Picosecond time-resolved infrared spectroscopic investigation into electron localisation in the excited states of Re(I) polypyridyl complexes with bridging ligands
Marina K.
Kuimova
a,
Keith C.
Gordon
*b,
Sarah L.
Howell
b,
Pavel
Matousek
c,
Anthony W.
Parker
c,
Xue-Zhong
Sun
a,
Michael
Towrie
c and
Michael W.
George
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: Mike.George@nottingham.ac.uk
bDepartment of Chemistry, University of Otago, PO Box 56, Dunedin, New Zealand. E-mail: kgordon@alkali.otago.ac.nz
cCentral Laser Facility, CCLRC Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire, UK OX11 0QX
First published on 21st November 2005
Abstract
Mono- and binuclear complexes of {Re(CO)3Cl} with dipyrido[2,3-a:3′,2′-c]-6,7-dimethylphenazine (ppbMe2) were synthesised and their photophysical properties probed using picosecond time-resolved infrared spectroscopy (TRIR). Excitation of these complexes in solution at 400 nm produces short-lived excited states. The IR spectrum of the excited state of the mononuclear [Re(CO)3Cl(ppbMe2)] have ν(CO) bands shifted to higher wavenumber relative to those of the ground state. This is consistent with formation of a 3MLCT excited state. The IR spectrum of the excited state of the bimetallic [(Re(CO)3Cl)2(μ-ppbMe2)] shows the formation of two distinct groups of ν(CO) bands. This is interpreted as the formation of two distinct Re sites arising from a localised MLCT state with formally oxidised Re centre and a formally reduced bridging ligand. The ν(CO) bands of the adjacent Re centre are affected by the reduction of the bridging ligand. On the IR timescale the excited state structure is best formulated as [Cl(CO)3ReII(μ-ppbMe2˙−)ReI(CO)3Cl].
Introduction
An understanding of the mechanisms of the photoinduced processes in complex systems, containing covalently linked donor and acceptor units, are of interest due to the variety of applications such as photocatalysis, solar energy conversion and storage.1,2 Such systems are also of interest as they provide a route towards the understanding of mechanisms of the electron and energy transfer. Mixed-valence compounds can be used as units of supramolecular systems which may provide an opportunity to probe electron and energy transfer phenomena.3 The mixed-valence concept has important ramifications for organic systems such as conducting polymers.4,5 However, most of the systems studied are polynuclear transition metal complexes with heteronuclear aromatic bridging ligands.6–8 The properties in such systems can be tuned by synthetic modification of the ligand, or changing the metal. The bridging ligand not only plays a role in the covalent link between the adjacent metal centres but also mediates electronic communication between them and changes in its structure can have a profound effect on the energy and electron transfer rates.Mixed-valence states are classified as class I, II or III, depending of the level of valence trapping.9 In a complex in which there is no communication between adjacent valence sites the resulting mixed-valence species would show class I behaviour but when the communication is effective, class II or III states may be formed.9–13 Class III systems show complete delocalisation so that the extra charge is equally shared by both metal centres. The class II compounds are intermediate between I and III, there is a barrier that prevents full charge delocalisation between the adjacent centres. The characteristic timescale of the method used for the monitoring of such mixed-valence states determines whether one equivalent state or two localised centres are observed.
A variety of experimental methods have been used to determine the extent of charge-delocalisation in mixed-valence states14 including cyclic voltammetry15,16 and electronic absorption spectroscopy in the near-infrared region.17–20 A valuable probe for electron delocalisation is the Stark effect spectroscopy (electroabsorption technique).21,22 The transient absorption (TA) technique can be used to monitor the energy and electron transfer in dyads and polyads of metal complexes following photoexcitation.23 However, the interpretation of electronic absorption data can be complicated because of the presence of more than one chromophore.20
