res., 1993, 26, 63 CrossRef CAS.
- E. Dickinson and Y. Matsumura, Colloids Surf., B, 1994, 3, 1 CrossRef CAS.
- E. Dickinson, J. Dairy Sci., 1997, 80, 2607 CrossRef CAS.
- E. Dickinson, J. Chem. Soc., Faraday Trans., 1998, 94, 1657 RSC.
- E. Dickinson, D. S. Horne, J. S. Phipps and R. M. Richardson, Langmuir, 1993, 9, 242 CrossRef CAS.
- P. J. Atkinson, E. Dickinson, D. S. Horne and R. M. Richardson, J. Chem. Soc., Faraday Trans., 1995, 91, 2973 Search PubMed.
- T. Kull, F. Nylander, F. Tiberg and N. M. Wahlgren, Langmuir, 1997, 13, 5141 CrossRef CAS.
- N. Puff, A. Cagna, V. Aguié-Béghin and R. Douillard, J. Colloid Interface Sci., 1997, 208, 405.
- F. A. M. Leermakers, P. J. Atkinson, E. Dickinson and D. S. Horne, J. Colloid Interface Sci., 1996, 178, 681 CrossRef CAS.
- E. Dickinson, D. S. Horne, V. J. Pinfield and F. A. M. Leermakers, J. Chem. Soc., Faraday Trans., 1997, 93, 425 RSC.
- G. J. Fleer, M. A. Cohen Stuart, J. M. H. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at Interfaces, Chapman & Hall, London, 1993 Search PubMed.
- T. Milner and T. A. Witten, Macromolecules, 1992, 25, 5495 CrossRef CAS.
- E. Dickinson, M. G. Semenova and A. S. Antipova, Food Hydrocolloids, 1998, 12, 227 CrossRef CAS.
- E. Dickinson, V. J. Pinfield, D. S. Horne and F. A. M. Leermakers, J. Chem. Soc., Faraday Trans., 1997, 93, 1785 RSC.
- H. Casanova and E. Dickinson, J. Agric. Food Chem., 1998, 46, 72 CrossRef CAS.
- E. Dickinson, J. Chem. Soc., Faraday Trans., 1997, 93, 2297 RSC.
- S. R. Euston, S. R. Finnigan and R. L. Hirst, Food Hydrocolloids, 2000, 14, 155 CrossRef CAS.
- H.-J. Kim, E. A. Decker and D. J. McClements, Langmuir, 2002, 18, 7577 CrossRef CAS.
- E. Dickinson and E. L. Parkinson, Int. Dairy J., 2004, 14, 635 CrossRef CAS.
- E. L. Parkinson and E. Dickinson, Colloids Surf., B, 2004, 39, 23 CrossRef CAS.
- E. L. Parkinson and E. Dickinson, Int. Dairy J., 2006 DOI:10.1016/j.idairyj.2006.01.010.
- J. A. de Feijter, J. Benjamins and M. Tamboer, Colloids Surf., 1987, 27, 243 CrossRef CAS.
- J.-L. Courthaudon, E. Dickinson and D. G. Dalgleish, J. Colloid Interface Sci., 1991, 145, 390 CrossRef CAS.
- J.-L. Courthaudon, E. Dickinson and W. W. Christie, J. Agric. Food Chem., 1991, 39, 1365 CrossRef CAS.
- S. E. Euston, H. Singh, P. A. Munro and D. G. Dalgleish, J. Food Sci., 1995, 60, 1124 CrossRef CAS.
- N. M. Barfod, N. Krog, G. Larsen and W. Buchheim, Fett Wiss. Technol., 1991, 93, 24 Search PubMed.
- J.-L. Gelin, L. Poyen, J.-L. Courthaudon, M. Le Meste and D. Lorient, Food Hydrocolloids, 1994, 8, 299 CrossRef CAS.
- S. Kerstens, B. S. Murray and E. Dickinson, J. Colloid Interface Sci., 2006, 296, 332 CrossRef CAS.
- E. Dickinson and S.-T. Hong, J. Agric. Food Chem., 1995, 43, 2560 CrossRef CAS.
- J. Chen and E. Dickinson, J. Agric. Food Chem., 1998, 46, 91 CrossRef CAS.
- E. Dickinson and J. Chen, J. Dispersion Sci. Technol., 1999, 20, 197 CrossRef CAS.
- J. Chen, E. Dickinson, M. Langton and A.-M. Hermansson, Lebensm.-Wiss. Technol., 2000, 33, 299 CrossRef CAS.
- C. M. Wijmans and E. Dickinson, Langmuir, 1999, 15, 8344 CrossRef CAS.
- L. A. Pugnaloni, R. Ettelaie and E. Dickinson, Colloids Surf., B, 2003, 31, 149 CrossRef CAS.
