Bioinspired colloidal systems via layer-by-layer assembly
Alexandra S.
Angelatos
,
Kiyofumi
Katagiri†
and
Frank
Caruso
*
Centre for Nanoscience and Nanotechnology, Department of Chemical and Biomolecular Engineering, The University of Melbourne, Victoria 3010, Australia. E-mail: fcaruso@unimelb.edu.au
First published on 25th November 2005
Abstract
This paper provides an overview of our recent work in the area of bioinspired colloidal particles. We highlight how modifying the basic polyelectrolyte multilayer shell with materials such as light-absorbing gold nanoparticles, lipid bilayer membranes, and targeting ligands can functionalize colloids prepared via the layer-by-layer assembly technique. These nanoengineered colloids are expected to show promise in areas ranging from drug and gene delivery to cell membrane modeling.
 Alexandra Angelatos | Alexandra Angelatos (born in Melbourne, Australia in 1979) is a PhD student at the Centre for Nanoscience and Nanotechnology, Department of Chemical and Biomolecular Engineering, The University of Melbourne. She completed her BEng/BSc degree at The University of Melbourne in 2003, and commenced her postgraduate studies under the supervision of Professor Frank Caruso in 2004. |
 Kiyofumi Katagiri | Kiyofumi Katagiri (born in Gifu, Japan in 1975) is a postdoctoral research fellow in the Department of Materials Science, Toyohashi University of Technology (supervisor: Professor A. Matsuda). He received his BEng degree from Osaka Prefecture University (supervisors: Professors M. Tatsumisago and T. Minami), and his MEng and PhD degrees from Nara Institute of Science and Technology (supervisors: Professors K. Ariga and J. Kikuchi). He spent two years as a postdoctoral research fellow under the supervision of Professor Frank Caruso at the Centre for Nanoscience and Nanotechnology, Department of Chemical and Biomolecular Engineering, The University of Melbourne. He was awarded a Research Fellowship for Young Scientists from the Japan Society for the Promotion of Science. His current research interests include biomimetic materials and organic–inorganic nanohybrid materials based on supramolecular chemistry, colloid and surface science, and sol–gel science and technology. |
 Frank Caruso | Frank Caruso (born in Platania, Italy in 1968) is a professor, Australian Research Council Federation Fellow, and Director of the Centre for Nanoscience and Nanotechnology in the Department of Chemical and Biomolecular Engineering at The University of Melbourne. He received his PhD degree from The University of Melbourne in 1994. He then moved to the CSIRO Division of Chemicals and Polymers in Melbourne studying the interfacial alignment of receptor molecules for biosensor applications. In 1997 he became an Alexander von Humboldt Research Fellow at the Max Planck Institute (MPI) of Colloids and Interfaces (Germany), and from 1998–2002 was group leader at the MPI. His main research interests are polymers at interfaces, biomaterials, nanostructured colloidal systems, and nanocomposite thin films. He has published over 150 papers in peer-reviewed scientific journals and is co-inventor of more than 15 patents. |
1. Introduction
Nanostructured colloidal materials show great potential for use in a wide variety of applications, hence the design and fabrication of such systems has attracted significant interest in recent years.1,2 Many of the systems currently under development, particularly those for bioapplications, draw their inspiration from naturally occurring structures. That is, by mimicking the supramolecular architecture of structures found in nature, one can prepare complex materials capable of highly sophisticated functions. Core–shell particles and their counterpart hollow capsules are two classes of colloids that form the basis for the generation of more complex colloidal systems that can be designed to mimic biocolloids. A variety of procedures have been employed for the manufacture of core–shell particles in the nanometre to micrometre size range.1,2 One of the simplest and most versatile approaches is the layer-by-layer (LbL) assembly technique, originally introduced in 1991 by Decher and Hong3 for the construction of polymer multilayer films on macroscopic planar