Infrared spectroscopy is an attractive method for determining the extent of charge delocalisation in mixed-valence systems possessing ancillary ligands which can act as IR reporters, such as CO and CN. These ligands show shifts in band frequencies in the IR spectrum, with the change of metal oxidation state. Ito et al.24,25 and others26–30 in a study of ruthenium cluster complexes, showed that the IR signature of the pendant CO moiety could be used to probe the formation of either class III or class II mixed-valence systems following electrochemical oxidation. The sensitivity of ν(CO) and ν(CN) to the electron density on the metal centre may be utilised in the examination of mixed-valence states which are created by photoexcitation. Such states could be formed in multinuclear metal polypyridyl complexes in which excitation through a metal-to-ligand charge-transfer (MLCT) transition creates adjacent metal centres with different oxidation states. Time resolved infrared (TRIR) spectroscopy could be used to probe the charge distribution in these molecules following photoexcitation (on the timescale of the IR experiment). Schoonover et al.7 showed that TRIR could be used to monitor intramolecular energy transfer among the excited states of [(phen)(CO)3ReI(CN)RuII(CN)(bpy)2]+. Excitation at 300 nm creates a ReII based MLCT excited state as evidenced by frequency upshifts in the CO ligand vibrations, however within 40 ps energy transfer to a second lower energy excited state based on the RuIII centre had occurred. The ReII based MLCT excited state signature disappeared to be replaced by the ν(CO) bands at a slightly higher wavenumber than these of the ground state. This shift in the ν(CO) bands was attributed to a [(phen)(CO)3ReI(CN)RuIII(CN)(bpy˙−)(bpy)]+ state.7 [(CO)5W(μ-BL)W(CO)5], where BL = pyrazine and 4,4′-bipyridine (4,4′-bpy), have been studied with TRIR spectroscopy to determine the degree of delocalisation taking place upon excitation in this complex.6 It was found that the excited state contained IR signatures from the CO ligands attributable to W(0) and W(I) centres; on the IR timescale there was no evidence of a delocalised W(+0.5) valency as would be observed in class III systems. A recent TRIR study of molecular squares containing {Re(CO)3Cl} units connected by 4,4′-bpy bridges in polymer medium at 77 K also gave evidence for formation of class II species following 355 nm excitation.31 In the excited state spectra two distinct carbonyl environments were observed, one associated with a Re(II) centre and the second with a Re(I) centre adjacent to a reduced ligand. The most relevant study to the work presented in this paper is the report of [Cl(CO)3Re(μ-BL)Re(CO)3Cl] (BL = 2,3-di-(2-pyridyl)quinoxaline (dpq) and its derivatives) where the two Re environments were observed by TRIR and a class II mixed-valence state was formed, on the IR timescale, upon generation of the MLCT excited state.8
The understanding of the electronic structure of the bridging ligand in assisting the charge delocalisation in mixed-valence systems is of great interest. In the systems where more than one MLCT state localised on the separate MOs within the bridging ligand are possible, electronic communication between two metal centres might be dependent on the interconversion between the two MLCT states. In mono- and heterometallic binuclear complexes of Ru(II) and Os(II) with tetrapyridophenazine32 interconversion of phenanthroline- and phenazine-type MLCT excited states plays an important role in the rate of energy/electron transfer processes.
In this paper we have studied the ReI mono- and binuclear systems containing dipyrido[2,3-a:3′,2′-c]-6,7-dimethylphenazine (ppbMe2), Scheme 1, as the bridging ligand. It was recently shown by DFT calculations and FT-Raman spectroscopy that the two manifolds of excited states are possible within such ligands and their Cu(I) complexes due to the two types of unoccupied MOs: bpy-like and phenazine-like orbitals. Although these complexes are related to those studied by Abbott et al.,8 in the present case the bridging ligand is fused and thus more planar. The greater planarity of the bridging ligand should result in the more extensive π-conjugation thus lowering the π* LUMO of the bridging ligand.33 This facilitates an inter-metal communication29 and could facilitate the formation of a class III mixed-valence system upon excitation into MLCT excited state. The mononuclear complex of ppbMe2 serves as a model for the localised system as the CO band signatures originate from a single MLCT state.
 | ||
| Scheme 1 | ||
Experimental
Synthesis of Re(I) complexes
Preparation of [Re(CO)3Cl(ppbMe2)] and [Cl(CO)3Re(μ-ppbMe2)Re(CO)3Cl] complexes was based on the method by Moya et al.34 [Re(CO)5Cl] was used as supplied (Aldrich). Details for the preparation of ppbMe2 are published elsewhere.35[Re(CO)3Cl(ppbMe2)]: [Re(CO)5Cl] (0.149 g, 0.41 mmol) and ppbMe2 (0.178 g, 0.63 mmol) were combined in argon purged methanol (75 ml) and refluxed under an argon atmosphere for 2.5 h. The solution was cooled and the precipitate collected. The solid was washed with methanol, diethyl ether and finally pentane. The crude solid was dissolved in dichloromethane (∼1000 ml) and any remaining solid precipitate was removed by filtration. The dilute solution was loaded onto a partially deactivated (10% v/v H2O) silica gel column (25 cm × 5 cm diameter, 200–400 mesh, 40–63 μm). The mononuclear complex was eluted as an orange band, with dichloromethane–ethyl acetate mixture (1 ∶ 4 v/v). Yield (151 mg, 60%). Found: C, 44.93; H, 2.25; N, 9.11. C23H14N4O3ClRe requires C, 44.84; H, 2.29; N, 9.09%. m/z 639 ({MNa}+), 617 ({MH}+); νmax/cm−1 (CO) 2023, 1919 and 1906 (CH3CN).
