Synthesis and evaluation of the first cis-cyclobutane-containing receptor for lipid A†
Kevin M.
Bucholtz
cd,
Peter C.
Gareiss
bd,
Stephen G.
Tajc
bd and
Benjamin L.
Miller
*abd
aDepartment of Dermatology, University of Rochester, Rochester, New York, USA. E-mail: Benjamin_Miller@futurehealth.rochester.edu; Fax: +1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (1)5852731346; Tel: +1(1)5852759805
(1)5852731346; Tel: +1(1)5852759805
bDepartment of Biochemistry and Biophysics, University of Rochester, Rochester, New York, USA
cDepartment of Chemistry, University of Rochester, Rochester, New York, USA
dCenter for Future Health, University of Rochester, Rochester, New York, USA
First published on 29th September 2006
Abstract
The first example of a designed receptor containing a cis-1,3-disubstituted cyclobutane ring has been synthesized. This molecule binds diphosphoryl lipid A (a conserved portion of the Gram-(−) bacterial cell membrane, and the causative agent of septic shock) with an affinity comparable to previously described ter-cycloalkane based lipid A-binding compounds.
The design and synthesis of receptors for carbohydrates and carbohydrate derivatives represents a key frontier for bioorganic chemistry.1 In addition to providing new insights into the basic processes underlying molecular recognition of carbohydrates,2 novel synthetic receptors can serve as important tools in the elucidation and control of carbohydrate-mediated cellular activities, as components of biosensors3 and, potentially, as therapeutic agents.4
Lipid A, the conserved headgroup of Gram-(−) bacterial lipopolysaccharide5 and a primary causative agent of bacterial septic shock,6 is a particularly interesting target for molecular recognition studies. Several naturally-occurring compounds, including the well-studied antibiotic polymyxin B,7 are known to target lipid A. As part of a general effort directed towards developing receptors for carbohydrates, we initiated a program to examine the ability of stereoregular oligocycloalkane derivatives to serve as lipid A receptors. We chose the oligocycloalkane scaffold in part because of its similarity (in terms of size and anticipated rigidity) to cholic acid, a compound that has proven useful as a scaffold for the construction of lipid A binders.8 In contrast to cholic acid, however, we anticipated that the ter-cycloalkane scaffold would permit more extensive variation, allowing for subtle changes on structure and conformation to be examined. As we reported previously, compound 1 (“TWTCP”) binds lipid A in buffered aqueous solution with an affinity comparable to lipid A-binding natural products.9 In this paper, we report the synthesis and preliminary evaluation of the first structural variant of TWTCP incorporating a central cis-1,3-disubstituted cyclobutane ring (“TW545”, 2).
Molecular mechanics analysis of the core scaffold 2 suggests that it should have a slightly more linear structure than 1, without dramatically altering the conformational energy profile (Fig. 1). Therefore, we anticipated that 2 would likely retain lipid A binding ability, while potentially displaying subtle differences in conformational dynamics. While trans-1,3-cyclobutanes have been examined in the context of molecular recognition by other researchers,10 to our knowledge the use of a cis-1,3-disubstituted cyclobutane as a core structural element is unprecedented.
 | ||
| Fig. 1 Left: overlay of minimum-energy conformers of a ter-cyclopentane scaffold (blue) and a cyclobutane analog (red). Right: inter-ring dihedral angle drive (MMFFs force field,11 GB/SA CHCl3,12 Maestro/Macromodel13) for the cyclopentane–cyclopentane (black) and cyclopentane–cyclobutane (grey) torsion. | ||
Our intent from the outset was to synthesize 2 using a bidirectional route analogous to that employed previously for the synthesis of 1 (Scheme 1). Thus, cis-cyclobutane-1,3-dialdehyde 4 represented a key initial synthetic target, by analogy to the synthesis of 1 from dialdehyde 3. Compounds of this type are deceptively simple in appearance; however, relatively few examples of cis-1,3-disubstituted cyclobutanes appear in the literature. The two primary approaches to such compounds have either employed a one-bond formation (cyclization) strategy,14 or cleavage of the highly strained (and highly volatile) benzvalene15,16 for the construction of the cyclobutane ring.
 | ||
| Scheme 1 Oligocycloalkane containing receptors 1 and 2 and precursors. | ||
Because neither of these approaches seemed ideal, we initially sought to adapt an intramolecular photochemical cycloaddition of diacrylimides (Scheme 2). An analogous system derived from methacrylimide was reported by Lalonde and Aksentijevitch as providing the cis-1,3 isomer (6).17 This assertion was subsequently confirmed spectroscopically by Bellus and coworkers.18 Preparation of the required diacrylimide (5) by alkylation of acryloyl chloride and phenethyl amine proceeded smoothly in 73% yield. Subsequent photolysis and hydrolysis of the imide to a monoamide–monocarboxylic acid provided a single cyclobutane product; however, single crystal X-ray analysis‡ revealed that this was not the desired 1,3-substituted compound, but rather the cis-1,2-cycloadduct 7.19 Presumably, the reduced steric demand of 5 (X = H) switches the mode of the cycloaddition, and we are currently exploring alternative syntheses employing removable groups at the α-position of the acryloyl functionality to overcome this problem.
