RAFT mediated polymerisation in heterogeneous media
J. B.
McLeary
a and
B.
Klumperman
*ab
aUNESCO Centre for Macromolecules and Materials, Department of Chemistry and Polymer Science, University of Stellenbosch, Private Bag X1, Matieland 7602, South Africa
bLaboratory of Polymer Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: l.klumperman@tue.nl; Fax: +31 (40) 2463966; Tel: +31 (40) 2472339
First published on 28th November 2005
Abstract
Reversible addition fragmentation chain transfer (RAFT) mediated polymerisation is one of a number of living radical polymerisation processes that were developed over the last decade. The RAFT process is facilitating the synthesis of controlled macromolecular architectures via radical polymerisation in homogeneous and heterogeneous media. Here we discuss the progress in the use of the RAFT process, the past, the state of the art and potential directions for the future with specific emphasis on heterogeneous reactions.
 James McLeary | James McLeary completed his doctoral studies under the leadership of Bert Klumperman in 2004. His primary research interests are the mechanism and applications of the RAFT process. James is currently working for MONDI business Paper (SA) as a research project manager based at the University of Stellenbosch. |
 Bert Klumperman | Bert Klumperman obtained his MSc degree at the University of Twente (The Netherlands) in 1985. He worked at DSM Research from 1985–1994, and obtained his PhD degree in 1994 under the supervision of Prof. A.L. German (Eindhoven University) and Prof. K.F. O'Driscoll (University of Waterloo, Canada). Since 1995 he has been employed by the Eindhoven University (The Netherlands) where he currently holds a position as Associate Professor. Next to that he holds a part-time position as Extraordinary Professor at the University of Stellenbosch (South Africa) since 1998. |
1 Introduction
The use of radical polymerisation for the efficient design of materials has traditionally been limited due to some characteristic features of this technique. The continuous generation of new chains in the initiation process, and the concomitant formation of dead chains in chain transfer and bimolecular termination processes, leads to polymers with broad molar mass distributions. The synthesis of polymers with more complex architectures is restricted due to the same phenomena. On the other hand, radical polymerisation is quite popular for the large-scale synthesis of polymers due to high rates of reaction and tolerance to functional groups and impurities. Radical polymerisation in heterogeneous media is commonly applied in industry, where emulsion polymerisation is in widespread use for the production of latex-based products. Emulsion polymerisation has the added advantages of environmental friendliness, excellent heat dissipation through the water phase and high rates of reaction due to radical compartmentalisation effects.Over the last decade the development of effective modifications of the free radical polymerisation process1–3 has allowed the technique to become a useful polymer construction tool without losing the benefits inherent to radical chemistry. The modifications of the free radical polymerisation process are known collectively as either living or controlled radical polymerisation techniques.4 All of these techniques have the ability to create a dormant chain by reversibly end-capping the radical species.
2 The RAFT process
The reversible addition fragmentation chain transfer (RAFT) process3,5,6 is probably the most versatile living radical polymerisation4,7,8 process.In Scheme 1 the elementary reactions for the central exchange process of the RAFT mechanism are depicted. The relatively stable intermediate radical that is formed (for some RAFT agent/monomer combinations) by the addition process can fragment to release one of two radical species. These are the original incoming radical species (Pm˙, a propagating radical of degree of polymerisation m), or the homolytic leaving group (Pn˙) that was previously a part of the RAFT agent.
 | ||
| Scheme 1 The elementary RAFT equilibrium | ||
Some terminology has become common in referring to specific portions of RAFT agents. The stabilizing group is commonly refered to as the Z group and the leaving groups are generically classed as R groups.
The RAFT process allows polymers of narrow molecular weight distribution and advanced architecture to be prepared. These materials include star,9–12 comb13,14 or brush structures, and end-functional polymers with application as telechelic materials.15
One of the interesting advantages that is offered by the RAFT technique is the ability to form architectures that can easily be disassembled for detailed study. The labile dithioester functionality is readily removed.16,17 This means that depending on whether a multifunctional R (leaving) or Z (stabilizing) group is utilized a polymer of similar structure may be prepared but of different stability. The disassembly approach has been followed by a number of authors to demonstrate the successful synthesis of complex macromolecular designs12,18,19 by disassembly and analysis of the building blocks of the macrostructure.
3 Surfaces
Heterogeneous polymerisations cover a number of classes including the modification of insoluble materials. Surface initiated polymerisation to provide modified materials, whether organic20–23 or inorganic surfaces24,25 is a field in which the RAFT process is being widely applied. The ability to supply a surface layer of a multiblock copolymer provides materials that are highly versatile. One of the problems that is typically encountered in surface intiated polymerisation is the restriction of the growing radical centres to a limited volume and the increased termination that occurs as a result. Techniques that restrict the radical concentration in the reaction such as atom transfer radical polymerisation (ATRP) have traditionally been more applicable in this environment. However, an unexpected phenomenon was discovered in polymerisations mediated by dithiobenzoates. This surprising phenomenon is rate retardation as a result of a reduced propagating radical concentration.26–28 For the purposes of surface initiated polymerisation, this is an unexpected, but pleasant advantage. However, one of the advantages of the RAFT procedure as conceived by the CSIRO group was the fact that the rate of polymerisation should ideally not be affected by the addition of a RAFT agent to a reaction.4 Two schools of thought
A critical issue with the development of every new technique is an in depth understanding of the mechanisms by which the process operates. The RAFT process as originally published was conceptually elementary, but additions have been required to explain the laboratory results which have led to two schools of thought developing with quite different understandings of the process.26,27,29–31 The difference in opinion as to the origin of the rate retardation is due to the fact that electron spin resonance determinations of the concentration of intermediate radicals in RAFT mediated polymerisations have not proven acceptable to all researchers in the field. The discussion centres on the nature of the intermediate radical and the sulfur atoms that form part of its structure. The school that accepts the ESR results considers the rate retardation to be a function of radical termination of the intermediate radical species. The school that discounts the ESR evidence suggests that slow fragmentation of intermediate radicals leads to a buildup of intermediate radical species in polymerisations, resulting in a decreased propagating radical concentration.The two hypotheses can both be used to explain polymerisation results via modelling, however they are fundamentally incompatible. The discussion is ongoing in the literature.