supports. As the method typically exploits electrostatic interactions between oppositely charged polyelectrolytes, it can be readily transferred to colloidal particles.1,2,4,5 Colloidal cores of various composition (latexes, metal nanoparticles, enzymes, low molecular weight species, and cells) and size (nanometre–micrometre–millimetre range) have been coated with multilayers of diverse composition and controllable thickness.2,4,5 A common application of core–shell particles produced via the LbL strategy is the formation of hollow multilayer capsules, obtained after removal of the templating cores by either chemical or thermal means.2,4–6 The physicochemical characteristics of the capsule shell, such as permeability, elasticity, and stability, depend upon the building blocks and the networks they form. Thus, multilayer capsules have the potential to impact a broad range of disciplines, in particular the biosciences, as their properties may be readily tailored to specific applications by simply varying the shell composition, thickness, and structure. Research on the fabrication and properties of core–shell particles and multilayer capsules has been reviewed elsewhere.1,2,6 Here, we highlight our recent work on the modification of core–shell particles and multilayer capsules with functional materials to produce intelligent colloids for various bioapplications (Fig. 1).7–10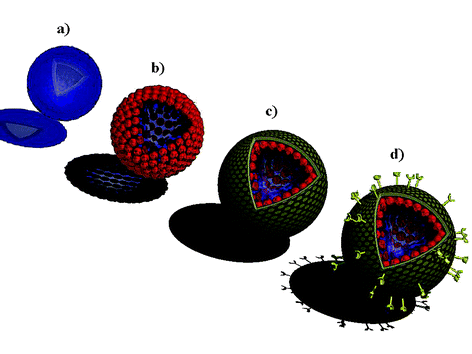 | ||
| Fig. 1 Illustration of bioinspired colloidal systems prepared via LbL assembly: (a) polyelectrolyte multilayer capsules; (b) light-responsive polyelectrolyte–gold nanoparticle capsules for controlled delivery; (c) polyelectrolyte-supported lipid vesicles for cell membrane modeling and drug/gene delivery; (d) biofunctionalized colloids for targeted delivery. | ||
2. Light-responsive polyelectrolyte–gold nanoparticle capsules
Optically addressable delivery systems7,8,11–16 have been the subject of much research in recent years because irradiation with light promises to be a more direct means of releasing the active materials than an environmental trigger such as a change in pH, temperature, ionic strength, or enzyme concentration.17 For example, Sershen and co-workers11 developed a photothermally responsive macroscopic hydrogel (i.e., a copolymer of N-isopropylacrylamide and acrylamide incorporating gold–gold sulfide nanoshells). Although the hydrogel undergoes a dramatic volume collapse upon illumination with light in the near-infrared (NIR) region (700–1500 nm) where most tissues show only weak light absorption,18 the system has not as yet been translated to colloidal delivery vehicles. Recently, Tao et al.15 and Skirtach et al.16 reported polyelectrolyte multilayer capsules doped with congo red dye molecules and silver nanoparticles, respectively. These light-responsive colloids, however, rely on irradiation with light outside the ‘biological window’ (i.e., non-NIR laser light) to induce release. To this end, we prepared optically addressable colloidal carriers that show potential for bioapplications by infiltrating light-absorbing gold nanoparticles into the shell of polyelectrolyte multilayer capsules (Fig. 1b).7,8 The gold nanoparticles within the capsule shell absorb in the NIR region, and because the pulses of NIR laser light required to induce release are short, the laser light energy is effectively confined to the capsule shell (Fig. 2a). Consequently, laser-induced release may be achieved in vivo without significantly damaging surrounding tissue and the encapsulated biomaterials. We investigated the release of various materials from polyelectrolyte–gold nanoparticle capsules upon irradiation at 1064 nm with a series of 10 ns pulses from a Q-switched neodymium∶yttrium–aluminium–garnet (Nd∶YAG) laser. It was demonstrated that lysozyme (which was encapsulated within the polyelectrolyte–gold nanoparticle shell via LbL assembly on the surface of lysozyme crystals) can be released on demand without a significant loss of bioactivity following irradiation with short pulses of NIR laser light.7 The enzyme activity following laser irradiation is comparable to that caused by mechanical rupturing of the capsules (Fig. 2b). We also showed that fluorescein isothiocyanate (FITC)-labelled dextran (a convenient model of DNA which was post-loaded into preformed capsules by exploiting the pH-dependence of the shell permeability) is released upon laser irradiation, provided the capsules contain gold nanoparticles within their shell.8 Electron microscopy confirmed that the laser irradiation has no apparent effect on capsules without the light-absorbing gold nanoparticles (Fig. 3). Based on our findings7,8 and those of previous optical studies,19–21 we propose that the laser-induced release results from the gold nanoparticle-mediated heating of the capsule shell to extreme temperatures, which produces significant thermal stresses that ultimately cause the shell to rupture (Fig. 2a). It is believed that phase explosions18 around the gold nanoparticles and electron ejection22 from the gold nanoparticles may also play a role in the laser-induced release.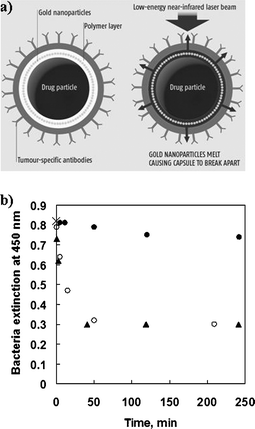 | ||
| Fig. 2 (a) Illustration of laser-induced drug release from biofunctionalized polyelectrolyte–gold nanoparticle capsules (diagram courtesy of New Scientist, Issue 8, January 5, 2005). (b) Extinction at 450 nm of the bacterium micrococcus lysodeikticus as a function of time after mixing with various poly(sodium 4-styrenesulfonate) (PSS)–poly(allylamine hydrochloride) (PAH)–gold nanoparticle-encapsulated lysozyme crystals: intact encapsulated crystals (closed circles); irradiated encapsulated crystals (open circles); crushed encapsulated crystals (triangles). The initial data point (0 min, 0.82), denoted by a cross, applies to all three systems. The bacterium is a substrate for the enzymatic action of lysozyme, with its extinction being a measure of the amount of bacteria in the sample. Hence, the amount of bacteria digested by the lysozyme correlates to the activity of the released lysozyme. When no release is induced, the extinction is essentially constant over the time range investigated; this implies that the lysozyme-loaded PSS–PAH–gold nanoparticle capsules are effectively leak-proof. When release is induced via laser irradiation, the extinction decreases abruptly and then levels out; this indicates that a significant proportion of the bacteria are digested and that the lysozyme activity remains stable, following the laser-induced release. When release is induced via crushing, the decay in the extinction is very similar to that for the laser-induced release case; this indicates that any loss in lysozyme activity induced by the laser pulse is comparable to that caused by mechanical rupturing of the capsules. | ||
 | ||
| Fig. 3 (a) (i) Scanning electron microscope (SEM) and (ii) transmission electron microscope (TEM) images of PSS–PAH–gold nanoparticle capsules prior to laser exposure. The dried capsules resemble deflated balloons, and there is a homogeneous dense packing of gold nanoparticles (which appear bright in TEM) within the capsule shells. (b) (i) SEM and (ii) TEM images of PSS–PAH–gold nanoparticle capsules following laser exposure. The outlines of individual capsules can no longer be readily discerned, and there is evidence of gold nanoparticle fusion among the remnants of the capsules. (c) (i) SEM and (ii) TEM images of PSS–PAH capsules prior to laser exposure. These images are indistinguishable from those of PSS–PAH capsules following laser exposure, indicating that the laser irradiation has no apparent effect on the morphology of PSS–PAH capsules without the light-absorbing gold nanoparticles in their shell. | ||
3. Polyelectrolyte-supported lipid vesicles
Liposomes comprise an aqueous core surrounded by a spherical lipid bilayer or multilayer shell and typically form via the self-assembly of lipid molecules in aqueous solution.23–27 Liposomes find application in cell membrane modeling, drug delivery, and transfection,28–30 however their use is limited by difficulties associated with controlling their size and monodispersity, and the thickness, permeability, and stability of the lipid membrane. Therefore, there is considerable interest in the preparation of polyelectrolyte-supported lipid vesicles via LbL assembly9,10,31–36 since they offer several key advantages over liposomes. Firstly, the size and monodispersity of the vesicles can be readily controlled through the choice of particle template. Secondly, the thickness, permeability, and stability of the vesicle wall can be tuned for specific applications by varying the composition, number, and arrangement of the polyelectrolyte and lipid layers. Moya and coworkers have studied the coating of core-shell particles with various phospholipid membranes.33,34 We recently reported the functionalization of colloids with robust inorganic-based lipid coatings9 and asymmetric lipid bilayer membranes (Fig. 1c).103.1 Robust inorganic-based lipid coatings
Polystyrene (PS) and melamine formaldehyde (MF) particles with narrow size distributions were LbL-coated with poly(diallyldimethylammonium chloride) (PDDA) and PSS multilayers, followed by the inorganic-based synthetic lipid N-[N-(3-triethoxysilyl)propylsuccinamoyl]dihexadecylamine (Si-lipid). The coated MF particles were subsequently exposed to hydrochloric acid (HCl) to decompose the templating cores, thereby yielding monodisperse polyelectrolyte-supported lipid vesicles.9 Microelectrophoretic measurements provided qualitative proof of film formation on the particles and indicated that the surface electrical state of the lipid-coated colloids resembles that of Cerasomes (i.e., bilayer liposomes of the same Si-lipid),37–42 which suggests that the Si-lipid coating is in the form of a bilayer membrane. Microelectrophoresis also demonstrated that the Si-lipid coating is not significantly delaminated during the core removal step and hence must be sufficiently permeable to allow removal of the MF decomposition products, which are several nanometres in size.43 In a related study,8 we observed that MF decomposition products did not permeate through PSS–PAH multilayers coated with the phospholipid dilauroylphosphatidylethanolamine (DLPE). This difference in the permeability of the Si-lipid and DLPE membranes may be attributed to the different phase transition temperatures of the two membranes. The phase transition temperature of the Si-lipid membrane is 10.5 °C,37 therefore it is in the relatively permeable liquid crystalline state during the core removal process (performed at room temperature), whereas the phase transition temperature of the DLPE membrane is 30.5 °C,44 so it is in the relatively impervious gel state during core removal. To further probe the stability of the Si-lipid coating on the colloids, the homogeneity of which was confirmed via microscopy, we treated the PS particles with Triton X-100 (TX-100) (i.e., a nonionic surfactant widely used as a lipid membrane-lysing agent) and ethanol (EtOH).9 It was found that, relative to PS particles LbL-coated with PDDA and PSS multilayers followed by the phospholipid dimyristoylphosphatidic acid sodium salt (DMPA), the colloids coated with the Si-lipid membrane exhibited high morphological resistance to both TX-100 (Fig. 4a) and EtOH (Fig. 4b). The superior stability of the Si-lipid coating is ascribed to cross-linking of the Si-lipid molecules to form a siloxane network during the deposition process.9 Colloids functionalized with such a robust inorganic-based lipid membrane show potential for bioapplications where they promise biocompatibility (e.g., the silanol groups present within the membrane have an affinity for, and promote the formation of, bone)45–47 and stability during further processing, such as the insertion of proteins into the membrane to form ion-channels or the attachment of receptor molecules to the membrane surface for targeted delivery.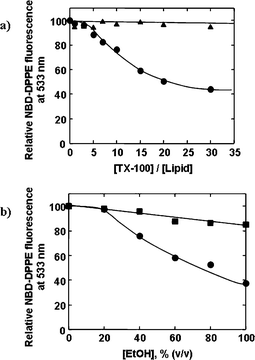 | ||
| Fig. 4 (a) Relative N-(7-nitro-2,1,3-benzoxadiazol-4-yl)-1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (NBD-DPPE) fluorescence at 533 nm as a function of the TX-100/lipid concentration ratio for PS core–PDDA–PSS shell particles coated with Si-lipid (containing 3 mol% NBD-DPPE) (triangles) or DMPA (containing 3 mol% NBD-DPPE) (circles). In the case of the core–shell particles coated with DMPA, the lipid probe fluorescence decreases, indicating disassembly of the DMPA membrane when the particles are exposed to five or more equivalents of TX-100. In the case of the core–shell particles coated with Si-lipid, the lipid probe fluorescence remains constant, and hence the Si-lipid membrane remains stable in the presence of up to 30 equivalents of TX-100. (b) Relative NBD-DPPE fluorescence at 533 nm as a function of EtOH concentration for PS core–PDDA–PSS shell particles coated with Si-lipid (containing 3 mol% NBD-DPPE) (squares) or DMPA (containing 3 mol% NBD-DPPE) (circles). In the presence of pure EtOH, the decrease in the lipid probe fluorescence for the core–shell particles coated with DMPA is more than three times that for the core–shell particles coated with Si-lipid, demonstrating that the Si-lipid membrane is considerably more resistant to EtOH than the DMPA membrane. | ||
3.2 Asymmetric lipid bilayer membranes
MF particles with a narrow size distribution were LbL-coated with several precursor polyelectrolyte (PDDA, PSS) layers, followed by an asymmetric lipid bilayer membrane comprising the cationic lipid dimethyldioctadecylammonium bromide (DDAB) and the anionic lipid dihexadecyl phosphate, sodium salt (DHP), followed by several additional polyelectrolyte (PDDA, PSS) layers (Fig. 5a).10 Subsequent treatment of the coated MF particles with HCl to decompose the templating cores yielded monodisperse polyelectrolyte-supported asymmetric lipid bilayer vesicles. Film formation on the particles was monitored via microelectrophoresis, while fluorescence microscopy verified the homogeneity of the asymmetric lipid bilayer membrane, even after removal of the templating cores. In order to confirm that the lipid membrane is indeed asymmetric (i.e., that the inner and outer layers of the bilayer comprise different lipid molecules), we performed a fluorescence-quenching assay. The data obtained indicated that not only is the lipid membrane asymmetric, but that its asymmetry is maintained for days after preparation, which suggests that lipid flip–flop does not occur in this system (Fig. 5b).10 The high morphological stability of the asymmetric lipid bilayer membrane most likely arises from complexation between the lipid molecules in each close-packed monolayer (as determined by quartz crystal microgravimetry) and the polyelectrolyte molecules in the adjacent multilayers. The fact that the colloids are resistant to TX-100 supports the notion that the polyelectrolyte multilayers on either side of the asymmetric lipid bilayer membrane play an important role in preserving the membrane's stability. On the basis of these results, we showed, by means of fluorescence spectroscopy, that it is possible to coat particles with multiple asymmetric lipid bilayer membranes (with intermediate polyelectrolyte multilayers).10 The ability to control the thickness, permeability, and stability of the vesicle wall via the composition, number, and arrangement of the polyelectrolyte and lipid layers renders these polyelectrolyte-supported asymmetric lipid bilayer vesicles attractive for a range of applications, notably cell membrane modeling (since the plasma membrane of a cell comprises an asymmetric distribution of phospholipids), drug delivery, and transfection.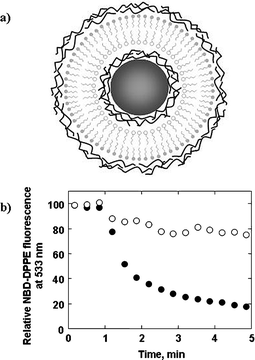 | ||
| Fig. 5 (a) Illustration of a templating core particle LbL-coated with several precursor polyelectrolyte layers, followed by an asymmetric lipid bilayer membrane (i.e., a lipid membrane where the inner and outer layers of the bilayer comprise different lipid molecules), followed by several additional polyelectrolyte layers. (b) Relative NBD-DPPE fluorescence at 533 nm as a function of time for MF particles coated with a polyelectrolyte-supported asymmetric lipid bilayer where DHP (containing 3 mol% NBD-DPPE) forms the inner (open circles) or outer (closed circles) layer of the bilayer membrane. In both cases, NBD-DPPE is embedded in the DHP layer and DDAB forms the alternate lipid layer. Sodium hydrosulfite (Na2S2O4), a compound capable of quenching the fluorescence of lipid probe molecules located in the outer layer of lipid membranes only as it cannot diffuse across lipid bilayers, was added to the particle dispersions after 1 min. For the particles where DHP forms the outer layer of the asymmetric lipid bilayer, the lipid probe fluorescence undergoes a rapid decrease upon Na2S2O4 addition (ca. 80% within 4 min), which confirms that the majority of the NBD-DPPE, and in turn the DHP, resides in the outer layer of the lipid membrane. For the particles where DHP forms the inner layer of the asymmetric lipid bilayer, the lipid probe fluorescence undergoes a relatively minor decrease upon Na2S2O4 addition (ca. 20% within 4 min), confirming that the majority of the NBD-DPPE, and hence the DHP, resides in the inner layer of the lipid membrane. No apparent change in the emission properties of the two systems was observed after 72 h, indicating that the asymmetry of the lipid bilayers is maintained for days after preparation. | ||
4. Biofunctionalized colloids
Targeted delivery may be achieved through either passive targeting or active targeting. The former approach is based on colloids with prolonged circulation and selective target localization properties, while in the latter approach specific targeting ligands (e.g., antibodies, peptides, polysaccharides, aptamers) are coupled to the colloid surface to enhance interaction with target cell membranes.48 We recently reported the biofunctionalization of core–shell particles with monoclonal immunoglobulin G (IgG) antibodies for the purposes of targeted delivery (Fig. 1d).7,8 First, to impart biospecificity, a primary antibody (mouse IgG) was coupled via electrostatic interaction to the surface of MF core–PSS–PAH shell particles coated with the phospholipid DLPE. Then, to probe the nature of the lipid/primary antibody coupling, the particles were incubated with a FITC-labelled secondary antibody (rabbit anti-mouse IgG). Fluorescence microscopy demonstrated that the surface coverage of the FITC-labelled secondary antibody, and hence that of the primary antibody, was homogeneous (Fig. 6). Successfully functionalizing the surface of colloidal carriers with targeting ligands represents a key step towards efficient drug/gene delivery. | ||
| Fig. 6 Fluorescence microscope images of MF core–PSS–PAH shell particles coated with DLPE and functionalized with antibodies, mouse IgG (primary antibody) and FITC-labelled rabbit anti-mouse IgG (secondary antibody). The fluorescence observed originates from the FITC label on the secondary antibody. | ||
5. Conclusions and outlook
This paper has highlighted some of our recent work in the area of bioinspired colloidal systems via LbL assembly, namely light-responsive polyelectrolyte/gold nanoparticle capsules for controlled delivery,7,8 polyelectrolyte-supported lipid vesicles for cell membrane modeling and drug/gene delivery,9,10 and biofunctionalized colloids for targeted delivery.7,8 Currently, we are investigating porous materials as a general method for biomolecule loading,49,50 hydrophilic–hydrophobic copolymers51,52 and DNA block polymers53 for thermoresponsive and biocompatible/biodegradable LbL film formation, respectively, blends of weak and strong polyelectrolyte components for enhanced control of film properties,54–56 and core–shell nanoparticles functionalized with transferrin for receptor-mediated endocytosis. We have also developed a new method for measuring the permeability of polyelectrolyte multilayer capsules, an understanding of which is vital for the preparation of delivery vehicles with designed release characteristics.57 Toward the next generation of complex colloidal containers, we recently reported the preparation of nanoporous polyelectrolyte spheres with interconnected polymer networks, which show promise for the facile uptake of high quantities of biomolecules.58 We anticipate that, with further development, these systems will make an important contribution not only to the understanding of biosystems, but also in the future application of bioinspired colloids in nano- and biotechnology.Acknowledgements
Funding from the Australian Research Council (Discovery Project and Federation Fellowship Schemes) and the Victorian State Government (STI Initiative) is gratefully acknowledged. KK acknowledges the Japan Society for the Promotion of Science for a Research Fellowship for Young Scientists and the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant-in-Aid for JSPS Fellow, No. 16·5511).References
- F. Caruso, Adv. Mater., 2001, 13, 11 CrossRef CAS.
- Colloids and Colloid Assemblies: Synthesis, Modification, Organization and Utilization of Colloidal Particles, ed. F. Caruso, Wiley-VCH, Weinheim, 2003 Search PubMed.
- G. Decher and J.-D. Hong, Ber. Bunsen-Ges. Phys. Chem., 1991, 95, 1430 CAS.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111 CrossRef CAS.
- E. Donath, G. B. Sukhorukov, F. Caruso, S. A. Davis and H. Möhwald, Angew. Chem., Int. Ed., 1998, 37, 2201 CrossRef.
- C. S. Peyratout and L. Dähne, Angew. Chem., Int. Ed., 2004, 43, 3762 CrossRef.
- B. Radt, T. A. Smith and F. Caruso, Adv. Mater., 2004, 16, 2184 CrossRef CAS.
- A. S. Angelatos, B. Radt and F. Caruso, J. Phys. Chem. B, 2005, 109, 3071 CrossRef CAS.
- K. Katagiri and F. Caruso, Macromolecules, 2004, 37, 9947 CrossRef CAS.
- K. Katagiri and F. Caruso, Adv. Mater., 2005, 17, 738 CrossRef CAS.
- S. Sershen, S. L. Westcott, N. J. Halas and J. L. West, J. Biomed. Mater. Res., 2000, 51, 293 CrossRef CAS.
- P. Shum, J.-M. Kim and D. H. Thompson, Adv. Drug Delivery Rev., 2001, 53, 273 CrossRef CAS.
- N. K. Mal, M. Fujiwara and Y. Tanaka, Nature, 2003, 421, 350 CrossRef CAS.
- B. I. Ipe, S. Mahima and K. G. Thomas, J. Am. Chem. Soc., 2003, 125, 7174 CrossRef CAS.
- X. Tao, J. Li and H. Möhwald, Chem.–Eur. J., 2004, 10, 3397 CrossRef CAS.
- A. G. Skirtach, A. A. Antipov, D. G. Shchukin and G. B. Sukhorukov, Langmuir, 2004, 20, 6988 CrossRef CAS.
- L. Yang and P. Alexandridis, Curr. Opin. Colloid Interface Sci., 2000, 5, 132 Search PubMed.
- A. Vogel and V. Venugopalan, Chem. Rev., 2003, 103, 577 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729 CrossRef CAS.
- C. M. Pitsillides, E. K. Joe, X. Wei, R. R. Anderson and C. P. Lin, Biophys. J., 2003, 84, 4023 CrossRef CAS.
- C. M. Aguirre, C. E. Moran, J. F. Young and N. J. Halas, J. Phys. Chem. B, 2004, 108, 7040 CrossRef CAS.
- T. E. McGrath, A. C. Beveridge and G. J. Diebold, Angew. Chem., Int. Ed., 1999, 38, 3353 CrossRef CAS.
- A. D. Bangham and R. W. Horne, J. Mol. Biol., 1964, 8, 660 CrossRef CAS.
- G. Sessa and G. J. Weissmann, J. Biol. Chem., 1968, 243, 4364 CAS.
- T. Kunitake and Y. Okahata, J. Am. Chem. Soc., 1977, 99, 3860 CrossRef CAS.
- H. Ringsdorf, B. Schlarb and J. Venzmer, Angew. Chem., Int. Ed. Engl., 1988, 27, 113 CrossRef.
- Y. Murakami and J. Kikuchi, in Bioorganic Chemistry Frontiers, ed. H. Duga, Springer-Verlag, Berlin, 1991, vol. 2 Search PubMed.
- D. D. Lasic, Liposomes: from Physics to Applications, Elsevier, Amsterdam, 1993 Search PubMed.
- Liposome Technology, ed. G. Gregoriadis, CRC Press, Boca Raton, 1993, vols. 1–3 Search PubMed.
- Artificial Self-Assembling Systems for Gene Delivery, ed. P. L. Felgner, M. J. Heller, P. Lehn, J. P. Behr and F. C. Szoka Jr., The American Chemical Society, Washington D.C., 1996 Search PubMed.
- B. Lindholm-Sethson, Langmuir, 1996, 12, 3305 CrossRef CAS.
- T. Cassier, A. Sinner, A. Offenhäuser and H. Möhwald, Colloids Surf., B, 1999, 15, 215 CrossRef CAS.