 | ||
| Scheme 2 Intramolecular photocycloaddition products. | ||
We next turned to the literature synthesis of cis-1,3-cyclobutane carboxylic acid 8, originally reported by Allinger and coworkers.20 While not expedient (9 steps), this route permitted sufficient production of cyclobutane 8 to permit carrying the synthesis forward (Scheme 3). Reduction of the cis-1,3-cylobutane dicarboxylic acid 8 with a borane–THF complex afforded the diol (9) in 83% yield. Conversion of diol 9 to a dialdehyde was accomplished via PCC oxidation. Subsequent Horner–Wadsworth–Emmons olefination by analogy to our previous efforts yielded several products; separation and HPLC analysis indicated that a substantial fraction of the material had epimerized under the reaction conditions to provide a mixture of the cis- and trans-1,3-cyclobutane isomers. This epimerization had not been a complicating factor in our prior synthesis of TWTCP. One potential factor in this difference is the higher s-character in the exocyclic bonds of cyclobutane rings relative to cyclopentane rings, postulated as a factor in the increased acidity of cyclobutyl protons in other studies,21 may allow for more facile deprotonation of this system. Substitution of Roush–Masamune conditions22 in the olefination reaction provided the desired cis compound (10) in 32% yield over two steps from 9.
 | ||
| Scheme 3 (a) 1 M BH3–THF (5 eq), THF, 12 h, RT, 83%; (b) PCC (3 eq.), CH2Cl2, RT, 4 h; (c) (i-PrO)2POCH2COCH2CH2Ph (2.4 eq.), DBU (2.4 eq.), LiCl (2.0 eq.), CH3CN, RT, 8 h, 32% (over two steps); (d) Me3Al (0.05 eq.), AlCl3 (0.5 eq.), CH2Cl2, 0 °C, 1 h, then cyclopentadiene (10 eq.), CH2Cl2, 4 °C, 12 h, 60%; (e) LiAlH4 (6 eq.), THF, RT, 4 h; (f) BzCl (8 eq.), Et3N (9.25 eq.), CH2Cl2, RT, 4 h, 61% (over two steps); (g) O3, MeOH–CH2Cl2 (1 : 1), −78 °C, 1 h, then NaBH4 (14 eq.), RT, 6 h, 58%; (h) Boc–L-Trp (5.5 eq.), DCC (6.4 eq.), DMAP (3.6 eq.), CH2Cl2–DMF (7 : 3) RT, 12 h; (i) TFA, triethylsilane, CH2Cl2, RT, 12 h, 14% (over two steps). | ||
Double Diels–Alder reaction of 10 proceeded smoothly to provide 11 (two inseparable diastereomers, only one of which is shown) in 60% yield following chromatography. Interestingly, compound 11 (and all subsequent compounds in the synthesis) elutes as a single peak on normal phase and gradient reverse-phase HPLC, as well as on several chiral HPLC columns. However, 13C NMR spectra clearly show doubling of several resonances, indicating that 11 is most likely a mixture of meso and pseudo C2-symmetric diastereomers. Because the primary goal of this study was simply to demonstrate that compounds incorporating the cis-1,3-disubstituted cyclobutane are able to bind lipid A, we set the issue of diastereomeric purity aside for the time being and proceeded with the synthesis. Subsequent reduction of 11 and derivatization with benzoyl chloride afforded 12 in 61% yield over two steps. Ozonolysis of 12 followed by reductive workup with NaBH4 provided a 58% yield of the tetra alcohol, 13. Finally, DCC-mediated coupling of N-Boc-L-tryptophan and global deprotection with trifluoroacetic acid provided the desired receptor 2 in 14% yield over two steps.
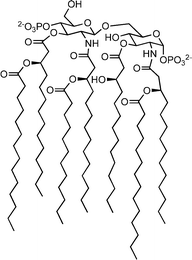
Like 1, 2 is sufficiently soluble in buffered aqueous solution to permit binding analysis by spectrophotometric methods. We evaluated its affinity for diphosphoryl lipid A (14, from E. coli F583, Rd mutant, and 15, from Salmonella minnesota Re595) by UV-Vis titration in HEPES buffer at pH 7.0. An example titration is shown in Fig. 2. We also confirmed the affinity of 1 and 2 for E. coli F583, Rd mutant lipid A by fluorescence titration. In all cases, changes in the absorbance (λmax = 280 nm) or fluorescence (λem = 357 nm) of indole substitutents as a function of added lipid A were observed. Both UV-Vis and fluorescence titration measurements were corrected for sample dilution; UV-Vis titrations were also corrected for the concentration-dependent absorbance of lipid A. As shown in Table 1, 2 binds lipid A with affinity comparable to 1. We observed only a slight difference in affinity between E. coli and S. minnesota derived lipid A; these structures differ only in the number of lipid chains (6 vs 7, respectively). This observation supports a binding model in which 2 interacts primarily with the diphosphoryl disaccharide headgroup of lipid A, rather than with the lipid tails. Analysis of the interaction using the method of continuous variation (Job plot) showed an inflection point at approximately 50% mole fraction of 2; this provides strong support for the existence of a 1 : 1 binding interaction (Fig. 3). This also suggests that the lipid A binding constants for the two diastereomers of 2 are similar, since one would expect to see deviation from 1 : 1 binding behavior if the two diastereomers present in the mixture had substantially different affinities for lipid A.