The appearance of inhibition is also quite common in RAFT mediated polymerisations (Scheme 2). The slow fragmentation approach suggests that this behaviour is due to a buildup of intermediate radicals.28,32 Additional evidence has been presented that the initial steps of the RAFT process show quite different behaviour to the long chain behaviour of the process.33–35 The term initialization was introduced to describe the period in the polymerisation when conversion is driven by initial radicals, whether derived from the RAFT agent or from the initiator in the system. It has been shown that the differences in radical propagation rates can create the illusion of inhibition in polymerisations. The increased understanding of the RAFT process is gradually making the implementation of the RAFT process on an industrial scale more realistic.
![The RAFT process in a homopolymerization of styrene mediated by cumyl dithiobenzoate. Three experiments with identical reactant concentrations are combined in the scheme: in situ1H NMR Spectroscopic,2913C NMR spectroscopic30 and ESR spectroscopic30 experiments. The integration of signals provides a qualitative description of the behaviour within the polymerization reaction. The data presented in the scheme are supportive of the intermediate radical termination model. The decrease of the intermediate radical concentration (after the pre-equilibrium at circa 110 min) scales with [I]0.5. The integrations of the terminated species are integrations over an area of the spectrum and only provide an indication of the increase in concentration of these species.30](/image/article/2006/SM/b513575c/b513575c-s2.gif) | ||
| Scheme 2 The RAFT process in a homopolymerization of styrene mediated by cumyl dithiobenzoate. Three experiments with identical reactant concentrations are combined in the scheme: in situ1H NMR Spectroscopic,2913C NMR spectroscopic30 and ESR spectroscopic30 experiments. The integration of signals provides a qualitative description of the behaviour within the polymerization reaction. The data presented in the scheme are supportive of the intermediate radical termination model. The decrease of the intermediate radical concentration (after the pre-equilibrium at circa 110 min) scales with [I]0.5. The integrations of the terminated species are integrations over an area of the spectrum and only provide an indication of the increase in concentration of these species.30 | ||
The greatest advantage of the living radical polymerisation techniques is their ability to allow nanostructures to be constructed of highly specific architecture and chemical composition. One of the advantages of conventional free radical polymerisation is the ease with which it is applied to heterogeneous polymerisations. Heterogeneous polymerisations provide convenient and accessible routes to nanostructuring of materials. Combining the advantages of living and conventional free radical polymerisation, techniques such as suspension polymerisation have been pursued with the RAFT process.36 The most dominant heterogeneous technique is however emulsion polymerisation.
5 Emulsion polymerisation
Nanoparticulate polymers have traditionally been produced in emulsion polymerisation.37,38 Emulsion polymerisation consists of discrete nanoreactors, suspended in a continuous aqueous phase. These nanoreactors are formed during the early stages of the polymerisation, and polymerisation largely takes place in these nanoreactors. These polymerizing particles are stabilized by surface active molecules or surfactants. The initial research and theoretical background for emulsion polymerisation was laid by Harkins,39 Smith and Ewart,40 among others. The classical description of an emulsion polymerisation process, above the critical micelle concentration (CMC), for a monomer with low water solubility, begins with three intervals of polymerisation (Scheme 3).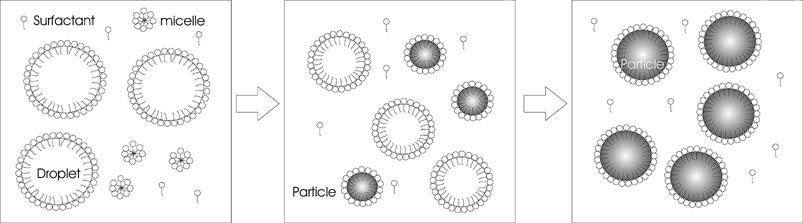 | ||
| Scheme 3 Emulsion intervals (I, II, III) showing the presence of droplets, micelles, free surfactant, nucleated particles and final latex particles. | ||
Interval I
Large monomer droplets are present in the reaction vessel with many small micelles (surfactant aggregates). Surface-active radicals generated in the water phase enter the micelles and continue to polymerize, creating a monomer diffusion gradient from the aqueous phase, leading to a gradual depletion of the droplets as they replenish the aqueous phase. The key aspects of interval I are that there are micelles and droplets present, the particle number and particle size are increasing, and the rate of the reaction is increasing.Interval II
Particle formation has ceased and micelles have disappeared either by becoming particles or by acting as monomer and surfactant reservoirs and being depleted by adsorption to the increasing surface area of the particles. The key aspects of interval II are that there are no longer micelles present, but monomer droplets are still present, although decreasing in size due to depletion by the aqueous phase, (which is being depleted by the particles). The particle number is constant, while the particle size is increasing, and the concentration of monomer within the droplet is constant. The rate of polymerisation during this interval is relatively constant.Interval III
No more monomer droplets are present. The remaining monomer is in the particles and is consumed without any dependence on transport phenomena. The key aspects of interval III are that there are no micelles or droplets, the particle size and particle numbers remain constant while the concentration of monomer within the droplets is decreasing and thus the rate of the reaction is decreasing. It should be noted that in the case of a glassy system, the rate of termination would decrease as diffusion decreases due to increased viscosity. It is possible that an autoacceleration will occur due to an increase in the number of radicals in the particle.This classical theory is typically true for monomers such as styrene where homogeneous nucleation above the critical micelle concentration (CMC) is limited. In the case of relatively water-soluble monomers such as methyl methacrylate, homogeneously nucleated particles play an important role, even above the CMC.