- L. A. Pugnaloni, E. Dickinson, R. Ettelaie, A. R. Mackie and P. J. Wilde, Adv. Colloid Interface Sci., 2004, 107, 27 CrossRef CAS.
- C. M. Wijmans and E. Dickinson, Langmuir, 1998, 14, 7278 CrossRef CAS.
- C. M. Wijmans and E. Dickinson, Phys. Chem. Chem. Phys., 1999, 1, 2141 RSC.
- A. R. Mackie, A. P. Gunning, P. J. Wilde and V. J. Morris, J. Colloid Interface Sci., 1999, 210, 157 CrossRef CAS.
- A. R. Mackie and P. J. Wilde, Adv. Colloid Interface Sci., 2005, 117, 3 CrossRef CAS.
- B. S. Murray and E. Dickinson, Food Sci. Technol. Int., Tokyo, 1996, 2, 131 CrossRef CAS.
- S. Roth, B. S. Murray and E. Dickinson, J. Agric. Food Chem., 2000, 48, 1491 CrossRef CAS.
- J. T. Petkov, T. D. Gurkov, B. E. Campbell and R. P. Borwankar, Langmuir, 2000, 16, 3703 CrossRef CAS.
- M. A. Bos and T. van Vliet, Adv. Colloid Interface Sci., 2001, 91, 437 CrossRef CAS.
- G. B. Bantchev and D. K. Schwartz, Langmuir, 2003, 19, 2673 CrossRef CAS.
- E. M. Freer, K. S. Yim, G. G. Fuller and C. J. Radke, J. Phys. Chem. B, 2004, 108, 3835 CrossRef CAS.
- P. Cicuta and E. M. Terentjev, Eur. Phys. J. E, 2005, 16, 147 CrossRef CAS.
- P. A. Wierenga, H. Kosters, M. R. Egmond, A. G. J. Voragen and H. H. J. de Jongh, Adv. Colloid Interface Sci., 2006, 119, 131 CrossRef CAS.
- N. E. Hotrum, M. A. Cohen Stuart, T. van Vliet and G. A. van Aken, Langmuir, 2003, 19, 10210 CrossRef CAS.
- L. A. Pugnaloni, R. Ettelaie and E. Dickinson, J. Colloid Interface Sci., 2005, 287, 401 CrossRef CAS.
- L. A. Pugnaloni, R. Ettelaie and E. Dickinson, Langmuir, 2004, 20, 6096 CrossRef CAS.
- S. Huang, K. Minami, H. Sakaue, S. Shingubara and T. Takahagi, Langmuir, 2004, 20, 2274 CrossRef CAS.
- E. Dickinson, C. Ritzoulis and M. J. W. Povey, J. Colloid Interface Sci., 1999, 212, 466 CrossRef CAS.
- E. Dickinson, M. Golding and M. J. W. Povey, J. Colloid Interface Sci., 1997, 185, 515 CrossRef CAS.
- E. Dickinson and M. Golding, Food Hydrocolloids, 1997, 11, 13 CrossRef CAS.
- S. J. Radford and E. Dickinson, Colloids Surf., A, 2004, 238, 71 CrossRef CAS.
- Y. Cao, E. Dickinson and D. J. Wedlock, Food Hydrocolloids, 1991, 5, 443 CrossRef CAS.
- T. Moschakis, B. S. Murray and E. Dickinson, J. Colloid Interface Sci., 2005, 284, 714 CrossRef CAS.
- K. Koczo, A. D. Nikolov, D. T. Wasan, R. P. Borwankar and A. Gonsalves, J. Colloid Interface Sci., 1996, 178, 694 CrossRef CAS.
- E. Dickinson and M. Golding, J. Colloid Interface Sci., 1998, 197, 133 CrossRef CAS.
- S. J. Radford, E. Dickinson and M. Golding, J. Colloid Interface Sci., 2004, 274, 673 CrossRef CAS.
- E. Dickinson, S. J. Radford and M. Golding, Food Hydrocolloids, 2003, 17, 211 CrossRef CAS.
- E. Dickinson and M. Golding, Colloids Surf., A, 1998, 144, 167 CrossRef CAS.
- C. Eliot and E. Dickinson, Int. Dairy J., 2003, 13, 679 CrossRef CAS.
- E. Dickinson, Colloids Surf., A, 2006 DOI:10.1016/j.colsurfa.2006.01.012.
- M. Srinivasan, H. Singh and P. A. Munro, Food Hydrocolloids, 2000, 14, 497 CrossRef CAS.
- E. Dickinson, in Food Structure—Its Creation and Evaluation, ed. J. M. V. Blanshard and J. R. Mitchell, Butterworths, London, 1988, p. 41 Search PubMed.
- T. Moschakis, B. S. Murray and E. Dickinson, Langmuir, 2006, 22, 4710 CrossRef CAS.
- E. Dickinson, M. I. Goller and D. J. Wedlock, Colloids Surf., A, 1993, 75, 195 CrossRef CAS.