 | ||
| Fig. 2 UV-Vis titration of 6.0 µM TW545 (2) with 300 µM E. coli diphosphoryl lipid A in 20 mM HEPES at pH 7.0. Extracted A280 values are corrected for lipid A absorbance and dilution of 2. A Kd of 5.9 µM is derived from nonlinear least-squares fit (dashed line) to a one-site binding model. | ||
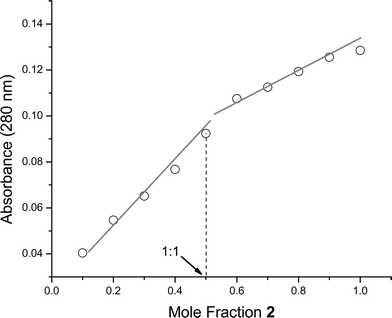 | ||
| Fig. 3 Job plot of E. coli diphosphoryl lipid A into 2 (total concentration 12.0 µM in HEPES, pH 7.00) is consistent with a 1 : 1 binding stoichiometry. | ||
Conclusions
In conclusion, we have reported the first synthesis of 2, a cis-1,3-cyclobutane analog of our previously disclosed lipid A receptor, TWTCP (1). This represents the first example of the synthesis of a designed receptor incorporating a cis-1,3-disubstituted cyclobutane moiety. The affinity of 2 for diphosphoryl lipid A in aqueous solution is similar to that observed for TWTCP, suggesting that this subtle structural modification does not diminish the binding ability of the receptor. Additional experiments designed to explore structural and conformational differences between TWTCP and TW545, as well as synthetic studies designed to improve the diastereoselectivity of the bidirectional Diels–Alder reaction, are in progress.Experimental
All nonaqueous reactions were conducted in flame dried glassware, under an atmosphere of N2, and were stirred with a Teflon coated magnetic stir-bar, unless otherwise stated. Air-sensitive reagents and solutions were transferred via syringe (unless otherwise stated) and were introduced to the reaction vessel through a rubber septum. Room temperature (RT) refers to ambient temperatures, which is approximately 25 °C. Unless otherwise stated, temperatures other than RT denote the temperature of the cooling/heating bath. All distillations were performed under an N2 atmosphere, or under at reduced pressure aspirator (15–30 mm Hg), or vacuum pump (≫1 mm Hg). The phrase “reduced or concentrated in vacuo” refers to removal of solvent by means of a Buchi rotary-evaporator attached to an aspirator pump (10 mm Hg).Purification by flash chromatography was performed following the procedure of Still23 using the indicated solvent systems on EM Reagents silica gel 60 (230–400) mesh. Analytical thin layer chromatography (TLC) was performed using EM silica gel 60 F-254 pre-coated glass plates (0.25 mm). Visualization was effected by short-wave UV illumination and/or by dipping the plate into a solution of KMnO4, ceric ammonium molybdate (CAM), or 2,4-dinitrophenyl hydrazine (2,4-DNPH), followed by heating on a hot plate.
Analytical purification using high pressure liquid chromatography (HPLC) was performed on a Shimadzu LC-2010A (reverse phase) and a Beckman 112 Solvent Delivery System and Waters 450 Variable Wavelength Detector set at 254 nm (normal phase).
Reagent-grade solvents were used without further purification for all extractions and work-up procedures. Deionized water was used for all aqueous reactions, work-ups, and for the preparation of all aqueous solutions. Reaction solvents diethyl ether (Et2O), tetrahydrofuran (THF), methylene chloride (CH2Cl2), triethylamine (TEA), and dimethyl formamide (DMF) were obtained from a Glass Contour solvent purification system.
Proton nuclear magnetic resonance (NMR) spectra were obtained on a Bruker WH-400 (400 MHz), or an Avance 400 (400 MHz), or a Bruker-500 (500 MHz) instrument. Carbon NMR spectra were obtained on an Avance 400 (75 MHz), or on a Bruker-500 (75 MHz). Chemical shifts are reported in ppm (δ) relative to the appropriated deuturated solvent. Multiplicity was designated by the following abbrevations and combinations thereof: singlet (s), doublet (d), doublet of doublet (dd), doublet of triplets (dt), multiplet (m). Infrared (IR) spectra were recorded on a Perkin Elmer 1610 FT-IR spectrophotometer and are reported in wavenumbers (cm−1) with a polystyrene standard. High resolution mass spectrometry (HRMS) was performed at the mass spectrometry facility at the State University of New York at Buffalo, Buffalo, NY. The X-ray crystallographic data‡ were collected on a standard Siemens SMART CCD Area Detector System equipped with a normal focus molybdenum-target X-ray tube operated at 2.0 kW (50 kV, 40 mA).
Photochemical irradiations were carried out using a 450 W Hanovia medium pressure mercury vapor arc lamp. The lamp operates at an internal pressure between 1 to 10 atm and emits radiation over the region of 200–1400 nm. Particularly intense emissions are at 254, 313, 366, 435.8, and 546.1 nm. All solvents used were degassed via bubbling nitrogen gas through the solution for 30 min. The irradiations were carried out in immersion well-type reaction vessels. The outer well was a three neck flask that held 250 ml of solvent. For larger scale reaction a 2000 ml graduated cylinder was used. The inner well was a quartz glass immersion well with a water cooled jacket. To select the lower wavelength output from the lamp only quartz glassware was used. To filter out 254 nm light and allow only wavelengths greater than 300 nm a pyrex filter was used that surrounded the lamp.
N-Phenethyl diacrylimide (5)
Acryloyl chloride (4.63 ml, 56.97 mmol) was added to 55 ml of dry methylene chloride at 0° C. To this cooled and stirred solution was added dropwise over 20 min 2,6 lutidine (7.29 ml, 62.67 mmol). The solution became orange in color. Phenethylamine (2.50 ml, 19.99 mmol) was then added dropwise via an addition funnel over 1 h. After the addition was complete, the reaction was stirred at 0° C for 1 h, and then allowed to warm to room temperature. After 16 h at room temperature, the reaction was quenched with the addition of 50 ml of 10% HCl. The organic layer was separated and washed with 3 × 50 ml of 10% HCl, 1 × 50 ml water, 3 × 50 ml saturated sodium bicarbonate solution. The organic layer was then washed with brine solution, dried with magnesium sulfate, filtered, and concentrated in vacuo to give a yellow oil. Purification of the yellow oil via flash chromatography (silica, 75 : 25 hexanes–ethyl acetate) yielded 5 as a light yellow oil (3.34 g, 14.59 mmol, 73% yield). νmax/cm−1 (film) 3023, 1680, 1651, 1620, 1401, 1150; δH (400 MHz, CDCl3) 7.32 (2H, t); 7.28–7.22 (3H, m); 6.59 (2H, dd, J = 3.2, 13.6 Hz); 6.42 (2H, d, J = 9.2 Hz); 5.79 (2H, d, 5.8 Hz); 4.00 (2H, t, J = 7.6 Hz); 2.93 (2H, t, J = 7.6 Hz); δC (75 MHz, CDCl3) 168.8, 138.1, 130.5, 129.9, 128.9, 128.6, 126.7, 46.3, 35.2; HRMS m/z (EI) 229.1098 (M+. C14H15NO2 requires 229.1103).1,2-Monocarboxylic acid, mono N-phenethyl amide cyclobutane (7)
N-Phenethyl diacrylimide (5) (1.661 g, 7.22 mmol) was dissolved in 1400 ml of acetonitrile and was then degassed by bubbling nitrogen through the solution. The solution was poured into a 2000 ml graduated cylinder and a quartz cooled water jacketed photochemical immersion well was submerged into the solution. A 450 W Hanovia medium pressure mercury vapor lamp was used and the solution was irradiated for 18 h. The solution was then concentrated in vacuo to yield a dark brown oil, and 75 ml of 3.5 M KOH was added. The reaction was stirred at RT for 20 min and then the solid was filtered off with suction filtration. The solid was then washed with 3 × 25 ml of 3.5 M KOH to yield a light tan solid. This was dissolved in 50 ml of 10% HCl, concentrated, and dried in vacuo to yield a light tan solid (1.159 g, 4.693 mmol, 65% yield over two steps). Crystals suitable for X-ray analysis‡ were obtained by dissolution in methanol followed by slow evaporation of the solvent. νmax/cm−1 (film) 3323, 2940, 1718, 1615, 1548, 1168; δH (400 MHz, CDCl3) 7.32 (2H, t); 7.28–7.20 (3H, m); 5.63 (1H, bs); 3.58 (2H, m); 3.42 (1H, m); 3.30 (1H, m); 2.84 (2H, t, J = 5.8 Hz), 2.49 (1H, m); 2.30–2.17 (3H, m); δC (75 MHz, CDCl3) 172.5, 169.6, 134.7, 124.8, 124.7, 122.5, 38.4, 36.8, 36.7, 31.5, 18.7, 18.2; HRMS m/z (EI) 247.1197 (M+. C14H17NO3 requires 247.1208).Ester 10
Pyridinium chlorochromate (3.724 g, 17.28 mmol) was dissolved in 35 ml of CH2Cl2 and the resulting orange slurry was stirred vigorously. Diol 9 (668 mg, 5.76 mmol) was dissolved in 6 ml of CH2Cl2 and was then added dropwise to the solution over 20 min. The reaction was allowed to run for 4 h, at which time monitoring via TLC indicated disappearance of starting material and formation of an aldehyde (2,4-DNPH stain). The reaction was then quenched with 150 ml of diethyl ether, and was stirred for 1 h. The ether layer was decanted away from the black, tarry chromium residue, filtered through a pad of Florisil, and flushed with 5 × 50 ml of ether. The solution was then concentrated very carefully in an ice bath in vacuo to remove the majority of solvent. When the total weight of the crude mixture became ∼1 g, concentration was stopped, and carried on to the next step.Reagent quantities for the olefination step were calculated based on an assumption that the maximum yield in the foregoing reaction was 75%. To a stirred suspension of lithium chloride (440 mg, 10.37 mmol) in 100 ml of dry acetonitrile was added (i-PrO)2POCH2COCH2CH2Ph (5.10 g, 10.37 mmol) and then 10 min later was added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.29 ml, 8.64 mmol). The solution was stirred for 10 min, and the crude cyclobutane cis-1,3-dialdehyde (∼484 mg, 4.32 mmol) was then added. After the addition of all of the reagents the lithium chloride began to dissolve. The reaction was monitored via TLC and after 8 h was complete. The reaction solution was quenched with the addition of water (20 ml). The solution was extracted with diethyl ether (4 × 100 ml). The organics were pooled and washed with brine solution, dried with magnesium sulfate, filtered and concentrated in vacuo to yield a yellow oil. Purification of the yellow oil via flash column chromatography (silica, 80 : 20 hexanes–diethyl ether) afforded 10 as a light yellow oil (743 mg, 1.84 mmol, 32% over two steps, E,E : E,Z > 20 : 1). νmax/cm−1 (film) 2977, 1703, 1646, 1465, 1380, 1303, 1266, 1158, 1093; δH (400 MHz, CDCl3) 7.31 (4H, t); 7.23 (6H, m); 7.10 (1H, dd, J = 15.6 Hz); 6.94 (1H, dd, J = 15.6 Hz); 5.75 (2H, dd, J = 15.6 Hz ); 4.35 (4H, q, ); 3.11 (1H, q); 3.03 (5H, m); 2.42 (1H, q); 2.21 (2H, t ); 1.88 (1H, q); δC (75 MHz, CDCl3) 166.7, 151.7, 151.4, 137.9, 128.9, 128.5, 126.6, 119.9, 119.7, 64.9, 35.2, 33.8, 33.6, 32.0; HRMS m/z (EI) 404.1975 (M+. C26H28O4 requires 404.1988).
Diels–Alder adduct 11
Bis-dienophilic ester 10 (565 mg, 1.40 mmol) was dissolved into 5 ml methylene chloride. The solution was cooled to 0 °C for 15 min. Addition of (CH3)3Al (0.045 ml, 0.07 mmol, 2.0 M in hexanes) produced the evolution of small amounts of gas. After allowing this mixture to stir at 0 °C for 10 min, AlCl3 (0.70 ml, 0.70 mmol, 1.0 M in CH3NO2) was then added to the solution via syringe, and the reaction was stirred for 5 min at 0 °C. Cyclopentadiene (924 mg, 14 mmol, 4.0 M in CH2Cl2) was added via a syringe over 30 min (0.5 ml portions every 5 min). The reaction was then warmed to 4 °C and allowed to proceed with stirring for 12 h. The reaction was then quenched with pyridine (0.2 ml) and warmed to RT. The resulting white slurry was filtered through a plug of silica gel, and washed with diethyl ether (5 × 20 ml). The organics were reduced in vacuo. The pyridine and CH3NO2 were removed azeotropically via treatment with heptane (4 × 10 ml). Final concentration in vacuo yielded a yellow residue. Purification via flash chromatography (silica, 95 : 5, hexanes–diethyl ether) afforded 11 as a yellow oil (449 mg, 0.84 mmol, 60% yield) νmax/cm−1 (film) 2961, 1721, 1468, 1380, 1168, 1091; δH (400 MHz, CDCl3) 7.35–7.29 (4H, m); 7.26–7.22 (6H, m); 6.23 (2H, m); 5.83 (2H, apparent q); 4.35–4.21 (4H, m); 3.07 (2H, bs); 2.93 (4H, t, J = 7 Hz); 2.50 (2H, bs); 2.32 (2H, m); 2.17–2.04 (2H, m); 1.93–1.83 (3H, m); 1.81 (1H, m); 1.67 (1H, m); 1.57 (1H, m); 1.48–1.33 (4H, m); δC (75 MHz, CDCl3) 174.8, 174.5, 138.7, 138.5, 137.9, 133.5, 128.8, 128.4, 126.4, 64.6, 50.3, 49.5, 49.4, 46.2, 46.2, 46.1, 45.8, 45.0, 36.8, 36.1, 35.1, 33.0, 31.1; HRMS m/z (EI) 536.2908 (M+: C36H40O4 requires 536.2927).Dibenzoate 12
LiAlH4 (140 mg, 3.774 mmol) was slurried into 12 ml of THF at RT. Ester 11 (337 mg, 0.629 mmol) in 2 ml of THF was added dropwise via syringe to this slurry over 15 min at RT. During the addition, hydrogen gas was evolved. The mixture was stirred for 4 h at RT. It was then carefully quenched sequentially with water (0.14 ml), 15% aq. NaOH (0.14 ml), and more water (0.42 ml). The reaction formed a white precipitate, which stirred vigorously for an additional 2 h at RT. The reaction mixture was filtered through a plug of Celite and subsequently washed with ether (5 × 10 ml). The filtrate was dried over magnesium sulfate, filtered, and reduced in vacuo to give a yellow oil that was used without further purification. The crude oil was an inseparable mixture of pheneythyl alcohol (1.258 mmol) and the desired diol. The diol was slurried into 6 ml of CH2Cl2 at RT. Sequential addition of benzoyl chloride (706 mg, 5.032 mmol), and Et3N (0.80 ml, 5.832 mmol) at RT caused the reaction to become a pale yellow. After stirring for 1 h at RT, the reaction began to form a white precipitate. After 4 h, the reaction mixture was quenched with water (5 ml), and poured into diethyl ether (20 ml). The layers were separated, and the organic layer was washed sequentially with water (2 × 20 ml), 10% HCl (3 × 20 ml), water (1 × 20 ml), and saturated aq. Na2CO3 (3 × 20 ml). The organic layers were combined, washed with brine, dried over magnesium sulfate, filtered, and reduced in vacuo to afford a tan oil. Purification of the oil via flash column chromatography (silica, 95 : 5 hexanes–ether) yielded 12 (194 mg, 0.382 mmol, 61% yield over two steps) as a pale yellow oil νmax/cm−1 (film) 2961, 1711, 1445, 1385, 1272, 1114; δH (400 MHz, CDCl3) 8.07 (4H, t); 7.62–7.40 (6H, m); 6.30–6.15 (2H, m) 6.10–6.03 (2H, m); 4.32–4.06 (2H, m); 3.97–3.81 (2H, m); 2.90–2.70 (2H, m); 2.56–2.20 (2H, m); 2.11 (1H, m), 2.03–1.85 (5H, m); 1.47–1.37 (5H, m); 1.31 (1H, m), 1.10 (1H, dd); 0.86 (1H, dd); δC (75 MHz, CDCl3) 162.7, 134.5, 129.8, 128.9, 126.5, 125.6, 124.4, 64.9, 64.8, 46.6, 45.1, 45.1, 42.3, 42.1, 41.2, 40.6, 40.5, 40.4, 40.2, 32.7, 30.1, 28.0; HRMS m/z (EI) 508.2607 (M+. C34H36O4 requires 508.2614).Alcohol 13
The dibenzoate 12 (158 mg, 0.311 mmol) was dissolved into 3 ml of a solution of 1 : 1 CH2Cl2–MeOH. This solution was then cooled to −78 °C for 10 min. O3 was bubbled through the solution at a moderate rate, and after 1 h the solution became a persistent blue color. After another 20 min of O3 treatment, O3 production was discontinued, and O2 was bubbled through the solution for approximately 5 min to remove any excess O3. After the blue color disappeared, the reaction was warmed from −78 to 0 °C and maintained at 0 °C for 10 min. NaBH4 (165 mg, 4.354 mmol) was added slowly. (Caution: vigorous evolution of H2 gas.) After all of the NaBH4 had been added, the reaction was stirred and warmed slowly to RT over 30 min. Once at RT, the reaction was stirred for an additional 6 h. 10% aq. HCl was then added until a pH of 1.0 was reached, as measured by pH paper. The mixture was diluted with ethyl acetate, and the layers were separated. The aqueous layer was extracted with more ethyl acetate (3 × 20 ml) and the organic extracts were combined and then washed sequentially with water (20 ml), sat. aq. Na2CO3 (3 × 20 ml), and brine solution (1 × 20 ml). The organic solution was then dried over magnesium sulfate, filtered, and reduced in vacuo to afford a white solid. The solid was purified via flash chromatography (silica, 95 : 5 CH2Cl2–MeOH) to afford 13 (105 mg, 0.181 mmol, 58% yield) as a colorless oil νmax/cm−1 (film) 3400, 2923, 1715, 1558, 1272, 668, 609; δH (400 MHz, CDCl3) 8.17–8.02 (4H, m); 7.77–7.61 (2H, m); 7.60–7.48 (4H, m); 4.51–4.11 (4H, m); 3.83–3.40 (8H, m); 2.61–2.42 (1H, m); 2.42–2.10 (7H, m); 2.10–1.73 (7H, m); 1.608–1.57 (1H, m); 1.50–1.30 (2H, m); δC (75 MHz, CDCl3) 161.7, 132.8, 130.2, 130.1, 129.1, 128.2, 66.2, 65.6, 65.4, 65.2, 65.1, 62.1, 62.1, 49.9, 49.8, 48.8, 48.7, 46.1, 45.9, 44.1, 43.9, 43.7, 43.5, 43.4, 43.2, 42.9, 42.6, 37.1, 36.9, 36.6, 36.5, 32.7, 32.6, 31.7, 29.7; HRMS m/z (MALDI) 603.2903 (M + Na+. C34H44O8Na requires 603.2953).Receptor 2
Alcohol 13 (32 mg, 0.055 mmol) was dissolved into 1 ml of DMF. Boc–L-tryptophan (92 mg, 0.303 mmol) was added to the solution, followed 10 min later by DCC (73 mg, 0.352 mmol), and by DMAP (24 mg, 0.196 mmol) after another 10 min. This solution was stirred at RT for 21 h, at which point a significant amount of white solid had precipitated. The mixture was filtered through Celite to remove the precipitant, and the pad was washed with ethyl acetate (5 × 10 ml). The organics were combined, and were washed sequentially with 1% HCl (4 × 10 ml), water (1 × 10 ml), sat. aq. NaHCO3 solution (4 × 10 ml), and brine solution, dried over sodium sulfate, and then filtered through filter paper. The solution was then filtered through a pad of Celite to remove any remaining white solid, and was reduced in vacuo affording a light tan residue (117 mg) that was used without further purification. The crude Boc-protected