Structuring these materials via core–shell techniques, i.e., multiple layers of polymer being added discretely to the particles to form complex polymer beads has become critical for many applications such as impact modifiers,41 adhesives, coatings, biomedical applications, etc.
6 RAFT in emulsion
RAFT polymerisation, a transfer technique, appears to be the best choice for implementing living radical polymerisation in water borne systems, as it does not require significantly more stringent reaction conditions than ordinary free radical polymerisation. The Le et al.42 patent of the RAFT process includes a number of examples of RAFT in heterogeneous media. The guidelines provided for emulsion polymerisation suggested that the RAFT agent used should partition favourably to the organic phase while having sufficient aqueous phase solubility that transport between monomer droplets and particles was possible. The examples provided for emulsion polymerisation suggested that high instantaneous conversions—i.e. a removal of monomer droplets from the systems—were required for successful polymerisation. Successful emulsion polymerisation of butyl methacrylate with cumyl dithiobenzoate was also reported as a table entry in the initial RAFT paper.3The results reported by the CSIRO group however suggested that implementation in emulsion could be problematic. The conditions used required high instantaneous conversions and polymerisation in the presence of monomer droplets was avoided.42 Uzulina et al. reported attempts at RAFT mediated polymerisation of styrene, methyl methacrylate and vinyl acetate.43 Using an amide functional RAFT agent, styrene was successfully polymerized, albeit with PDI between 1.5 and 2. The other monomers used, proved less successful. The polydispersity of the polymer in the latex was much higher than should be expected from the RAFT process. The nature of the RAFT agent appeared critical as more hydrophobic RAFT agents proved unsuitable due to phase separation.
Monteiro et al. examined the seed polymerisation of styrene mediated by cumyl dithiobenzoate. They proposed that the excessive retardation observed was due to exit of the RAFT leaving group radicals from the seeded particles. They also observed large-scale phase separation of the monomer into RAFT end-capped oligomer rich phases.44 The transport of the RAFT agent as well as the RAFT agent solubility were discussed as possible influences on the success of the polymerisation.
Moad et al. later discussed some of the important factors for successful use of RAFT polymerisation in emulsion, however starved feed implementations in emulsion were still necessary for low polydispersity index polymer.45 Moad et al. also confirmed that the use of cumyl dithiobenzoate as RAFT agent in ab initio emulsion provided problems of inhomogeneous distribution of the RAFT agent.45 The aqueous phase transport of oil soluble RAFT agents to the polymerisation locus was dealt with by Prescott et al. who used acetone as a phase transfer agent.46 This allowed the successful growth of living polymer on a preexisting seed latex.
Charmot et al.47 and Monteiro et al.48–51 were able to prepare latex particles using xanthate agents, which operate via an inefficient RAFT mechanism for many monomers. There are plausible reasons for the fact that xanthates are capable of being used directly in emulsion systems. The most important is the fact that long chains are formed quite rapidly in the polymerisation and particle nucleation is consequently substantially more efficient. This phenomenon is termed “hybrid behavior” by some researchers.52 The control that is sought has however remained elusive unless special techniques are used, e.g. controlled-feed,42 or the use of phase transfer agents.46 The surface activity of xanthates (see the canonical forms in Scheme 4) has also been postulated as playing a significant role in the implementation of the RAFT process in emulsion.50
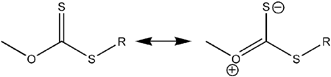 | ||
| Scheme 4 The canonical forms proposed for xanthate species that provide ionic character to these RAFT agents. | ||
A problem that was observed with RAFT, unlike other degenerative controlled polymerisation techniques in organic dispersions in aqueous media, was that phase separation occurred at the beginning of the polymerisation in both emulsion and miniemulsion systems.44,53 A number of reasons for this behaviour have been suggested, but it seems that the more important reasons include the transport of the RAFT agent to the polymerisation sites, the exit of short radicals from the polymerisation sites and the slow formation of higher molar mass polymer. In the case of emulsion polymerisation, there is another potential explanation that has not been published before.54 In a conventional (non-RAFT) emulsion polymerisation, micellar nucleation accounts for the largest fraction of particle formation due to the large difference in surface area between the two oil phase domains (micelles and monomer droplets). The primary reason for this is that entry events dominate the distribution of radicals between the different domains, as exit of radicals is substantially less probable than propagation beyond the critical length for exit. In the case of RAFT-mediated polymerisation, the probabilities are quite different. Polymer chains grow slowly, and have the opportunity to enter into and exit from a micelle many times. Growing chains that enter droplets are much less likely to exit than growing chains in micelles. The net result of this behaviour is that entry, which is dependent on the surface area differential between micelles and monomer droplets, is no longer the primary factor driving the distribution of growing chains between the different types of oil phase domains. Exit behaviour begins to dominate the distribution of growing chains. The distribution of the oligomers is now based rather on the volume fractions of the various organic phases, and no longer on the available surface area. Hence, the oligomers will end up in the monomer droplets, and not in the micelles. The transfer of monomer and RAFT agent to micelles is prevented by the increase in osmotic pressure generated by the formation of polymer in the droplets and as a result, micelles that do nucleate do not contain the RAFT agent. The behaviour can be seen as the difference between kinetic control (conventional emulsion polymerisation) and thermodynamic control (RAFT-mediated emulsion polymerisation). The formation of polymer and rate at which it occurs are crucial for emulsion polymerisation.