[Cl(CO)3Re(μ-ppbMe2)Re(CO)3Cl]: As for the mononuclear product but with a 2 ∶ 1 ratio of [Re(CO)5Cl] to ligand. A mixture of dichloromethane–ethyl acetate (4 ∶ 1 v/v) has been used to elute a purple band due to the binuclear complex. Yield (281 mg, 74%). Found: C, 33.13; H, 1.47; N, 5.83. C26.5H15N6O6Cl3Re2 requires C, 33.01; H, 1.57; N, 5.81%. m/z 945 ({MNa}+), 923 ({MH}+); νmax/cm−1 (CO) 2025, 2021, 1926 and 1915 (CH3CN).
The picosecond TRIR experiments were carried out on the PIRATE apparatus at the Central Laser Facility of the CCLRC Rutherford Appleton Laboratory. This apparatus has been described in detail previously.36 Part of the output from a 1 kHz, 800 nm, 150 fs, 2 mJ Ti-sapphire oscillator/regenerative amplifier (Spectra Physics Tsunami/Spitfire) was used to pump a white light continuum seeded β-BaB2O4 (BBO) optical parametric amplifier (OPA). The signal and idler produced by this OPA were difference frequency mixed in a type I AgGaS2 crystal to generate tunable mid-infrared pulses (ca. 150 cm−1 FWHM, 1 μJ), which were split to give probe and reference pulses. Second harmonic generation of the residual 800 nm light provided 400 nm pump pulses. Both the pump and probe pulses were focused to a diameter of 200–300 μm in the sample. Changes in infrared absorption at various pump–probe time delays were recorded by normalising the outputs from a pair of 64 element MCT infrared linear array detectors on a shot-by-shot basis.
In all TRIR experiments dry, degassed solutions were saturated with argon and flowed through a home-built flow system incorporating an IR cell (Harrick Scientific Corp.) and a re-circulating pump (Micropump) or Teflon peristaltic pump (Cole Palmer).
Results and discussion
The mononuclear complex I displays three CO stretching vibrations at 2023, 1919 and 1906 cm−1 (Fig. 1a). The spectrum is indicative of a facial disposition of the CO ligands in Cs symmetry. TRIR spectra obtained at the series of time delays following 400 nm excitation are shown in Fig. 1b. Excitation results in the depletion of the ground state bands at 2023, 1919 and 1906 cm−1 within the instrument response. A number of new transient features appear at higher wavenumber. Transient bands initially formed after excitation are broad. They narrow and shift to higher wavenumber within several tens of picoseconds. This narrowing and shifting can be attributed to the vibrational cooling of IR bands often observed on the ps timescale for transition metal carbonyl complexes.37–40 A fast initial vibrational relaxation (τ ≈ 12 ps), in which the transient bandshape evolves, is followed by a slower relaxation to the ground state (τ = 3 ± 0.2 ns). The TRIR spectrum (Fig. 1) obtained after the initial bandshape relaxation, e.g. at Δt = 100 ps, may be fitted using 6 Lorentzian curves to reveal transient bands positioned at 2058, 2000 and 1963 cm−1. The shift to higher wavenumber upon photoexcitation is consistent with the population of an MLCT excited state.41,42 In such a state the Re(I) centre is formally oxidised to Re(II) while the acceptor ligand ppbMe2 is formally reduced. The decrease in the electron density on the metal centre results in a significant reduction in back-bonding from the dπ(Re) to the π*CO orbitals, increasing the bond order of ν(CO) stretches. The ν(CO) bandshifts observed for the monometallic complex I are typical for MLCT excited states of ReI tricarbonyls.41,43![(a) FTIR spectrum of fac-[Re(Cl)(CO)3{(CH3)2ppb}] (I) in CH3CN at room temperature. (b) Series of TRIR spectra obtained between 2 and 1000 ps after 400 nm excitation of this solution.](/image/article/2006/PP/b508335d/b508335d-f1.gif) | ||
| Fig. 1 (a) FTIR spectrum of fac-[Re(Cl)(CO)3{(CH3)2ppb}] (I) in CH3CN at room temperature. (b) Series of TRIR spectra obtained between 2 and 1000 ps after 400 nm excitation of this solution. | ||
The magnitude of the ν(CO) shift directly characterises the electron density on the metal centre. Therefore the magnitude of the frequency shift of the CO modes following photoexcitation of the binuclear complex, in comparison with the mononuclear complex, should allow the extent of communication between the two metal centres to be characterised.