tetratryptophaninate was dissolved into 0.84 ml of CH2Cl2. In a separate vial, 0.361 ml of trifluoroacetic acid, 0.180 ml of triethyl silane, and 0.3 ml of CH2Cl2 were mixed and then added dropwise via a syringe over 15 min to the Boc-tryprophaninate solution. The reaction was allowed to run for 12 h and was then concentrated in vacuo to afford a yellow oil. The residue was purified via reverse-phase HPLC using gradient elution (linear gradient of 90 : 10 0.1% TFA in water–0.1% TFA in CH3CN to 100% of 0.1% TFA in CH3CN over 30 min) to give 2 as a light tan–yellow viscous oil (10 mg, 0.0076 mmol, 14% yield over two steps) νmax/cm−1 (film) 3357, 2924, 1684, 1272, 1204, 1134, 1025; δH (400 MHz, CD3OD) 8.00 (4H, bs); 7.67–7.32 (15H, m); 7.25 (11H, m); 4.44–4.24 (6H, m); 4.24–3.87(12H, m); 3.87–3.77 (2H, m); 3.71–3.65 (2H, unresolved t); 3.63–3.54 (2H, m); 3.39 (8H, bs); 2.43–2.25 (2H, m); 2.14–1.89 (8H, m); 1.88–1.47 (12H, m); 1.44–1.27 (4H, m); 1.04–0.86 (1H, bm), 0.75–0.61 (1H, bm); δC (75 MHz, CD3OD) 169.2, 166.4, 136.8, 136.8, 133.1, 129.9, 129.9, 129.2, 129.1, 128.3, 126.8, 124.2, 124.0, 121.6, 118.9, 117.5, 111.4, 111.4, 66.4, 66.2, 66.2, 53.3, 43.9, 43.5, 43.2, 43.0, 41.8, 40.1, 40.1, 39.6, 39.4, 36.2, 36.0, 29.3, 28.5, 26.9, 26.6, 26.5; LRMS m/z (ES) 1325.4 (M + H+. C78H85N8O12 requires 1325.6).Lipid A and LPS preparation
Diphosphoryl lipid A Escherichia coli F583 (Rd Mutant) or LPS, was obtained from Sigma Chemical Company and used without purification. The diphosphoryl lipid A was first suspended in doubly distilled water, then diluted by half in 40.0 mM HEPES buffer, pH 7.00. The diluted diphosphoryl lipid A sample was then sonicated for 30 min at room temperature. Finally, the diphosphoryl lipid A sample was heat cycled no fewer than four times between 4 and 70 °C, then stored at 4 °C for 12 h before use.General procedure for UV-vis titrations
A 600 µl, 12.0 µM solution of 2.1 or 2.14 in 20.0 mM HEPES, pH = 7.00 was allowed to equilibrate in a 1.0 ml quartz UV cell at ambient temperature in a Shimadzu 1600-PC UV spectrophotometer. Tryptophan indole absorbance was monitored at 280 nm to ensure a completely equilibrated solution had been obtained. Aliquots of 2.0 µl of a 150 µM diphosphoryl lipid (prepared as described above) were sequentially added to the cuvette. After each addition, the solution was allowed to equilibrate for a minimum of 10 min, then scanned for absorbance from 500–220 nm. The control experiment of diphosphoryl lipid A titrated into HEPES buffer was subtracted from each of the titration experiments. Other titrations discussed in the text were carried out analogously.General procedure for fluorescence titrations
A 200 µl, 12.0 µM solution of 2.1 or 2.14 in 20.0 mM HEPES, pH = 7.00 was allowed to equilibrate in a 0.3 ml quartz fluorescence cell at ambient temperature in a Aminco-Bowman Series 2 Luminescence Spectrometer Fluorometer. Tryptophan indole emission was monitored at 357 nm to ensure a completely equilibrated solution had been obtained. Aliquots of 1.0 µl of a 150 µM diphosphoryl lipid A were sequentially added to the cuvette. After each addition, the solution was allowed to equilibrate for a minimum of 10 min, then excited at 280 nm and emission was monitored between 300–400 nm. The control experiment of diphosphoryl lipid A titrated into HEPES buffer showed zero fluorescence; thus, was not subtracted from the fluorescence data. Other fluorescence titrations discussed in the text were carried out analogously.We thank Dr Rene Lachicotte for carrying out X-ray crystallographic analysis of compound 7. This research was supported by the NIH-NIGMS (5RO1-GM62825). S. G. T. was supported via an NIH training grant in Dermatology (T32AR007472).
References
- Selected reviews include: A. P. Davis and T. D. James, in Functional Synthetic Receptors, ed. T. Schrader and A. D. Hamilton, Wiley-VCH, Weinheim, 2005. p. 47 Search PubMed; A. P. Davis and R. S. Wareham, Angew. Chem., Ind. Ed., 1999, 38, 2978 Search PubMed.
- A. Laederach and P. J. Reilly, Proteins: Struct., Funct., Bioinf., 2005, 60, 591 Search PubMed; C. A. Aarnoudse, J. J. Garcia-Vallejo, E. Saeland and Y. van Kooyk, Curr. Opin. Immunol., 2006, 18, 105 CrossRef CAS; E. Yuriev, W. Farrugia, A. M. Scott and P. A. Ramsland, Immunol. Cell Biol., 2005, 83, 709 CrossRef CAS.