7 RAFT in miniemulsion
An aqueous heterogeneous polymerisation process that avoids transport issues is miniemulsion polymerisation.55–58The process of miniemulsion formation requires mixing of all the components of the emulsion followed by a preshearing step in which the droplet size is mechanically reduced to provide an emulsion with large droplets and a wide droplet size distribution (Scheme 5). At this point, ultrasonic homogenization is carried out via a fission–fusion process in such a manner that a narrow distribution of small droplet size is formed. After the application of the homogenization process, the system will relax to a homogenized state that is stablized against particle growth, and at this point the system can be polymerized.
 | ||
| Scheme 5 The five steps that occur during the formation of a miniemulsion latex: mixed surfactant and oil phase forming micelles as well as stabilized macro droplets, pre-sheared pre-emulsion with smaller droplets of various sizes, sonicated pre-emulsion with a fission–fusion process allowing the formation of a homogenous droplet size, homogenized droplets after relaxation, polymerized particles. Adapted from Landfester et al.57 | ||
For a miniemulsion to be formed, a significant amount of energy is required to predisperse the system to a theoretical minimum particle size for each specific surfactant concentration. The precise time to reach the minimum particle size for each experimental setup needs to be determined, as machine differences as well as other environmental variables play a role. To experimentally determine the dispersion period required involves surface tension or turbidity measurements with time. When these values have reached a constant level, (in the case of turbidity measurements, a maximum is reached, after which a decrease in turbidity occurs to reach a plateau of constant value, and in the case of surface tension the measurement reaches an upper limit after which there is no change), the system has reached equilibrium, and homogenization can be halted.59 At this point the particle stability is dependent on molecular diffusion degradation (Ostwald ripening, a process dependent on the individual particle) as well coalescence by collision (a process dependent on two particles).
The Le et al.42 patent of the RAFT process includes examples for miniemulsion polymerisation but these were only reported below 40% conversion. Moad et al. discussed some of the important factors for successful use of RAFT polymerisation in miniemulsion, however the conditions provided did not lead to an improvement in implementation with conversions below 30% reported.45
Monteiro et al.53 provided detailed studies of efficient RAFT agents in miniemulsion polymerisation, observing a number of difficulties. The polymerisations of styrene showed a linear increase of molecular weight until approximately 30% conversion after which deviations from the expected values became excessive. Butyl methacrylate and (2-ethyl)hexyl methacrylate were polymerized more successfully but still with unexpectedly large polydispersity indexes occurring in the polymer.53
The investigations into miniemulsion polymerisation mediated with dithiobenzoates led to the use of polymeric stabilizers which allowed the formation of block copolymers in heterogeneous media via an efficient RAFT process.60 Here the concept of two polymerisation mechanisms competing in one RAFT mediated reaction was introduced. The nature of the RAFT process meant that it was possible for particles containing dithioester end-capped polymers to be polymerized in the presence of particles that followed a conventional free radical polymerisation process. This provided latexes that contained two different distributions of particles and polymer.
Luo et al.61 provided a theoretical basis for the failure of the ionic surfactant stabilized polymerisations to provide living characteristics. Monomer diffusion in an emulsion system is governed by the monomer chemical potential difference between droplets and particles. After nucleation, the monomer chemical potential of a particle is lower than that of a monomer droplet due to the formation of polymer in the particle. The rate at which monomer diffusion occurs in the system will affect the rate at which particles increase in size, and the sizes that are reached in the system. Based on this model, Luo et al.61 predicted that emulsion systems would exhibit a super-swelling state if no early equilibrium was reached with respect to the chemical potential of droplets and particles. The model uses two concepts to explain instabilities in the reaction system:
• Super-swelling, which is the swelling of a droplet or particle in a rapid fashion to a size many times that of the original droplet.
• Swelling capacity, which is a measure of the relative amount of swelling in a system, i.e. a ratio of the droplet swollen size to unswollen size.
A simple display of the model is presented in Scheme 6. The droplet diameter was chosen as 60 nm. The weight percentage ultrahydrophobe was set at 4%, the fractional conversion in the particles was set at 0.1%. The temperature was set to 75 °C. The interaction parameters were taken from Landfester et al,61 and the monomer-ultrahydrophobe combination is methyl methacrylate and hexadecane. The RAFT agent to monomer ratio was set at 0.01.