- E. Dickinson, Food Hydrocolloids, 2003, 17, 25 CrossRef CAS.
- E. Dickinson, in Gums and Stabilisers for the Food Industry—12, ed. P. A. Williams and G. O. Phillips, Royal Society of Chemistry, Cambridge, 2004, p. 394 Search PubMed.
- A. Parker, P. A. Gunning, K. Ng and M. M. Robins, Food Hydrocolloids, 1995, 9, 333 CrossRef CAS.
- H. Quian, M. P. Sheetz and E. L. Elson, Biophys. J., 1991, 60, 910 Search PubMed.
- A. Kusimi, Y. Sako and M. Yamamoto, Biophys. J., 1993, 65, 2021 CrossRef CAS.
- F. C. MacKintosh and C. F. Schmidt, Curr. Opin. Colloid Interface Sci., 1999, 4, 300 CrossRef CAS.
- A. Mukhoppadhyay and S. Granick, Curr. Opin. Colloid Interface Sci., 2001, 6, 423 Search PubMed.
- M. T. Valentine, Z. E. Perlman, M. L. Gardel, J. H. Shin, P. Matsudaira, T. J. Mitchison and D. A. Weitz, Biophys. J., 2004, 86, 2021.
- H. Tanaka, Y. Nishikawa and T. Koyama, J. Phys.: Condens. Matter, 2005, 17, L143 CrossRef CAS.
- E. Dickinson, M. G. Semenova, A. S. Antipova and E. G. Pelan, Food Hydrocolloids, 1998, 12, 425 CrossRef CAS.
- L. Matia-Merino and E. Dickinson, in Gums and Stabilisers for the Food Industry—12, ed. P. A. Williams and G. O. Phillips, Royal Society of Chemistry, Cambridge, 2004, p. 461 Search PubMed.
- C. Bonnet, M. Corredig and M. Alexander, J. Agric. Food Chem., 2005, 53, 8600 CrossRef CAS.
- J. Chen, E. Dickinson and M. Edwards, J. Texture Stud., 1999, 30, 377 Search PubMed.
- J. Chen and E. Dickinson, Int. Dairy J., 2000, 10, 541 CrossRef CAS.
- E. Dickinson and L. Matia-Merino, Food Hydrocolloids, 2002, 16, 321 CrossRef CAS.
- S. Gohtani, C. Ritzoulis and E. Dickinson, in Food Colloids, Biopolymers and Materials, ed. E. Dickinson and T. van Vliet, Royal Society of Chemistry, Cambridge, 2003, p. 100 Search PubMed.
- A. M. Puertas, M. Fuchs and M. E. Cates, Phys. Rev. Lett., 2002, 88, 098301 CrossRef.
- H. Tanaka, S. Jabbari-Farouji, J. Meunier and D. Bonn, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 021402 Search PubMed.
- E. Dickinson, in Food Polysaccharides and their Applications, ed. A. M. Stephen, Marcel Dekker, New York, 1995, p. 501 Search PubMed.
- D. G. Dalgleish and A.-L. Hollocou, in Food Colloids: Proteins, Lipids and Polysaccharides, ed. E. Dickinson and B. Bergenståhl, Royal Society of Chemistry, Cambridge, 1997, p. 236 Search PubMed.
- E. Dickinson, Trends Food Sci. Technol., 1998, 9, 347 CrossRef CAS.
- D. J. McClements, Langmuir, 2005, 21, 9777 CrossRef CAS.
- D. J. McClements, T. Aoki, E. A. Decker, Y.-S. Gu, D. Guzey, H.-J. Kim, U. Klinkesorn, L. Moreau, S. Ogawa and P. Tanasukam, in Food Colloids: Interactions, Microstructure and Processing, ed. E. Dickinson, Royal Society of Chemistry, Cambridge, 2005, p. 326 Search PubMed.
- E. Dickinson and V. B. Galazka, Food Hydrocolloids, 1991, 5, 281 CrossRef CAS.
- M. Akhtar and E. Dickinson, Colloids Surf., B, 2003, 31, 125 CrossRef CAS.
- R. Ettelaie, B. S. Murray and E. L. James, Colloids Surf., B, 2003, 31, 195 CrossRef CAS.
- C. G. de Kruif, F. Weinbreck and R. de Vries, Curr. Opin. Colloid Interface Sci., 2004, 9, 340 Search PubMed.
- R. Shepherd, A. Robertson and D. Ofman, Food Hydrocolloids, 2000, 14, 281 CrossRef CAS.
Footnote |
| † Based on the author’s Rideal Lecture, delivered in London on 11th April 2006, at a symposium on ‘Colloid Science of Mixed Ingredients’ organized jointly by the SCI Colloid and Surface Chemistry Group (UK) and the RSC Colloid and Interface Science Group (UK). |
| This journal is © The Royal Society of Chemistry 2006 |