An efficient carbonyl-alkene metathesis of bicyclic oxetanes: photoinduced electron transfer reduction of the Paternò–Büchi adducts from 2,3-dihydrofuran and aromatic aldehydes
Raúl Pérez-Ruiza, Miguel A. Miranda*a, Ronald Alleb, Klaus Meerholzb and Axel G. Griesbeck*c
aDepartamento de Quimica/Instituto de Tecnologia Quimica UPV-CSIC, Universidad Politecnica de Valencia, Camino de Vera s/n, Apdo. 22012, 46022, Valencia, Spain. E-mail: mmiranda@qim.upv.es
bInstitute of Physical Chemistry, University of Cologne, Luxemburgerstr. 116, 50939, Köln, Germany
cInstitute of Organic Chemistry, University of Cologne, Greinstr. 4, 50939, Köln, Germany. E-mail: griesbeck@uni-koeln.de
First published on 2nd December 2005
Abstract
The bicyclic oxetanes 1 and 2 resulting from photocycloaddition of aromatic aldehydes to 2,3-dihydrofuran, were efficiently cleaved by means of electron-transfer reduction, photoinduced by the electronically excited reductants 1-methoxynaphthalene (MN) and 2,7-dimethoxynaphthalene (DMN) in acetonitrile. The fluorescence quenching rates of DMN/MN by 1 and 2 were determined by static methods, the triplet quenching rates were determined by means of laser flash photolysis (LFP). The product analysis established a “photo-photo metathesis” where both cycloaddition and cycloreversion processes are induced by photochemical processes.
Introduction
The [2 + 2] photocycloaddition of electronically excited carbonyl components to alkenes, termed Paternò–Büchi reaction, is a versatile and synthetically valuable synthetic route to oxetanes and secondary products derived from chemical modifications of these strained heterocycles.1–3 Among numerous methods for thermal ring-opening, by nucleophilic substitution or by electrophilic addition, the metathesis of oxetanes is an attractive tool for the synthesis of new carbonyl-ene pairs. The thermal ring cleavage has been described as carbonyl-olefin metathesis by G. Jones.4In the recent years, photoinduced electron transfer (PET) activation has come in the focus also of oxetane chemistry. One important biological background of these investigations is the fact that photoinduced DNA lesions can be initiated by Paternò–Büchi photocycloaddition between two adjacent pyrimidine bases followed by rearrangement to 6-4 photoproducts.5 The repair mechanism of these defects is postulated to be initiated by a reductive electron transfer thus generating oxetane radical anions. Subsequently, the oxetane radical anions are cleaved to restore the substrates in neutral and reduced form, respectively.6
Reports on the donor-facilitated PET of 1,3-dimethylthymine with benzaldehyde and benzophenone as model oxetanes have been studied in order to elucidate a possible mechanism.7 More recently, 2,3-diarylated oxetanes have been reported resulting in intermolecular8 and intramolecular versions (donor–acceptor dyads)9 using methoxy-substituted naphthalenes as electron-donor photosensitizers.
We have described in numerous publications, the photochemical cycloaddition of cyclic enolethers to aromatic aldehydes.10 This triplet reaction leads to bicyclic oxetanes with high endo(aryl)-selectivity. These oxetanes, bearing only one aryl substituent at C-2 of the four-membered ring, can be cleaved in two ways: either by way of reforming the Paternò–Büchi substrates or by metathetical ring opening to give enolether aldehydes. The latter way is a synthetically appealing thermal route to electron-rich alkenes.11
Therefore, we were interested if the mild PET-route for ring-opening of oxetanes, that has been successfully demonstrated for diaryl-substituted compounds,8,9 is also feasible for structurally more simple oxetanes. Hence, the behaviour of the two substrates (Fig. 1) 6-phenyl-4,7-dioxabicyclo[3.2.0]heptane (1) and 6-(p-cyanophenyl)-4,7-dioxabicyclo[3.2.0]heptane (2), available in high diastereoselectivity from the photocycloaddition of 2,3-dihydrofuran with benzaldehyde and 4-cyanobenzaldehyde, respectively, has been examined under electron transfer conditions using 1-methoxynaphthalene (MN) and 2,7-dimethoxynaphthalene (DMN) as photosensitizers.