- S. Moya, E. Donath, G. B. Sukhorukov, M. Auch, H. Bäumler, H. Lichtenfeld and H. Möhwald, Macromolecules, 2000, 33, 4538 CrossRef CAS.
- S. Moya, W. Richter, H. Leporatti, H. Bäumler and E. Donath, Biomacromolecules, 2003, 4, 808 CrossRef CAS.
- L. Ge, H. Möhwald and J. Li, Biochem. Biophys. Res. Commun., 2003, 303, 653 CrossRef CAS.
- L. Ge, H. Möhwald and J. Li, Chem.–Eur. J., 2003, 9, 2589 CrossRef CAS.
- K. Katagiri, K. Ariga and J. Kikuchi, Chem. Lett., 1999, 28, 661 CrossRef.
- K. Katagiri, K. Ariga and J. Kikuchi, Stud. Surf. Sci. Catal., 2001, 132, 599 CAS.
- K. Katagiri, R. Hamasaki, K. Ariga and J. Kikuchi, J. Am. Chem. Soc., 2002, 124, 7892 CrossRef CAS.
- K. Katagiri, R. Hamasaki, K. Ariga and J. Kikuchi, Langmuir, 2002, 18, 6709 CrossRef CAS.
- K. Katagiri, R. Hamasaki, K. Ariga and J. Kikuchi, J. Sol-Gel Sci. Technol., 2003, 26, 393 CrossRef CAS.
- M. Hashizume, S. Kawanami, S. Iwamoto, T. Isomoto and J. Kikuchi, Thin Solid Films, 2003, 438–439, 20 CrossRef CAS.
- C. Gao, S. Moya, H. Lichtenfeld, A. Casoli, H. Fiedler, E. Donath and H. Möhwald, Macromol. Mater. Eng., 2001, 286, 355 CrossRef CAS.
- M. Benz, T. Gutsmann, N. Chen, R. Tadmor and J. Israelachvili, Biophys. J., 2004, 86, 870 CrossRef CAS.
- M. M. Pereira and L. L. Hench, J. Sol-Gel Sci. Technol., 1996, 7, 59 CAS.
- A. Osaka, S. Hayakawa, C. Ohtsuki, in Bioceramics, ed. R. Z. Legeros and J. P. Legeros, World Scientific, Singapore, 1998, vol. 11 Search PubMed.
- K. Tsuru, M. Kubo, S. Hayakawa, C. Ohtsuki and A. Osaka, J. Ceram. Soc. Jpn., 2001, 109, 409.
- S. P. Vyas, A. Singh and V. Sihorkar, Crit. Rev. Ther. Drug Carrier Syst., 2001, 18, 1 Search PubMed.
- Y. Wang and F. Caruso, Chem. Commun., 2004, 1528 RSC.
- Y. Wang and F. Caruso, Chem. Mater., 2005, 17, 953 CrossRef CAS.
- J. F. Quinn and F. Caruso, Langmuir, 2004, 20, 20 CrossRef CAS.
- J. F. Quinn and F. Caruso, Macromolecules, 2005, 38, 3414 CrossRef CAS.
- A. P. R. Johnston, E. S. Read and F. Caruso, Nano Lett., 2005, 5, 953 CrossRef CAS.
- J. Cho, J. F. Quinn and F. Caruso, J. Am. Chem. Soc., 2004, 126, 2270 CrossRef CAS.
- J. F. Quinn, J. C. C. Yeo and F. Caruso, Macromolecules, 2004, 37, 6537 CrossRef CAS.
- H. P. Yap, J. F. Quinn, S. M. Ng, J. Cho and F. Caruso, Langmuir, 2005, 21, 4328 CrossRef CAS.
- A. P. R. Johnston and F. Caruso, J. Am. Chem. Soc., 2005, 127, 10014 CrossRef CAS.
- Y. Wang, A. Yu and F. Caruso, Angew. Chem., Int. Ed., 2005, 44, 2888 CrossRef CAS.
Footnote |
| † Current address: Department of Materials Science, Toyohashi University of Technology, Toyohashi, Aichi 441-8580, Japan |
| This journal is © The Royal Society of Chemistry 2006 |