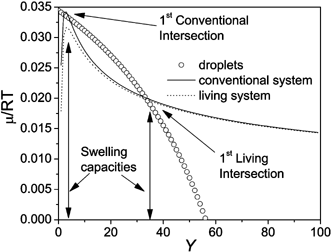 | ||
| Scheme 6 The chemical potential versus swelling capacity (Y) of droplets, conventional and living particles of 60 nm diameter containing 4 wt% cosurfactant at 0.1% conversion. | ||
If we examine the chemical potential plot produced by the model under these conditions there are three intersections of the chemical potentials of the conventional polymerisation without a controlling agent, and the droplet chemical potential as the swelling ratio Y increases. When the system arrives at the first intersection, the monomer transport is effectively stopped, as there is no longer a potential difference between the particles and the droplets. Luo et al. termed this the normal swelling state.
In the case of a living system however, the lower mixing energy in the case of the oligomeric chains produced in the living system leads to a lower chemical potential, and only a single intersection point is observed. An equilibrium swelling is reached at a much higher swelling capacity leading to a large amount of monomer transport between unnucleated droplets and particles. This “super swelling” effect could lead to particles large enough to be shear sensitive (1–10 µm),61 as well as a range of particle sizes making coagulation between particles by a heterocoagulation mechanism more probable.
Superswelling of the particles in the absence of high polymer also suggested that increased stabilization should lead to improvements in the performance of these systems.61 Luo et al. concluded that larger particles, higher cosurfactant levels and lower molecular weight controlling agent concentrations would all lead to a more stable latex. An interesting conclusion reached by Luo et al. was that nucleation efficiency was likely to be low. This was further addressed by Schork et al.62
The obstacles that were encountered by Monteiro et al. when using ionic surfactants were further explored by Butté et al. and Lansalot et al. but with mixed success.63,64 Specific agents were found to be suitable for the preparation of stable latexes, but best results were obtained when using a polymeric RAFT agent, which suggested that the instability problems were related to the exit characteristics of the leaving group radical species as well as the RAFT agents themselves.62
McLeary et al. implemented the recommendations of Luo et al. and found that ionically stabilized miniemulsions mediated by the RAFT process with reasonable conversions and low polydispersities could be prepared.65 Luo et al. implemented similar formulations, also showing successful polymerisation with predictable results.66
Some of the theoretical aspects of RAFT mediated miniemulsions have been explored by Tonge et al.67 Knowledge of the number of propagating radicals per particle in these systems appears to be a key factor in harnessing the potential of these systems. The two conflicting hypotheses on the nature of retardation in the RAFT process were examined and the possibility of very high numbers of intermediate radicals per particle (∼104) required by the slow fragmentation model was discounted. The results obtained suggested that intermediate radical termination or the lack thereof could allow unusual kinetic behaviour in RAFT mediated emulsions. If ![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) in the system is considered as the total reactive radical concentration and intermediate radical termination does not occur, “zero–one kinetics” may occur in systems with total radical numbers per particle exceeding 0.5. Tonge et al. investigated two different systems, one of which did not conform to “zero–one” type kinetics. The second system is reported as possibly “zero–one” but kinetic analysis was not conclusive.
in the system is considered as the total reactive radical concentration and intermediate radical termination does not occur, “zero–one kinetics” may occur in systems with total radical numbers per particle exceeding 0.5. Tonge et al. investigated two different systems, one of which did not conform to “zero–one” type kinetics. The second system is reported as possibly “zero–one” but kinetic analysis was not conclusive.
The fact that Tonge et al. showed that the number of RAFT agents per particle were insufficient to support the suggested intermediate radical concentration of the slow fragmentation model is indicative of the important role that heterogeneous polymerisation can play in the elucidation of the RAFT mediated mechanism of polymerisation.
Copolymerisation66 in miniemulsion is now being addressed with promising results. The improvement in behaviour of miniemulsions when using cyclodextrins as transport agents has also been reported by Zhang et al.68 though the surfactant concentration used for reference experiments was lower than recommended by McLeary et al.65
The major issues with traditional miniemulsion polymerisation are the high levels of surfactant and co-stabilizer used. To avoid these problems, it seems sensible to use compounds that are incorporated into the latex. The use of polymerizable surfactants69 and cosurfactants is a route that is still being investigated.
The RAFT technique expands the options for structured particulate polymers by adding molecular weight and distribution control to the molecular toolbox available for tailored particles.70
The ab initio polymerisation of a RAFT mediated emulsion however, remains elusive. Some of the specific problems are known. Conventional emulsion polymerisations often exhibit inhibition periods in which very little monomer is consumed. Inhibition is a common artifact in aqueous based systems due to dissolved oxygen, which acts as an inhibitor, or trace inhibitors added for stabilisation of monomers.71 Once the period is complete however, the formation of particles is no different from an emulsion system that does not have an inhibition period. RAFT-mediated emulsion polymerisation has more significant inhibition than conventional emulsion polymerisation.