The ground state IR spectrum of the binuclear complex II can be fitted to four bands lying at 2025, 2021, 1926 and 1915 cm−1. The band at 2025 cm−1 is a shoulder with an area of ca. 20% of the 2021 cm−1 band. The binuclear complex may exist as two isomers with the chloro groups cis or trans to each other. The effect of each isomer on the IR spectrum for related complexes has been modelled qualitatively using semi-empirical methods.44 These suggest that the splitting between the CO vibrations across the binuclear structure is about 10 cm−1 for the higher wavenumber band and 1–3 cm−1 for the two lower wavenumber modes with the trans isomer. In the case of the cis isomer the lowest wavenumber mode shows an additional 12 cm−1 splitting. The experimental data are consistent with a predominance of trans isomer for the samples studied herein.45 However, the presence of the cis isomer cannot be completely excluded.
A series of TRIR spectra obtained for the binuclear complex II after 400 nm excitation is shown in Fig. 2. It is clear that the ground state bands are bleached and the new transient bands are formed after excitation. As with the monometallic complex, the TRIR spectra undergo a rapid change in which the transient absorption bands alter shape as vibrational cooling occurs. There are two sets of transient bands present in the spectra of the TRIR spectra at later times: one shifted to higher wavenumber relative to the parent bands and the other set shifted to lower wavenumber. This indicates that two Re environments with different electron density distributions are being formed. The positively shifted transients (2075, 1994 and 1972 cm−1) indicate the formation of an MLCT excited state, analogous to the mononuclear complex. The magnitude of the positive shift of the high frequency ν(CO) band in the excited state of II is more than that of I consistent with less electron density being available for back bonding to the oxidised Re i.e. some of the electron density is located on the other Re centre. The two bands 2006 and 1895 cm−1 shifting to lower wavenumber are consistent with a signal from Re(I) centre adjacent to a reduced bridging ligand. Thus, the spectrum of the relaxed MLCT excited state can be fitted with 8 Lorentzian bands: three ground state bands (used for simplicity) and five transient bands. For the five fitted transient bands three originate from the CO ligands bound to a Re(II) centre, as in an MLCT state, and two originate from a Re(I) centre which is bound to a reduced ligand (ppbMe2−˙) and is a spectator to the MLCT state. The excited state decays with a lifetime of ca. 120 ps (Fig. 3). The pattern of IR absorptions observed in the transient spectrum of II (Fig. 2) strongly suggests the formation of a mixed-valence state from an MLCT excitation in which only one of the metals is formally oxidised. The excited state may be formulated as: [Cl(CO)3ReII(μ-ppbMe2˙−)ReI(CO)3Cl], a class II system. Thus the present TRIR study gives unequivocal evidence that the more planar bridging ligand ppbMe2 compared to dpq8 did not lead to complete delocalisation between adjacent Re centres following photoexcitation on the infrared timescale.
![(a) FTIR spectrum of fac-[{Re(Cl)(CO)3}2{(CH3)2ppb}] (II) in CH3CN at room temperature. (b) Series of TRIR spectra obtained between 5 and 300 ps after 400 nm excitation of this solution.](/image/article/2006/PP/b508335d/b508335d-f2.gif) | ||
| Fig. 2 (a) FTIR spectrum of fac-[{Re(Cl)(CO)3}2{(CH3)2ppb}] (II) in CH3CN at room temperature. (b) Series of TRIR spectra obtained between 5 and 300 ps after 400 nm excitation of this solution. | ||
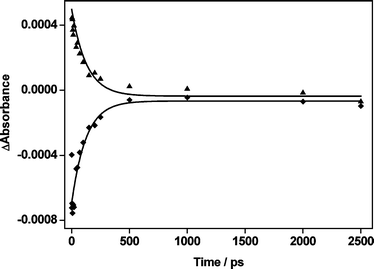 | ||
| Fig. 3 TRIR kinetic traces obtained at 1920 cm−1 (◆) and 1890 cm−1 (▲). Note all transient and parent bands decay/recover with the same lifetime (120 (±30) ps). | ||
The resonance Raman spectra of II indicate that excitation at 400 nm is resonant with a ππ* ligand-centred excited state.33 Furthermore, two MLCT states are identified absorbing at 575 and 510 nm. By analogy with the Cu(I) complexes with substituted ppb ligands46 we suggest that these two MLCT states have different LUMO orbitals localised either on bpy-like or phenazine-like parts of the ppbMe2 ligand. There is little evidence to suggest any interplay between these states in the time regime of this experiment. In fact the TRIR spectra provide no evidence for any species other than a single MLCT excited state, thus the relaxation from the ligand-centred state must be very rapid. The present TRIR data do not allow an unequivocal assignment of the observed MLCT state to either bpy- or phenazine-type. The electron density distribution observed for ppb ligands and their Cu(I) binuclear complexes46 suggests that the larger charge delocalisation is taking place in an MLCT (phenazine) excited state.