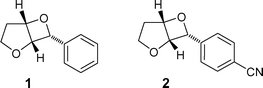 | ||
| Fig. 1 Substrates 1 and 2 used for the PET study. | ||
Results and discussion
Steady-state irradiations
The UV absorption of the oxetanes 1 and 2 in acetonitrile was weak at λ > 300 nm and did not interfere with the excitation of the PET-sensitizers MN and DMN. The singlet lifetime (τs) of MN was well-known in acetonitrile (13.6 ns),13 while the τs of DMN was not available in the literature and it was to be determined experimentally. The value was found to be 14.8 ns in acetonitrile. Thus, under the reaction conditions applied for reductive PET-cleavage of the oxetanes, the fluorescence lifetimes of both sensitizers were of comparable magnitude.Therefore, the oxetanes were irradiated (λmax = 300 nm) in acetonitrile, under argon, in the presence of catalytic amounts of MN or DMN. The reaction was followed by 1H NMR and GC-MS and resulted in the formation of the two cis/trans-diastereoisomeric enolethers 3-(styryloxy)propanal (3) and 4-(2-(2-formylethoxy)-vinyl)benzonitrile (4). In principle, oxetane 2 reacted in higher yields than 1 due to the substituted withdrawing group present in the para position of the phenyl group. The photoproduct structure determination was based on a thorough NMR analysis: for instance, in the case of compound 4, indicative were the 1H NMR shifts of the vinylic enolether hydrogen; for cis: 5.37 and 6.55 ppm (3JHH = 6.9 Hz); for trans: 5.98 and 7.34 ppm (3JHH = 13.0 Hz). The cis/trans-ratios after complete conversions were found to be 1 : 1 (Scheme 1).
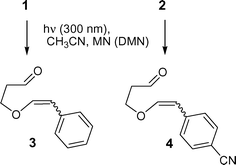 | ||
| Scheme 1 PET-reductive oxidative cleavage of 1 and 2. | ||
Excited state quenching
To evaluate the nature of the excited state involved in the PET process, the changes in fluorescence of MN and DMN, respectively, were studied in the presence of increasing amounts of 1 and 2. Fig. 2 shows the gradual decrease of the MN fluorescence in the presence of 2. The Stern–Volmer correlation (inset of Fig. 2) gave a value of 121 M−1 resulting in a fluorescence quenching constant of 8.9 × 109 M−1 s−1 (τF = 13.6 ns). The correlation for the fluorescence quenching of DMN by 2 (Fig. 3) gave a value of 46 M−1 resulting in a fluorescence quenching constant of 3.1 × 109 M−1 s−1 (τF = 14.8 ns). The analogous experiments (not shown here) with the phenyl-substituted oxetane 1 resulted in similar quenching properties: 5.6 × 109 M−1 s−1 for MN and 4.4 × 109 M−1 s−1 for DMN. From these data, oxetane 1 quenches the fluorescence of MN and DMN with nearly identical rates. Due to the 4-cyano-substituent, oxetane 2 was expected to behave as a better electron-acceptor and quench the photosensitizer fluorescences more efficiently. Actually, 2 was found to quench the fluorescence of MN near diffusion controlled conditions and that of DMN in a less efficient manner.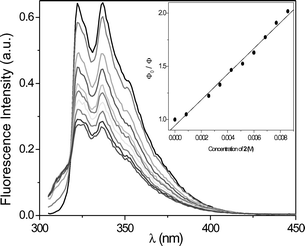 | ||
| Fig. 2 Fluorescence decay of MN (1.4 × 10−4 M) in acetonitrile in the presence of increasing amounts of 2. Inset: Stern–Volmer plot to obtain kq(S1). | ||
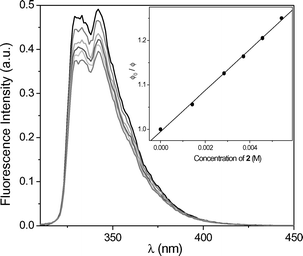 | ||