Butté et al. showed that the RAFT process should prove ideal for implementation in heterogeneous media, but suggested that although it was feasible in miniemulsion, obtaining the correct conditions, i.e., a rapid nucleation of particles in relation to RAFT agent diffusion, and a rapid diffusion of RAFT agent in relation to reaction time, for ab initio emulsion implementation would be complex.64
8 Retardation in RAFT mediated reactions
Retardation in polymerisation rate is observed with dithiobenzoate RAFT agents in miniemulsion and emulsion in much the same fashion as occurs in homogeneous media, although the retardation can be more pronounced due to the rapid rates of polymerisation typically expected in this medium.45,63 When the RAFT agent Z-group is replaced with a less stabilizing functionality than the phenyl ring, it is possible to obtain rates of polymerisation that are comparable to control polymerisations (although a reduction in gel effect is typically noticable).63,72The critical aspect for the implementation of living radical polymerisation techniques in heterogeneous aqueous media remains the rate of chain length development. Although polymerisation rates can approach those of conventional polymerisations, the relatively slow development of polymer with sufficient degrees of polymerisation to stabilize particles continues to be an obstacle to the implementation of heterogeneous polymerisation processes.
In the RAFT process it is also possible that rapid transfer of the radical leads to the generation of short leaving groups initially in the polymerisations and at later periods to a dominance of long chain radicals in the polymerisations. The short chains that are initially present are able to exit the particles quite easily, leading to a reduced rate of polymerisation. As has been pointed out, this will affect termination kinetics substantially.73
The use of gamma irradiation74 and simulation75 allowed Prescott et al. to determine that the chain length behaviour in RAFT mediated emulsion polymerisations differed substantially from conventional polymerisations with the propagating radical population consisting predominantly of long chains. The kinetic implications of this include slower termination and higher average numbers of radicals per particle.
9 Preventing exit?
In many successful implementations of the RAFT process in hetereogeneous aqueous media it has been necessary to prepolymerize in a homogeneous system and then gradually convert to a heterogeneous polymerisation. Vosloo et al. followed this methodology in a dithiobenzoate mediated styrene emulsion polymerisation.76Recently a number of papers have focused on the use of homogeneous polymerisation of a water soluble monomer and then feeding a second, water insoluble, monomer to the polymerisation to create a RAFT seed particle. This process has been used to create seeds for miniemulsion as well as emulsion polymerisation.77,72 The use of the homogeneous polymerisation and controlled feed system to provide a RAFT agent endcapped polymeric surfactant that can be chain extended in heterogeneous media provides an elegant route to avoiding uncontrolled particle formation.70 There are however still potential drawbacks to this technique. These include the the restrictions on the RAFT agent concentration, which is intertwined with the particle stabilization.
10 Future trends
The use of heterogeneous aqueous media is not the only environmentally friendly environment that has been explored for the production of RAFT mediated polymers. Ionic liquids, which are widely considered to be a “green” alternative78 due to their low volatility have also been investigated.79 Ionic liquids have drawbacks in terms of product recovery and polymer solubility that have yet to be solved, although distillable ionic liquids seem promising,80 but ionic liquids will possibly provide the long term solution to homogeneous polymerisation industrial and environmental requirements.A development that is beginning to generate interest in the field of radical polymerisation is the incorporation of not only chain length and chain length distribution control into the polymers that are being produced, but to simultaneously produce polymers of a specific stereochemistry. It is important to note that prior to the development of tacticity control in propylene polymerisation, this essential industrial monomer was very restricted in its use.81 In the case of the RAFT process tacticity control has been approached through the use of lewis acids.82–84 The use of fluoro alcohols as reaction medium for living radical polymerisation also appears to have promise.85
The potential to implement heterogeneous polymerisations in ionic liquids86,87 while controlling tacticity through the addition of other reagents suggests that radical polymerisation is on the edge of a major step forward.
This review has addressed the state of the art in the implementation of the RAFT process in heterogeneous media for the design of structured materials. The RAFT process continues to show the versatility that is required of an industrial process and as understanding and implementation continue to progress the large scale industrialization of this process seems ever more likely.
Acknowledgements
JM acknowledges the Dutch Polymer Institute, the Harry Crossley foundation and the National Research Foundation of South Africa for financial support. BK acknowledges Kris Matyjaszewski for discussions that led to the insight in emulsion destabilization during RAFT polymerisation as indicated in the text.References
- C. J. Hawker, Acc. Chem. Res., 1997, 30, 373–382 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- T. R. Darling, T. P. Davis, M. Fryd, A. A. Gridnev, D. M. Haddleton, S. D. Ittel, R. R. J. Matheson, G. Moad and E. Rizzardo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1706–1708 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- J. Jagur-Grdozinski, React. Funct. Polym., 2001, 49, 1–54 CrossRef CAS.
- E. L. Madruga, Prog. Polym. Sci., 2002, 27, 1879–1924 CrossRef CAS.
- M. Chen, K. P. Ghiggino, A. Launikonis, A. W. H. Mau, E. Rizzardo, W. H. F. Sasse, S. H. Thang and G. J. Wilson, J. Mater. Chem., 2003, 13, 2696–2700 RSC.
- V. Darcos, A. Dureault, D. Taton, Y. Gnanou, P. Marchand, A.-M. Caminade, J.-P. Majoral, M. Destarac and F. Leising, Chem. Commun., 2004, 2110–2111 RSC.
- H. T. Lord, J. F. Quinn, S. D. Angus, M. Whittaker, M. H. Stenzel and T. P. Davis, J. Mater. Chem., 2003, 13, 2819–2824 RSC.
- R. T. A. Mayadunne, J. Jeffery, G. Moad and E. Rizzardo, Macromolecules, 2003, 36, 1505–1513 CrossRef CAS.