- S. Chan, S. R. Horner, B. L. Miller and P. M. Fauchet, J. Am. Chem. Soc., 2001, 123, 11797 CrossRef CAS.
- K. A. Miller, E. V. K. Kumar, S. J. Wood, J. R. Cromer, A. Datta and S. A. David, J. Med. Chem., 2005, 48, 2589 CrossRef CAS; B. Ding, N. Yin, Y. Liu, J. Cardenas-Garcia, R. Evanson, T. Orsak, M. Fan, G. Turin and P. B. Savage, J. Am. Chem. Soc., 2004, 126, 13642 CrossRef CAS.
- C. R. H. Raetz, Annu. Rev. Biochem., 1990, 59, 129 CrossRef CAS; R. J. Ulevitch and P. S. Tobias, Curr. Opin. Immunol., 1994, 6, 125 CrossRef CAS.
- L. S. Young, W. J. Martin, R. D. Meyer, R. J. Weinstein and E. T. Anderson, Ann. Intern. Med., 1977, 86, 456 Search PubMed.
- T. M. Chapman and M. R. Golden, Biochem. Biophys. Res. Commun., 1972, 46, 2040 CrossRef CAS; M. E. Falagas and S. K. Kasiakou, Clin. Infect. Dis., 2005, 40, 1333 CrossRef CAS; M. D. Bruch, Y. Cajal, J. T. Koh and M. K. Jain, J. Am. Chem. Soc., 1999, 121, 11993 CrossRef CAS.
- C. Li, L. P. Budge, C. D. Driscoll, B. M. Willardson, G. W. Allman and P. B. Savage, J. Am. Chem. Soc., 1999, 121, 931 CrossRef CAS.
- R. D. Hubbard, S. R. Horner and B. L. Miller, J. Am. Chem. Soc., 2001, 123, 5810 CrossRef CAS.
- Y. Matsumura, T. Shimada, T. Nakayama, M. Urushihara, T. Asai, Y. Morizawa and A. Yasuda, Tetrahedron, 1995, 51, 8771 CrossRef CAS; S. K. Chung, S. H. Ban, S. H. Kim, B. E. Kim and S. H. Woo, Bioorg. Med. Chem. Lett., 1995, 5, 1097 CrossRef CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490 CrossRef CAS.
- W. C. Still, A. Tempczyk, R. C. Hawley and T. Hendrickson, J. Am. Chem. Soc., 1990, 112, 6127 CrossRef CAS.
- All molecular mechanics calculations and conformational analyses were conducted using Maestro 4.1.012/Batchmin 7.2 (Schroedinger, Inc.).
- N. L. Allinger and L. A. Tushaus, J. Org. Chem., 1965, 30, 1945 CAS; P. Pecquet, F. Huet, M. Legraverend and E. Bisagni, Heterocycles, 1992, 34, 739 CrossRef CAS.
- H. Leininger, F. Lanzendörfer and M. Christl, Chem. Ber., 1983, 116, 669 CrossRef CAS.
- R. G. Carlson and K. D. May, Tetrahedron Lett., 1975, 11, 947 CrossRef.
- R. T. Lalonde and R. I. Aksentijevich, Tetrahedron Lett., 1965, 6, 23 CrossRef.
- A. Alder, B. N. Buehler and C. Bellus, Helv. Chim. Acta, 1982, 65, 2405 CrossRef CAS.
- Selected X-ray crystallographic data: compound 7, C14H17NO3, formula weight 247.29, crystallized in an orthorhombic crystal system of unit cell dimensions a = 11.7699(8) Å, b = 9.9771(7) Å, c = 21.9102(15) Å, and angles α = 90°, β = 90°, and γ = 90°. Unit cell volume was 2572.9(3) Å3. Data were collected at 292 K. The, crystal was found to be of space group Pca2(1), with two molecules in the unit cell. Linear absorption coefficient was 0.090 mm−1. 10446 reflections were collected, 2830 of which were independent. Final R indices [I > 2σ(I)] were R1 = 0.0539, wR2 = 0.1474, while R indices (all data) are R1 = 0.0712, wR2 = 0.1582.
- N. L. Allinger and L. A. Tushaus, J. Org. Chem., 1965, 30, 1945 CAS.
- J. A. Chang, Y. Chiang, J. R. Keeffe, A. J. Kresge, V. A. Nikolaev and V. V. Popik, J. Org. Chem., 2006, 71, 4460 CrossRef CAS.
- M. A. Blanchette, W. Choy, J. T. Davis, A. P. Essenfeld, S. Masamune, W. R. Roush and T. Sakai, Tetrahedron Lett., 1984, 25, 2183 CrossRef CAS.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: X-Ray data for compound 7 and additional UV-Vis and fluorescence titration data for compounds 1 and 2. See DOI: 10.1039/b610727c |
| ‡ CCDC reference number 616053. For crystallographic data in CIF format see DOI: 10.1039/b610727c |
| This journal is © The Royal Society of Chemistry 2006 |