| Fig. 3 Fluorescence decay of DMN (1.4 × 10−4 M) in acetonitrile in the presence of increasing amounts of 2. Inset: Stern–Volmer plot to obtain kq(S1). | ||
Hence, the relative contributions of the different pathways for the deactivation of the singlet excited state of MN were calculated from eqn (1):
| kd(S1) = kq(S1) [oxetane] + kf + kisc | (1) |
Therefore, laser flash photolysis (LFP) experiments were carried out in order to check the reactivity of the triplet excited state. The T–T absorption spectra of MN and DMN were similar with absorption maxima at 435 and 430 nm, respectively. When the transient spectra were recorded with increasing amounts of oxetanes (e.g. in Fig. 4: MN and oxetane 2, excitation at 308 nm), the transient decays became faster and the triplet quenching rate constant could be determined from the lifetime/quencher concentration correlation (inset in Fig. 4). Thus, the corresponding triplet quenching rate constants kq(T1) were determined (Table 1). For all four combinations of quenchers and sensitizers, quenching rate constants were estimated and found to be about one order of magnitude lower than the singlet quenching rates. Under all conditions, however, the sensitizer radical cations could not be detected, probably due to their short lifetimes.
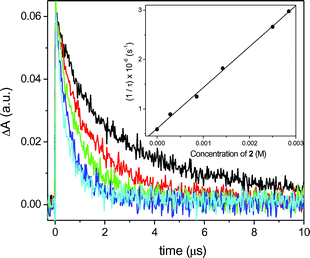 | ||
| Fig. 4 Decay traces of the T–T absorption of MN (10−4 M) measured at 435 nm in the presence of increasing amounts of 2: 0 M (black), 2.8 × 10−4 M (red), 9 × 10−4 M (green), 2 × 10−3 M (dark blue), 2.5 × 10−4 M (light blue). Inset: Plot of 1/τ against concentration of 2 to obtain kq(T1). | ||
Cyclic voltammetry
The redox potentials were measured by cyclic voltammetry (Fig. 5). The measurements were made in acetonitrile containing tetrabutylammonium hexafluorophosphate (0.1 M) as supporting electrolyte.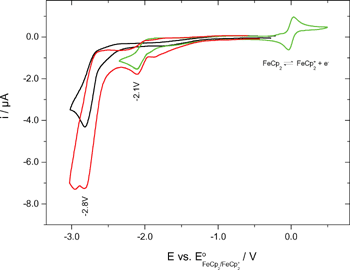 | ||
| Fig. 5 Cyclovoltammograms: 1 (black), 2 (green), 1 + 2-mixture (red). | ||
Because of the irreversibility of the reduction of the oxetanes 1 and 2 only peak potentials (−2.8 and −2.1 V [vs. E°(Fc/Fc+)]) could be determined. Conversion into SCE-based potentials gave −2.4 and −1.7 V (vs. SCE).15 Thus, the redox potentials (in fact the reduction potentials) of the oxetanes investigated herein are 0.2 and 0.9 V more negative than that of the 2-(p-cyanophenyl)-4-methyl-3-phenyloxetane originally investigated in the reductive PET ring opening.8
Electron transfer thermodynamics
Application of the Weller equation (eqn (2)16) was used for the estimation of the free energy changes (in kcal mol−1) associated with electron transfer processes:| ΔGET = 23.06 [E°(D+˙/D) − E°(A/A−˙)] − E*(S1 or T1) | (2) |
Due to the additional cyano-substituent in the oxetane 2, the reduction potential (−1.70 V) is strongly shifted to less negative values and this changes the thermodynamic situation for a possible PET process. The ΔGET(S1) and ΔGET(T1) values resulting from eqn (2) are −18.3 and +11.2 kcal mol−1 for MN and −18.4 and +11.6 kcal mol−1 for DMN. Hence, PET from the singlet excited state would be a strongly exergonic process, while the mechanism from the triplet excited state is still not thermodynamically feasible.