- J. F. Quinn, R. P. Chaplin and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2956–2966 CrossRef CAS.
- H. Shinoda and K. Matyjaszewski, Macromol. Rapid Commun., 2001, 22, 1176–1181 CrossRef CAS.
- V. Lima, X. Jiang, J. Brokken-Zijp, P. J. Schoenmakers, B. Klumperman and R. Van Der Linde, J. Polym. Sci., Part A: Polym. Chem., 2004, 43, 959–973.
- A. Postma, K. A. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38, 5371–5374 CrossRef CAS.
- G. Moad, B. Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2004, 1–11.
- R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 2002, 43, 6811–6814 CrossRef CAS.
- M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4498–4512 CrossRef CAS.
- M. Jesberger, L. Barner, M. H. Stenzel, E. Malmström, T. P. Davis and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3847–3861 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365–375 CrossRef CAS.
- L. Barner, N. Zwaneveld, P. Senake, Y. Pham and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4180–4192 CrossRef CAS.
- L. Ying, W. H. Yu, E. T. Kang and K. G. Neoh, Langmuir, 2004, 20, 6032–6040 CrossRef CAS.
- Y. Tsujii, M. Ejaz, K. Sato, A. Goto and T. Fukuda, Macromolecules, 2001, 34, 8872–8878 CrossRef CAS.
- M. Baum and W. J. Brittain, Macromolecules, 2001, 35, 610–615.
- Y. Kwak, A. Goto, Y. Tsujii, Y. Murata, K. Komatsu and T. Fukuda, Macromolecules, 2002, 35, 3026–3029 CrossRef CAS.
- M. J. Monteiro and H. de Brouwer, Macromolecules, 2000, 34, 349–352 CrossRef.
- P. Vana, T. P. Davis and C. Barner-Kowollik, Macromol. Theory Simul., 2002, 11, 823–835 CrossRef CAS.
- C. Barner-Kowollik, M. L. Coote, T. P. Davis, L. Radom and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2828–2832 CrossRef CAS.
- A. R. Wang, S. Zhu, Y. Kwak, A. Goto, T. Fukuda and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2833–2839 CrossRef CAS.
- C. Barner-Kowollik, J. F. Quinn, D. R. Morsley and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1353–1365 CrossRef CAS.
- S. Perrier, C. Barner-Kowollik, J. F. Quinn, P. Vana and T. P. Davis, Macromolecules, 2002, 35, 8300–8306 CrossRef CAS.
- J. B. McLeary, J. M. McKenzie, M. P. Tonge, R. D. Sanderson and B. Klumperman, Chem. Commun., 2004, 1950–1951 RSC.
- J. B. McLeary, F. M. Calitz, J. M. McKenzie, M. P. Tonge, R. D. Sanderson and B. Klumperman, Macromolecules, 2004, 37, 2383–2394 CrossRef CAS.
- J. B. McLeary, F. M. Calitz, J. M. McKenzie, M. P. Tonge, R. D. Sanderson and B. Klumperman, Macromolecules, 2005, 38, 3151–3161 CrossRef CAS.
- J. D. Biasutti, T. P. Davis, F. P. Lucien and J. P. A. Heuts, J. Polym. Sci., Part A: Polym. Chem., 2004, 43, 2001–2012.
- M. J. Monteiro and J. de Barbeyrac, Macromol. Rapid Commun., 2002, 23, 370–374 CrossRef CAS.
- S. Kirsch, A. Doerk, E. Bartsch, H. Sillescu, K. Landfester, H. W. Spiess and W. Maechtle, Macromolecules, 1999, 32, 4508–4518 CrossRef CAS.
- W. D. Harkins, J. Am. Chem. Soc., 1947, 69, 1428 CrossRef CAS.
- W. V. Smith and R. H. Ewart, J. Chem. Phys., 1952, 16, 592.
- K. Sparnacci, L. Tondelli and M. Laus, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3347–3354 CrossRef CAS.
- T. P. Le, G. Moad, E. Rizzardo and S. H. Thang, PCT Int. Appl., 1998 Search PubMed.
- I. Uzulina, S. Kanagasabapathy and J. Claverie, Macromol. Symp., 2000, 150, 33–38 CrossRef CAS.
- M. J. Monteiro, M. Hodgson and H. de Brouwer, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3864–3874 CrossRef CAS.
- G. Moad, J. Chiefari, B. Y. Chong, J. Krstina, R. T. Mayadunne, A. Postma, E. Rizzardo and S. H. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- S. W. Prescott, M. J. Ballard, E. Rizzardo and R. G. Gilbert, Macromolecules, 2002, 35, 5417–5425 CrossRef CAS.
- D. Charmot, P. Corpart, H. Adam, S. Z. Zard and B. G. Biadatti, Macromol. Symp., 2000, 150, 23–32 CrossRef CAS.
- M. J. Monteiro, M. Sjoberg, J. Van Der Vlist and C. M. Gottgens, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4206–4217 CrossRef CAS.
- M. J. Monteiro and J. de Barbeyrac, Macromolecules, 2001, 34, 4416–4423 CrossRef CAS.
- W. Smulders, R. G. Gilbert and M. J. Monteiro, Macromolecules, 2003, 36, 4309–4318 CrossRef CAS.