A phenazine-based MLCT state was previously observed with TRIR in mononuclear transition metal carbonyls containing the ppb isomer dipyrido[3,2a:2′,3′c]phenazine (dppz).47–49 Such states display short lifetimes of the order of several hundreds of ps and their TRIR spectra are characterised by the large positive shift of the ν(CO) bands (Δν > 90 cm−1) due to the lack of back-bonding between the π* MO of the reduced ligand radical anion and the dπ MOs of the rhenium centre. This is due to the fact that in the dppz ligand the phenazine-based MO has no wavefunction amplitude at the chelating (bpy) N atoms.50–52 The excited state lifetime of [ReCl(CO)3(ppbMe2)] and ν(CO) shift in the TRIR spectrum suggest the formation of an MLCT state in which the π* MO of the reduced ligand radical anion has appreciable wavefunction amplitude at the chelating N atoms thus facilitating back-bonding as reducing the magnitude of Δν. It should be noted that for ppb complexes the magnitude of the ν(CO) shift can not serve as an unambiguous indicator for the formation of either bpy MLCT or phenazine MLCT state as in each state at least one of the chelating N atoms will have wavefunction amplitude.
Conclusions
The time-resolved infrared spectra of mono- and binuclear complexes of ppbMe2 with the {ReI(CO)3Cl} unit have been measured. These complexes form short-lived MLCT excited states following 400 nm excitation. For binuclear [Re(CO)3Cl(μ-ppbMe2)Re(CO)3Cl] photoexcitation leads to the population of an MLCT excited state in which two Re sites are spectroscopically distinct, the excited state structure is best represented by the following scheme: [Cl(CO)3ReII(μ-ppbMe2˙−)ReI(CO)3Cl], thus forming a class II mixed-valence system.Acknowledgements
We thank Ms Amanda Dixon for synthesising the ligands used in this study. The support from ORS (MKK), the Foundation for Research, Science & Technology, the MacDiarmid Institute for Advanced Materials and Nanotechnology, the University of Otago and CCLRC, for funding access to the Central Laser Facility at the Rutherford Appleton Laboratory is gratefully acknowledged.References
- V. Balzani, M. Gomez-Lopez and J. F. Stoddart, Molecular machines, Acc. Chem. Res., 1998, 31, 405–414 CrossRef CAS.
- F. Scandola, M. T. Indelli, C. Chiorboli and C. A. Bignozzi, Photoinduced Electron and Energy-Transfer in Polynuclear Complexes, Top. Curr. Chem., 1990, 158, 73–149 CAS.
- K. D. Demadis, C. M. Hartshorn and T. J. Meyer, The localized-to-delocalized transition in mixed-valence chemistry, Chem. Rev., 2001, 101, 2655–2685 CrossRef CAS.
- C. Kitamura, S. Tanaka and Y. Yamashita, Design of narrow-bandgap polymers. Syntheses and properties of monomers and polymers containing aromatic-donor and o-quinoid-acceptor units, Chem. Mater., 1996, 8, 570 CrossRef.
- Q. T. Zhang and J. M. Tour, Alternating Donor/Acceptor Repeat Units in Polythiophenes. Intramolecular Charge Transfer for Reducing Band Gaps in Fully Substituted Conjugated Polymers, J. Am. Chem. Soc., 1998, 120, 5355–5362 CrossRef CAS.
- M. W. George, F. P. A. Johnson, J. J. Turner and J. R. Westwell, Excited-state properties of [(OC)5W(L)W(CO)5] [L = 4,4′-bipyridyl (4,4′-bipy) or pyrazine] and [(OC)5W(4,4′-bipy)], J. Chem. Soc., Dalton Trans., 1995, 2711–2718 RSC.
- J. R. Schoonover, K. C. Gordon, R. Argazzi, W. H. Woodruff, K. A. Peterson, C. A. Bignozzi, R. B. Dyer and T. J. Meyer, Application of Transient Infrared-Spectroscopy to Intramolecular Energy-Transfer in (Phen)(CO)3Re(I)(NC)Ru(II)(CN)(Bpy)2+, J. Am. Chem. Soc., 1993, 115, 10996–10997 CrossRef CAS.
- L. C. Abbott, C. J. Arnold, T. Q. Ye, K. C. Gordon, R. N. Perutz, R. E. Hester and J. N. Moore, Ultrafast time-resolved UV-visible and infrared absorption spectroscopy of binuclear rhenium(I) polypyridyl complexes in solution, J. Phys. Chem. A, 1998, 102, 1252–1260 CrossRef CAS.
- M. B. Robin and P. Day, Mixed Valence Chemistry - A Survey and Classification, Adv. Inorg. Chem. Rad., 1967, 10, 247 Search PubMed.
- J. H. Elias and R. S. Drago, Electronic structure of N,N′-bis[pentaammineruthenium(II,III)]pyrazine5+, Inorg. Chem., 1972, 11, 415–418 CrossRef CAS.
- N. S. Hush, Intervalence-transfer absorption. II. Theoretical considerations and spectroscopic data, Prog. Inorg. Chem., 1967, 8, 391–444 CAS.
- N. S. Hush, Distance dependence of electron transfer rates, Coord. Chem. Rev., 1985, 64, 135–157 CrossRef CAS.
- R. W. Callahan, G. M. Brown and T. J. Meyer, Effects of weak metal–metal interactions in ligand-bridged complexes of ruthenium. Dimeric complexes containing ruthenium ions in different coordination environments, Inorg. Chem., 1975, 14, 1443–1453 CrossRef.
- J. P. Sauvage, J. P. Collin, J. C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. de Cola and L. Flamigni, Ruthenium(II) and Osmium(II) Bis(Terpyridine) Complexes in Covalently-Linked Multicomponent Systems - Synthesis, Electrochemical-Behavior, Absorption-Spectra, and Photochemical and Photophysical Properties, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS.
- D. E. Richardson and H. Taube, Determination of E2-Degrees-E1-Degrees in Multistep Charge-Transfer by Stationary-Electrode Pulse and Cyclic Voltammetry-Application to Binuclear Ruthenium Ammines, Inorg. Chem., 1981, 20, 1278–1285 CrossRef CAS.
- D. E. Richardson and H. Taube, Electronic Interactions in Mixed-Valence Molecules as Mediated by Organic Bridging Groups, J. Am. Chem. Soc., 1983, 105, 40–51 CrossRef CAS.
- G. M. Tom, C. Creutz and H. Taube, Mixed-Valence Complexes of Ruthenium Ammines with 4,4′-Bipyridine as Bridging Ligand, J. Am. Chem. Soc., 1974, 96, 7827–7829 CrossRef CAS.
- C. Creutz, Mixed-Valence Complexes of d5-d6 Metal Centers, Prog. Inorg. Chem., 1983, 30, 1–73 CAS.
- K. D. Demadis, G. A. Neyhart, E. M. Kober, P. S. White and T. J. Meyer, Intervalence Transfer at the Localized-to-Delocalized, Mixed-Valence Transition in Osmium Polypyridyl Complexes, Inorg. Chem., 1999, 38, 5948–5959 CrossRef CAS.
- K. D. Demadis, G. A. Neyhart, E. M. Kober and T. J. Meyer, Vibrational Mapping at the Mixed-Valence, Localized-to-Delocalized Transition, J. Am. Chem. Soc., 1998, 120, 7121–7122 CrossRef CAS.
- D. H. Oh and S. G. Boxer, Stark-Effect Spectra of Ru(Diimine)32+ Complexes, J. Am. Chem. Soc., 1989, 111, 1130–1131 CrossRef CAS.
- D. H. Oh, M. Sano and S. G. Boxer, Electroabsorption (Stark-Effect) Spectroscopy of Monoruthenium and Biruthenium Charge-Transfer Complexes-Measurements of Changes in Dipole-Moments and Other Electrooptic Properties, J. Am. Chem. Soc., 1991, 113, 6880–6890 CrossRef CAS.
- C. Chiorboli, M. A. J. Rodgers and F. Scandola, Ultrafast processes in bimetallic dyads with extended aromatic bridges. Energy and electron transfer pathways in tetrapyridophenazine-bridged complexes, J. Am. Chem. Soc., 2003, 125, 483–491 CrossRef CAS.
- K.-I. Ota, H. Sasaki, T. Matsui, T. Hamaguchi, T. Yamaguchi, T. Ito, H. Kido and C. P. Kubiak, Syntheses and Properties of a Series of Oxo-Centered Triruthenium Complexes and Their Bridged Dimers with Isocyanide Ligands at Terminal and Bridging Positions, Inorg. Chem., 1999, 38, 4070–4078 CrossRef CAS.
- T. Ito, T. Hamaguchi, H. Nagino, T. Yamaguchi, H. Kido, I. S. Zavarine, T. Richmond, J. Washington and C. P. Kubiak, Electron Transfer on the Infrared Vibrational Time Scale in the Mixed Valence State of 1,4-Pyrazine- and 4,4′-Bipyridine-Bridged Ruthenium Cluster Complexes, J. Am. Chem. Soc., 1999, 121, 4625–4632 CrossRef CAS.
- C. H. Londergan and C. P. Kubiak, Electron transfer and dynamic infrared-band coalescence: It looks like dynamic NMR spectroscopy, but a billion times faster, Chem.–Eur. J., 2003, 9, 5962–5969 CrossRef CAS.
- C. H. Londergan, J. C. Salsman, S. Ronco, L. M. Dolkas and C. P. Kubiak, Solvent Dynamical Control of Electron-Transfer Rates in Mixed-Valence Complexes Observed by Infrared Spectral Line Shape Coalescence, J. Am. Chem. Soc., 2002, 124, 6236–6237 CrossRef CAS.
- C. H. Londergan, J. C. Salsman, S. Ronco and C. P. Kubiak, Infrared Activity of Symmetric Bridging Ligand Modes in Pyrazine-Bridged Hexaruthenium Mixed-Valence Clusters, Inorg. Chem., 2003, 42, 926–928 CrossRef CAS.
- C. H. Londergan and C. P. Kubiak, Vibronic Participation of the Bridging Ligand in Electron Transfer and Delocalization: New Application of a Three-State Model in Pyrazine-Bridged Mixed-Valence Complexes of Trinuclear Ruthenium Clusters, J. Phys. Chem. A, 2003, 107, 9301–9311 CrossRef CAS.
- C. H. Londergan, R. C. Rocha, M. G. Brown, A. P. Shreve and C. P. Kubiak, Intervalence Involvement of Bridging Ligand Vibrations in Hexaruthenium Mixed-Valence Clusters Probed by Resonance Raman Spectroscopy, J. Am. Chem. Soc., 2003, 125, 13912–13913 CrossRef CAS.
- T. P. Ortiz, J. A. Marshall, L. A. Emmert, J. Yang, W. Choi, A. L. Costello and J. A. Brozik, Transient Mixed-Valence Character of ReI4(CO)12(4,4′-bpy)4Cl4, Inorg. Chem., 2004, 43, 132–141 CrossRef CAS.
- C. Chiorboli, M. A. J. Rodgers and F. Scandola, Ultrafast processes in bimetallic dyads with extended aromatic bridges. Energy- and electron-transfer pathways in tetrapyridophenazine-bridged complexes, J. Am. Chem. Soc., 2003, 125, 483–491 CrossRef CAS.
- A. Flood, R. B. Girling, K. C. Gordon, R. E. Hester, J. N. Moore and M. I. J. Polson, Revealing the chromophoric composition of multichromophoric polypyridyl complexes of Re(I) and Os(II): a resonance Raman study, J. Raman Spectrosc., 2002, 33, 434–442 CrossRef CAS.
- S. A. Moya, R. Pastene, R. Schmidt, J. Guerrero and R. Sartori, Polyhedron, 1992, 11, 1665 CrossRef CAS.
- M. I. J. Polson, S. L. Howell, A. Flood, A. G. Blackman and K. C. Gordon, Synthesis and electronic properties of mononuclear osmium(II) and rhenium(I) complexes containing ligands derived from [2,3-a:3′,2′-c]dipyridophenazine (ppb), Polyhedron, 2004, 23, 1427–1439 CrossRef CAS.
- M. Towrie, D. C. Grills, J. Dyer, J. A. Weinstein, P. Matousek, R. Barton, P. D. Bailey, N. Subramaniam, W. M. Kwok, C. Ma, D. Phillips, A. W. Parker and M. W. George, Development of a broadband picosecond infrared spectrometer and its incorporation into an existing ultrafast time-resolved resonance Raman, UV/visible, and fluorescence spectroscopic apparatus, Appl. Spectrosc., 2003, 57, 367–380 CAS.
- T. P. Dougherty and E. J. Heilweil, Transient infrared spectroscopy of (η5-C5H5)Co(CO)2 photoproduct reactions in hydrocarbon solutions, J. Chem. Phys., 1994, 100, 4006–4009 CrossRef CAS.
- T. P. Dougherty, W. T. Grubbs and E. J. Heilweil, Photochemistry of Rh(CO)2(acetylacetonate) and Related Metal Dicarbonyls Studied by Ultrafast Infrared Spectroscopy, J. Phys. Chem., 1994, 98, 9396–9399 CrossRef CAS.
- T. Lian, S. E. Bromberg, M. Asplund, H. Yang and C. B. Harris, Femtosecond Infrared Studies of the Dissociation and Dynamics of Transition Metal Carbonyls in Solution, J. Phys. Chem., 1996, 100, 11994–12001 CrossRef CAS.
- J. B. Asbury, H. N. Ghosh, J. S. Yeston, R. G. Bergman and T. Lian, Sub-Picosecond IR Study of the Reactive Intermediate in an Alkane C–H Bond Activation Reaction by CpRh(CO)2, Organometallics, 1998, 17, 3417–3419 CrossRef CAS.
- M. W. George and J. J. Turner, Excited states of transition metal complexes studied by time-resolved infrared spectroscopy, Coord. Chem. Rev., 1998, 177, 201–217 CrossRef CAS.
- J. R. Schoonover, C. A. Bignozzi and T. J. Meyer, Application of transient vibrational spectroscopies to the excited states of metal polypyridyl complexes, Coord. Chem. Rev., 1997, 165, 239–266 CrossRef CAS.
- D. C. Grills, J. J. Turner and M. W. George, Time-resolved infrared spectroscopy, Compr. Coord. Chem. II, 2004, 2, 91–101 Search PubMed.
- S. E. Page, A. Flood and K. C. Gordon, Electron localisation in electrochemically reduced mono- and bi-nuclear rhenium(I) complexes with bridged polypyridyl ligands, J. Chem. Soc., Dalton Trans., 2002, 1180–1187 RSC.
- K. C. Gordon, A. K. Burrell, T. J. Simpson, S. E. Page, G. Kelso, M. I. J. Polson and A. Flood, Probing the nature of the redox products and lowest excited state of (bpy)2Ru(μ-bptz)Ru(bpy)24+: A resonance Raman study, Eur. J. Inorg. Chem., 2002, 554–563 CrossRef CAS.
- S. L. Howell, B. J. Matthewson, M. I. J. Polson, A. K. Burrell and K. C. Gordon, Structural Changes upon Reduction of Dipyrido[2,3-a:3′,2′-c]phenazine Probed by Vibrational Spectroscopy, ab Initio Calculations, and Deuteration Studies, Inorg. Chem., 2004, 43, 2876–2887 CrossRef CAS.
- M. K. Kuimova, W. Z. Alsindi, J. Dyer, D. C. Grills, O. S. Jina, P. Matousek, A. W. Parker, P. Portius, X. Z. Sun, M. Towrie, C. Wilson, J. Yang and M. W. George, Using picosecond and nanosecond time-resolved infrared spectroscopy for the investigation of excited states and reaction intermediates of inorganic systems, J. Chem. Soc., Dalton Trans., 2003, 3996–4006 RSC.
- M. K. Kuimova, D. C. Grills, P. Matousek, A. W. Parker, X.-Z. Sun, M. Towrie and M. W. George, Picosecond time-resolved infrared investigation into the nature of the lowest excited state of fac-[Re(Cl)(CO)3(CO2Et-dppz)] (CO2Et-dppz = dipyrido[3,2a:2′,3′c]phenazine-11-carboxylic ethyl ester), Vib. Spectrosc., 2004, 35, 219–223 CrossRef CAS.
- J. Dyer, W. J. Blau, C. G. Coates, C. M. Creely, J. D. Gavey, M. W. George, D. C. Grills, S. Hudson, J. M. Kelly, P. Matousek, J. J. McGarvey, J. McMaster, A. W. Parker, M. Towrie and J. A. Weinstein, The photophysics of fac-[Re(CO)3(dppz)(py)]+ in CH3CN: a comparative picosecond flash photolysis, transient infrared, transient resonance Raman and density functional theoretical study, Photochem. Photobiol. Sci., 2003, 2, 542–554 RSC.
- D. M. Dattelbaum, K. M. Omberg, J. R. Schoonover, R. L. Martin and T. J. Meyer, Application of Time-Resolved Infrared Spectroscopy to Electronic Structure in Metal-to-Ligand Charge-Transfer Excited States, Inorg. Chem., 2002, 41, 6071–6079 CrossRef.
- D. M. Dattelbaum, R. L. Martin, J. R. Schoonover and T. J. Meyer, Molecular and Electronic Structure in the Metal-to-Ligand Charge Transfer Excited States of fac-[Re(4,4′-X2bpy)(CO)3(4-Etpy)]+ (X = CH3, H, CO2Et). Application of Density Functional Theory and Time-Resolved Infrared Spectroscopy, J. Phys. Chem. A, 2004, 108, 3518–3526 CrossRef CAS.
- D. M. Dattelbaum, K. M. Omberg, P. J. Hay, N. L. Gebhart, R. L. Martin, J. R. Schoonover and T. J. Meyer, Defining Electronic Excited States Using Time-Resolved Infrared Spectroscopy and Density Functional Theory Calculations, J. Phys. Chem. A, 2004, 108, 3527–3536 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2006 |