Conclusions
The behaviour of bicyclic oxetanes 1 and 2, resulting from photocycloaddition of aromatic aldehydes to 2,3-dihydrofuran, has been described under PET conditions using methoxy-substituted naphthalenes as electron-donor photosensitizers. Product analysis revealed a “photo-metathesis”, obtaining the corresponding carbonyl-ene pairs. The rate constants for the quenching of fluorescence and triplet of photosensitizers have been determined. They showed a marked dependence on the substitution at the phenyl group. Further, free energy changes ΔGET(S1) and ΔGET(T1) have been calculated, and the reaction taking place from the singlet excited state was found to be exergonic in the case of 2.Experimental
Materials
1-Methoxynaphthalene (MN) and 2,7-dimethoxynaphthalene (DMN) were commercially available. Oxetanes 1 and 2 were obtained by literature methods12 and purified by silica gel column chromatography using hexane–ethyl acetate as eluent (gradient from 99 : 1 to 60 : 40). Acetonitrile was of HPLC grade and used without further purification.Cycloreversion reaction (CR)
Solutions of oxetane 1 (10−2 M) with photosensitizer MN (10−3 M) and DMN (10−3 M), separately, in acetonitrile, were placed in quartz tubes and bubbled with argon. Then, solutions were irradiated during 90 min in a multilamp photoreactor, using four 8 W lamps with emission maximum at λmax = 300 nm. The reactions were followed by GC-MS and 1H NMR spectroscopy. Control experiments showed that CR does not take place in the dark or in the absence of photosensitizer. The same conditions for oxetane 2 were performed. Spectral analysis of products 3 and 4 was based on NMR comparison with analogous compounds.21 Compound 4: 1H NMR data for the cis-isomer: δ (ppm, 300 MHz, CDCl3) 2.85–2.93 (m, 2H), 4.36 (t, 2H, J 5.7, 11.7 Hz), 5.37 (d, 1H, J 6.9 Hz), 6.55 (d, 1H, J 6.9 Hz), 9.82 (t, 1H, J 1.5 Hz); for the trans-isomer: δ (ppm, 300 MHz, CDCl3) 2.85–2.93 (m, 2H), 4.26 (t, 2H, J 6.0 Hz), 5.98 (d, 1H, J 13.0 Hz), 7.34 (d, 1H, J 13.0 Hz), 9.78 (t, 1H, J 1.5 Hz). GC-MS analyses revealed that cycloreversion to give 3, 4 was the dominant pathway (ca. 15% of the corresponding aromatic aldehydes were detected corresponding to the non-metathesis pathway). On a preparative scale, complete CR was observed (>90% product balance).Absorption and fluorescence spectroscopy
Absorption spectra were recorded using a Beckman Coulter UV-DU800 and a Perkin-Elmer Lambda 7 spectrophotometer. The steady-state fluorescence spectra were obtained with a FS 900 spectrofluorimeter equipped with a 450 W xenon lamp. The time-resolved fluorescence determinations were performed using a hydrogen–nitrogen flash lamp (2 ns pulse width). The samples were placed into quartz cells of 1 cm path length. Concentrations of 1-methoxynaphthalene and 2,7-dimethoxy-naphthalene were fixed adjusting the absorbance of the solution at an arbitrary value between 0.2 and 0.3. The fluorescence lifetime of DMN in acetonitrile was determined under nitrogen and under air (Fig. 6).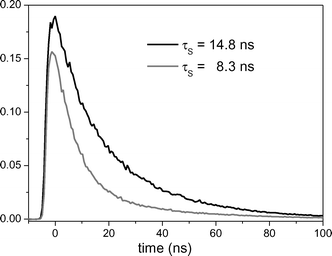 | ||
| Fig. 6 Fluorescence decay traces of DMN (λexc = 300 nm; λem = 342 nm) in acetonitrile under nitrogen (black) and air (grey). | ||
Cyclic voltammography
The electrochemical measurements of oxetanes 1 and 2 were performed in an air-tight cell under an argon atmosphere in anhydrous acetonitrile with an EG & G potentiostat model 283. It was used a three electrode arrangement. A simple Ag-wire, directly immersed in the acetonitrile solution of the substance of interest and tetrabutylammonium hexafluorophosphate as supporting electrolyte, acted as reference electrode. Because of the undefined potential of such an electrode the potentials are referenced to the formal potential of the redox couple ferrocene/ferricenium (Fc/Fc+) E°′(Fc) which was estimated under the same conditions as chosen for the actual measurements. This is in accordance with the IUPAC who recommends to give the formal potentials E°′ for non-aqueous solutions as E°′(Fc). If acetonitrile is used as solvent the last one is +0.40 V relative to the formal potential of the aqueous saturated calomel electrode (SCE) at 298 K.15A Pt-wire was sealed in a tube of soft glass to get a disc-shaped working electrode. A second Pt-wire winded around the shaft of this working electrode was used as counter electrode. All measurements were carried out at room temperature at a scan rate of 100 mV s−1.
Time-resolved absorption spectroscopy
The laser flash photolysis system was based on a pulsed XeCl Excimer laser, using 308 nm as excitation wavelength. The single pulses were ca. 17 ns duration and the energy was ca. 100 mJ pulse−1. A Lo255 Oriel xenon lamp was employed as detecting light source. The laser flash photolysis apparatus consisted of the pulsed laser, the Xe lamp, a monochromator, a photomultiplier (PMT) system, and an oscilloscope. The output signal from the oscilloscope was transferred to a personal computer for study.Acknowledgements
Financial support from the MCYT (Grant CTQ2004-03811 and FPI fellowship to Raúl Pérez-Ruiz) and the Deutsche Forschungs-gemeinschaft is acknowledged.References
- A. G. Griesbeck, Photocycloaddition of alkenes to excited carbonyls, Synthetic Organic Photochemistry, in Molecular and Supramolecular Photochemistry, ed. A. G. Griesbeck and J. Mattay, Marcel Dekker, New York, 2005, vol. 12, p. 89 Search PubMed
.
- J. A. Porco, Jr. and S. L. Schreiber, The Paternò–Büchi reaction, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and L. A. Paquette, Pergamon Press, Oxford, New York, 1991, vol. 5, p. 151 Search PubMed
- A. G. Griesbeck, M. Fiege, Stereochemistry of Photocyclizations and Photocycloadditions, in Molecular and Supramolecular Photochemistry, ed. V. Ramamurthy and K. S. Schanze, Marcel Dekker, New York, 2000, vol. 6, p. 33 Search PubMed
- G. Jones II, Synthetic applications of the Paternò–Büchi reaction, in Organic Photochemistry, ed. A. Padwa, Marcel Dekker, New York, 1981, vol. 5, p. 1 Search PubMed
- J.-S. Taylor and S. Nadji, Unraveling the origin of the major mutation induced by ultraviolet-light, the C → T transition at DTPDC sites- a DNA-synthesis building block for this cis-syn cyclobutane dimer of DTPDU, Tetrahedron Lett., 1991, 47, 2579 CAS
- Q. H. Song, X. Hei, Z. Xu, X. Zhang and Q. Guo, Model studies of the (6-4) photoproduct photoreactivation: synthesis and photosensitized splitting of uracil oxetane adducts, Bioorg. Chem., 2003, 31, 357 CrossRef CAS
-
(a) G. Prakash and D. E. Falvey, Model studies of the (6-4) photoproduct DNA photolyase: Synthesis and photosensitized splitting of a thymine-5,6-oxetane, J. Am. Chem. Soc., 1995, 117, 11375 CrossRef CAS
- R. Pérez-Ruiz, M. A. Izquierdo and M. A. Miranda, Reductive PET cycloreversion of oxetanes: singlet multiplicity, regioselectivity, and detection of olefin radical anion, J. Org. Chem., 2003, 68, 10103 CrossRef CAS
- R. Pérez-Ruiz, S. Gil and M. A. Miranda, Stereodifferentiation in the photochemical cycloreversion of diastereomeric methoxy-naphthalene-oxetane dyads, J. Org. Chem., 2005, 70, 1376 CrossRef CAS
- A. G. Griesbeck, M. Abe and S. Bondock, Selectivity Control in Electron Spin Inversion Processes: Regio- and Stereochemistry of Paternò–Büchi Photocycloadditions as a Powerful Tool for Mapping Intersystem Crossing Processes, Acc. Chem. Res., 2004, 37, 919 CrossRef CAS
- G. Jones, II, S. B. Schwartz and M. T. Marton, Regiospecific thermal cleavage of oxetane photoadducts. Carbonyl-olefin metathesis in sequential photochemical and thermal steps, J. Chem. Soc., Chem. Commun., 1973, 374 RSC
- A. G. Griesbeck and S. Stadtmüller, Regioselective and stereoselective photocycloaddition reactions of aromatic aldehydes to furan and 2,3-dihydrofuran, Chem. Ber., 1990, 123, 357 CrossRef CAS
- A. J. Maroulis and D. R. Arnold, Radical ions in photochemistry. 11. Photosensitized (electron-transfer) preparation of some 2,2,2-trifluoromethyl ethers, Synthesis, 1979, 819 CrossRef CAS
- M. Yamaji, T. Sekiguchi, M. Hocino and H. Shizuka, Proton-induced electron-transfer reaction from triplet methoxynaphthalenes to benzophenone via triplet exciplex, J. Phys. Chem., 1992, 96, 9353 CrossRef CAS
- N. G. Connelly and W. E. Geiger, Chemical redox agents for organometallic chemistry, Chem. Rev., 1996, 96, 877 CrossRef CAS
- A. Weller, Photoinduced electron transfer in solution: exciplex and radical ion pair formation free enthalpies and their solvent dependence, Z. Phys. Chem., 1982, 133, 93 CAS
- A. Zweig, A. H. Maurer and B. G. Roberts, Oxidation, reduction and electrochemiluminescence of donor-substituted polycyclic aromatic hydrocarbons, J. Org. Chem., 1967, 32, 1322 CrossRef CAS
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photo-chemistry, Marcel Dekker, New York, 1993, 2nd edn, pp. 33 and 115 Search PubMed
- A. A. Abdel-Shafi and F. Wilkinson, Electronic to vibrational energy conversion and charge transfer
contributions during quenching of molecular oxygen of electronically excited triplet states, Phys. Chem. Chem. Phys., 2002, 4, 248 RSC
- C. Schweitzer, Z. Mehrdad, F. Shafii and R. Schmidt, Common Marcus type dependence of the charge transfer induced processes in the sensitization and quenching of singlet oxygen by naphthalene derivatives, J. Phys. Chem. A, 2001, 105, 5309 CrossRef CAS
- I. Kadota, M. Kawada, V. Gevorgyan and Y. Yamamoto, Intramolecular reaction of (γ-alkoxyallyl)stannane with aldehyde: origin of the stereoselectivities, J. Org. Chem., 1997, 62, 7439 CrossRef CAS
| This journal is © The Royal Society of Chemistry and Owner Societies 2006 |