- W. Smulders and M. J. Monteiro, Macromolecules, 2004, 37, 4474–4483 CrossRef CAS.
- C. Barner-Kowollik, J. F. Quinn, T. L. U. Nguyen, J. P. A. Heuts and T. P. Davis, Macromolecules, 2001, 34, 7849–7857 CrossRef CAS.
- J. G. Tsavalas, F. J. Schork, H. de Brouwer and M. J. Monteiro, Macromolecules, 2001, 34, 3938–3946 CrossRef CAS.
- K. Matyjaszewski , personal communication.
- M. Antonietti and K. Landfester, Prog. Polym. Sci., 2002, 27, 689–757 CrossRef CAS.
- K. Landfester, Macromol. Rapid Commun., 2001, 22, 896–936 CrossRef.
- F. J. Schork, G. W. Poelein, S. Wang, J. L. Reimers, J. Rodrigues and C. J. Samer, Colloids Surf., A, 1999, 153, 39–45 CrossRef CAS.
- J. Asua, Prog. Polym. Sci., 2002, 27, 1283–1346 CrossRef CAS.
- K. Landfester, N. Bechthold, F. Tiarks and M. Antonietti, Macromolecules, 1999, 32, 5222–5228 CrossRef CAS.
- H. de Brouwer, J. G. Tsavalas, F. J. Schork and M. J. Monteiro, Macromolecules, 2000, 33, 9239–9246 CrossRef CAS.
- Y. Luo, J. G. Tsavalas and F. J. Schork, Macromolecules, 2001, 34, 5501–5507 CrossRef CAS.
- F. J. Schork, Y. Luo, W. Smulders, J. P. Russurn, A. Buttè and K. Fontenot, Adv. Polym. Sci., 2005, 175, 129–255 CAS.
- M. Lansalot, T. P. Davis and J. P. A. Heuts, Macromolecules, 2002, 35, 7582–7591 CrossRef CAS.
- A. Butté, G. Storti and M. Morbidelli, Macromolecules, 2001, 34, 5885–5896 CrossRef CAS.
- J. B. McLeary, M. P. Tonge, D. De Wet-Roos, R. D. Sanderson and B. Klumperman, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 960–974 CrossRef CAS.
- Y. Luo and X. Liu, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6248–6258 CrossRef CAS.
- M. P. Tonge, J. B. McLeary, J. J. Vosloo and R. D. Sanderson, Macromol. Symp., 2003, 193, 289–304 CrossRef CAS.
- F. Zhang, P. Ni, Q. Xiong and Z. Yu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2931–2940 CrossRef CAS.
- H. Matahwa, J. B. McLeary and R. D. Sanderson, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 427–442 CrossRef.
- A. J. P. Van Zyl, R. F. P. Bosch, J. B. McLeary, R. D. Sanderson and B. Klumperman, Polymer, 2005, 46, 3607–3615 CrossRef.
- K. Matyjaszewski and T. P. Davis, Handbook of Radical Polymerisation, John Wiley and Sons, Inc, New York, 2002 Search PubMed.
- B. T. T. Pham, D. Nguyen, C. J. Ferguson, B. S. Hawkett, A. K. Serelis and C. H. Such, Macromolecules, 2003, 36, 8907–8909 CrossRef CAS.
- M. F. Cunningham, Prog. Polym. Sci., 2002, 27, 1039–1067 CrossRef CAS.
- S. W. Prescott, M. J. Ballard, E. Rizzardo and R. G. Gilbert, Macromolecules, 2005, 38, 4901–4912 CrossRef CAS.
- S. W. Prescott, Macromolecules, 2003, 36, 9608–9621 CrossRef CAS.
- J. J. Vosloo, D. De Wet-Roos, M. P. Tonge and R. D. Sanderson, Macromolecules, 2002, 35, 4894–4902 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- K. Hong, H. Zhang, J. W. Mays, A. E. Visser, S. Brazel, D. Holbrey, W. M. Reichert and R. D. Rogers, Chem. Commun., 2002, 1368–1369 RSC.
- S. Perrier, T. P. Davis, A. J. Carmichael and D. M. Haddleton, Chem. Commun., 2002, 2226–2227 RSC.
- U. P. Kreher, A. E. Rosamilia, C. L. Raston, J. L. Scott and C. R. Strauss, Molecules, 2004, 9, 387–393 Search PubMed.
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 426 CrossRef CAS.
- B. Ray, Y. Isobe, A. Matsumoto, S. Habaue, Y. Okamoto, M. Kamigaito and M. Sawamoto, Macromolecules, 2004, 37, 1702–1710 CrossRef CAS.
- J.-F. Lutz, B. Kirci and K. Matyjaszewski, Macromolecules, 2003, 35, 3136–3145 CrossRef.
- B. Kirci, J.-F. Lutz and K. Matyjaszewski, Macromolecules, 2002, 35, 2448–2451 CrossRef CAS.
- Y. Miura, T. Satoh, A. Narumi, O. Nishizawa, Y. Okamoto and T. Kakuchi, Macromolecules, 2005, 38, 1041–1043 CrossRef CAS.
- K. A. Fletcher and S. Pandey, Langmuir, 2004, 29, 33–36 CrossRef.
- J. Eastoe, S. Gold, S. E. rogers, A. Paul, T. Welton, R. K. Heenen and I. Grillo, J. Am. Chem. Soc., 2005, 127, 7302–7303 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |