Pyrazolyl-functionalized 2-methylimidazolium-based ionic liquids and their palladium(II) complexes as recyclable catalysts†
Ruihu
Wang
,
Melissa M.
Piekarski
and
Jean'ne M.
Shreeve
*
Department of Chemistry, University of Idaho, Moscow, Idaho 83844-2343, USA. E-mail: jshreeve@uidaho.edu
First published on 20th April 2006
Abstract
A series of pyrazolyl- and 3,5-dimethylpyrazolyl-functionalized 2-methylimidazolium-based salts have been prepared through neat reactions of 1-pyrazolylmethylene-2-methylimidazole and 1-(3,5-dimethylpyrazolylmethylene)-2-methylimidazole with alkyl or polyfluoroalkyl iodides or 1-bromohexane, followed by anion exchange with LiN(SO2CF3)2 or KPF6. Their thermal properties were determined by DSC and TGA. Most of the bis(trifluoromethanesulfonyl)amide salts are room temperature ionic liquids. The influence of anions and of the structural variation in the 2-methylimidazolium-based cations on the physicochemical properties is discussed. These salts reacted easily with palladium(II) chloride to generate mononuclear palladium ionic liquid complexes. The catalytic activity and recyclability of the palladium complexes in the corresponding ionic liquids were preliminarily examined using Heck, Suzuki and Sonogashira cross-coupling reactions in the absence of phosphine ligands.
Introduction
Palladium-catalyzed coupling and cross-coupling reactions exemplify one of the most important processes in organic chemistry because they represent a powerful and popular method for the formation of carbon–carbon bonds.1–3 Significant innovations in the field have been made in the modifications of traditional phosphine ligands and in the discovery of novel phosphine systems.2,3 However, most of the phosphine ligands are sensitive to air and moisture, with conversion to, for example, phosphine oxides. This can lead to poisoning of the metal which may result in catalyst decomposition. Other problems frequently encountered in the use of phosphine ligands in catalysis, such as cost, high toxicity, laborious synthesis and catalyst loss during product extraction, have driven chemists to explore alternatives.Nitrogen-containing ligands have been widely used in coordination chemistry, supramolecular chemistry and material science.4–14 Some of their palladium complexes were applied in palladium-catalyzed coupling reactions and showed high stability and efficient catalytic activity.5–13 The preparation of these types of catalyst precursors can be carried out in air without detrimental effects on eventual catalytic reactions. Moreover, their coordinating ability with palladium(II) can be easily modified through the careful choice of N-containing coordination groups, which enhances the scope of catalysts for different catalytic applications. Some palladium(II) complexes prepared from nitrogen-containing ligands, such as 1) monodentate ligands with imine,6 oxime,7 pyrazolyl,8 nitrile,9 alkylimidazolyl,10,11a,b and pyridyl groups,11c and 2) chelating bidentenate ligands, including bisimidazolyl,12 diazabutanes13 and dipyridyl ligands,14 have been frequently used in numerous transformations for laboratory and industrial applications. Ionic liquids have been shown to be effective in solvating N-containing palladium(II) complexes.9–13 Catalyst precursors dissolved in ionic liquids can be effectively recovered and recycled. The organic products can also normally be easily separated from catalytic solution by simple distillation or extraction. During the last decade, many reactions were revisited in alternate solvents without significant loss of yields, and sometimes with improved catalytic activity11 and chemo- or regio-selectivity.15
Ionic liquids, especially those based on 1,3-dialkylimidazolium, have received growing interest as green alternatives to traditional volatile solvents in catalysis and organic synthesis owing to their specific physical and chemical properties.16 However, the presence of base is prone to deprotonate C2 of the imidazolium cation resulting in potential side-products17 or inactive polycarbene metal complexes.18 Moreover, the strong acidity of the proton at C2 also leads easily to extensive hydrogen bonding interactions.19 Subsequent investigations have suggested that the drawbacks could be avoided by utilization of 2-alkylimidazolium-based ionic liquids.18,20 However, this type of ionic liquid has been used rarely in palladium-catalyzed coupling reactions, despite the fact that catalysts in these media sometimes exhibited higher catalytic activity than corresponding imidazolium-based ionic liquids.21 This is mainly ascribed to the inability of 2-alkylimidazolium-based ionic liquids to effectively stabilize palladium catalysts during catalytic reactions and to leaching of catalysts during separation of target products in the absence of supporting ligands. Recently, metal-containing ionic liquids have been successfully employed in highly recyclable coupling reactions, in which ionic liquids serve as both immobilization solvent and ligand to the catalysts.9,11,22 Inspired by that strategy, we speculated that if appropriate nitrogen-containing coordinating groups were attached to a 2-alkylimidazolium cation, palladium catalysts might be immobilized effectively in the media by coordination, and the advantages of both nitrogen-containing coordinating groups and 2-alkylimidazolium-based ionic liquids could be realized. Most of all, further development of ionic liquids and their applications in other important processes may require the synthesis of new ionic liquids with suitable properties. As an extension of our research work in recyclable phosphine-free catalytic systems,11 we have been interested in the use of pyrazolyl-functionalized ionic liquids, since pyrazolyl-based palladium(II) compounds are excellent candidates as nitrogen-containing ligands in catalysis owing to their desirable coordinating ability with palladium.8 In this paper, we wish to report the syntheses and characterization of a family of pyrazolyl and dimethylpyrazolyl-functionalized 2-methylimidazolium-based ionic liquids, and to demonstrate their suitability as both ligands and solvents in palladium-catalyzed Heck, Suzuki and Sonogashira cross-coupling reactions.
Results and discussion
The synthetic pathway to salts 2a–2p is depicted in Scheme 1. Compounds 1-pyrazolylmethylene-2-methylimidazole (1a) and 1-(3,5-dimethylpyrazolylmethylene)-2-methylimidazole (1b) were easily prepared from reactions of 1-(chloromethyl)pyrazole hydrochloride and 1-(chloromethyl)-3,5-dimethylpyrazole hydrochloride with 2-methylimidazole in the presence of a base in DMF. In 1a and 1b, there are two basic nitrogen atoms available to participate in quaternization reactions. Since the nitrogen atom in the imidazolyl ring is a stronger electron donor than that in the pyrazolyl ring, we envisaged it would be more easily monoquaternized under appropriate conditions, and a pyrazolyl coordination center would remain after monoquaternization. The neat reactions of 1a and 1b with alkyl and polyfluoroalkyl iodides or 1-bromohexane were carried out at 80 °C for 12 h; subsequent metathesis reactions with either lithium bis(trifluoromethanesulfonyl)amide (LiNTf2) or potassium hexafluorophosphate (KPF6) led to the formation of the corresponding salts 2a–2p in excellent yields. | ||
| Scheme 1 | ||
All of the salts were stable in air and water. Since the replacement of the C2 proton of imidazolium ring by a methyl group precludes hydrogen bonding between the proton and water, these compounds tend to be immiscible with water. This allows purification by simple washing with water to remove water-soluble impurities. The salts are soluble in ethyl acetate and acetone, but they are immiscible with solvents of low polarity, such as ethers and alkanes.
The new salts were characterized by 1H, 13C, 19F NMR and elemental analyses. The 1H and 13C NMR data are routine with minimal or no change in the chemical shifts of the 2-methylimidazolium parent ring. The chemical shifts of the protons of the methylene bridge (–NCH2N–) in the pyrazolyl-functionalized and 3,5-dimethylpyrazolyl-functionalized salts are in the range 6.54–6.63 ppm and 6.41–6.46 ppm, respectively, which are downfield shifts compared to those in 1a (6.03 ppm) and 1b (5.90 ppm). Use of 19F NMR spectra helped to monitor the progress of metathesis reactions when introducing fluorine-containing anions in the form of polyfluoroalkyl chains. The relative areas of the resonance bands from the fluorine atoms of the anions (NTf2 and PF6) in 2c, 2e, 2g and 2i were readily compared with those of the polyfluoroalkyl substituents in the 2-methylimidazolium cation.
Phase transition temperatures (midpoints of glass transition and/or melting points) were determined by differential scanning calorimetry (DSC) and are given in Table 1.
| Compound | R | R′ | Y | T m/°Ca | T d/°Cb |
|---|---|---|---|---|---|
| a Melting point. b Thermal degradation. c Glass transition temperature. | |||||
| 2a | H | CH3 | NTf2 | −21c | 295 |
| 2b | H | (CH2)2CH3 | NTf2 | −32c | 288 |
| 2c | H | (CH2)2CF3 | NTf2 | 85 | 285 |
| 2d | H | (CH2)3CH3 | NTf2 | −44c | 318 |
| 2e | H | (CH2)3CF3 | NTf2 | 75 | 306 |
| 2f | H | (CH2)5CH3 | NTf2 | −50c | 304 |
| 2g | H | (CH2)2(CF2)3CF3 | NTf2 | 112 | 292 |
| 2h | H | (CH2)3CH3 | PF6 | −28c | 266 |
| 2i | H | (CH2)3CF3 | PF6 | 114 | 268 |
| 2j | H | (CH2)5CH3 | PF6 | −32c | 271 |
| 2k | CH3 | CH3 | NTf2 | 80 | 267 |
| 2l | CH3 | (CH2)3CH3 | NTf2 | −34c | 270 |
| 2m | CH3 | (CH2)5CH3 | NTf2 | −38c | 282 |
| 2n | CH3 | CH3 | PF6 | 163 | 242 |
| 2o | CH3 | (CH2)3CH3 | PF6 | −13c | 221 |
| 2p | CH3 | (CH2)5CH3 | PF6 | −25c | 231 |
As anticipated, the anion exhibits a major influence on the melting point. With the same substituents on the cations, changing the anion PF6 in 2h–2j and 2n–2p to NTf2 in 2d–2f and 2k–2m lowers the respective melting points greatly. Melting points of some salts containing PF6 anions, such as 2i and 2n, are above 100 °C which places them outside of the range for ionic liquids.23 Salts with the NTf2 anion, except 2g, fall into the ionic liquid class of compounds because of their lower melting points; most of them are room temperature ionic liquids. With the same anion, an increase of length and flexibility of alkyl groups on cations resulted in a lowering of the melting point, which is in keeping with related salts, such as alkyl-substituted imidazolium or 2-methylimidazolium cations.24 For example, for dimethylpyrazolyl-functionalized salts with NTf2 and PF6, variation of N-substituents from methyl to butyl to hexyl causes the melting points to decrease from 80 °C (2k) to −34 °C (2l) to −38 °C (2m) and from 163 °C (2n), to −13 °C (2o) to −25 °C (2p), respectively. A similar tendency can also be observed in pyrazolyl-functionalized salts; the effect of N-substituents in the NTf2 salts on the melting points is in the following order: methyl (−21 °C) > propyl (−32 °C) > butyl (−44 °C) > hexyl (−50 °C). This suggests decreased packing efficiency in the crystal lattice as the alkyl groups are elongated.
It should be pointed out that the dimethylpyrazolyl-functionalized ionic liquids 2k–2p have higher phase transition temperatures than their pyrazolyl-functionalized analogues. In general, replacement of alkyl substituents by polyfluoroalkyl groups results in an increase in melting point of the salts. For example, trifluoropropyl-substituted salt 2c and trifluorobutyl-substituted salts 2e and 2i have higher melting points than those with propyl- and butyl-substituents, 2b, 2d and 2h. The phase transition temperature of 1H,1H,2H,2H-nonafluorohexyl-substituted salt 2g (112 °C) is much higher than that of its non-fluoro analogue 2f (−50 °C). However, their thermal stabilities were essentially identical as determined by thermogravimetric analysis (TGA). The variation of the alkyl chain in the pyrazolyl-functionalized and dimethylpyrazolyl-functionalized cations has no obvious effect on the stability of the corresponding salts, but variations of anions and functional groups have an apparent influence on the thermal degradation of the salts. The decomposition temperatures of pyrazolyl-functionalized salts with NTf2 and PF6 anions are in the range 285–318 and 266–271 °C, respectively, which are higher than their dimethylpyrazolyl-functionalized analogues (267–282 °C for 2k–2m and 221–242 °C for 2n–2p). The salts with the NTf2 anion are more stable thermally than the corresponding salts with the PF6 anion.
The coordination abilities of the pyrazolyl- and 3,5-dimethylpyrazolyl-functionalized 2-methylimidazolium-based salts were also briefly studied. The reactions of palladium(II) chloride with 2 equiv. of 2d and 2l in methanol produced air stable yellow complexes 3a and 3b (Scheme 2), respectively. There were obvious downfield shifts in the 1H and 13C NMR spectra of pyrazolyl and methylimidazolium rings when compared with those of their respective reactants. Since the melting points of 3a and 3b are 132 and 137 °C, respectively, they can not be regarded as ionic liquids. Thermal decomposition occurred above 280 and 259 °C, which is lower than the analogous temperatures of the corresponding salts 2d (318 °C) and 2l (270 °C).
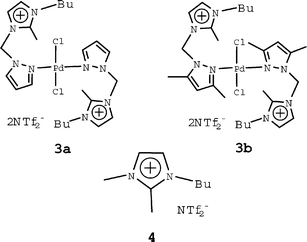 | ||
| Scheme 2 | ||
Palladium-catalyzed Heck cross-coupling reactions in ionic liquid 2d
Palladium-catalyzed Heck cross-coupling reactions usually take place under relatively mild conditions and tolerate a wide variety of functional groups on either coupling partner. Various strategies have been developed for recovery and reuse of notable palladium catalysts in the Heck reaction, especially in the absence of bulky phosphine ligands.9,11,22 One of the predominant aims involving metal-containing ionic liquids is that they can be demonstrated to be ideal immobilization and activation agents for transition-metal catalysts throughout the entire process. Some palladium-containing ionic liquids were reused more than ten times without significant loss of catalytic activity, and even led to the activation of aryl chlorides.11a In our work, the Heck reaction was initially evaluated using 3a as a catalyst precursor dissolved in pyrazolyl-functionalized ionic liquid 2d. As expected, it was possible to carry out the cross-coupling reactions of iodobenzene and n-butyl acrylate nine times in the catalytic solution without detectable loss of catalytic activity (Table 2, entry 1). Interestingly, using the same protocols, 3a-catalyzed cross-coupling reaction of iodobenzene and n-butyl acrylate in a non-functionalized ionic liquid, 1-butyl-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)amide (4), was also performed nine times with complete conversion of iodobenzene (Table 2, entry 2). This case is very different from the reported palladium-catalyzed cross-coupling reactions in that medium.21 This likely happens because 3a possesses a pendant 2-methylimidazolium tag which is similar to ionic liquid 4. The ionophilicity of 3a towards 4 was increased,25 which promoted the immobilization of 3a in 4. However, when PdCl2 was used as the catalyst instead of 3a in 4 under identical reaction conditions, palladium black was formed during the first cycle, causing a decrease of the catalytic activity after the first cycle, and the catalytic solutions to become completely inactive after 5 cycles (Table 2, entry 3). Not surprisingly, when the Heck reaction was examined in 2l using PdCl2 as the catalyst precursor following the same protocols, the catalytic system could be recycled nine times owing to pre-formation of the palladium-coordinating ionic liquid (Table 2, entry 4).|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | ILs | Catalyst | Cycle no. | ||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |||
| a The reactions were carried out using 1.0 mmol iodobenzene, 1.25 mmol n-butyl acrylate, 1.5 mmol Et3N, 2 mol% catalyst and 3.0 g 2d at 110 °C for 3 h. b GC yield. | |||||||||||
| 1 | 2d | 3a | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 |
| 2 | 4 | 3a | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 |
| 3 | 4 | PdCl2 | >99 | 86 | 45 | 12 | 3 | ||||
| 4 | 2l | PdCl2 | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 |

To further test the scope and recyclability of 3a in the ionic liquid 2d, the cross-coupling reactions between a wide array of aryl halides and n-butyl acrylate were examined using similar protocols (Table 3). The reactions of n-butyl acrylate with electron-rich 4-iodotoluene and 4-iodoanisole as well as electron-deficient 1-iodo-4-nitrobenzene and 1-fluoro-4-iodobenzene progressed smoothly. The corresponding n-butyl (E)-cinnamates were obtained in high yields with complete regioselectivity at the β-position. In addition, no cis coupled products were observed. Interestingly, a hetero-aromatic compound, 2-iodothiophene, also reacted with n-butyl acrylate to give the target product in a satisfactory isolated yield (entry 4). All coupled products were easily separated from the catalytic solution by simple extraction with ether and hexane. After extracting the products, the resulting solution was washed with water to remove ammonium salts and dried under vacuum for the next cycle. In the process, no apparent leaching of palladium species was observed. The catalytic solution was recovered and reused successfully twelve times without any detectable loss of catalytic activity.
|
|
||||
|---|---|---|---|---|
| Entry | R | X | (Cycle) Yield (%)b | |
| a Unless otherwise stated, the reactions were carried out using 1.0 mmol aryl halides, 1.25 mmol n-butyl acrylate, 1.5 mmol Et3N, 2 mol% catalyst and 3.0 g 2d at 110 °C for 3 h. b Isolated yield. c 2-Iodothiophene acts as a substrate. d Reaction time 15 h. e GC yield. | ||||
| 1 | H | I | (1) 91 | (2) 91 |
| 2 | Me | I | (3) 92 | (4) 93 |
| 3 | MeO | I | (5) 90 | (6) 92 |
| 4c | I | (7) 84 | (8) 87 | |
| 5 | NO2 | I | (9) 90 | (10) 89 |
| 6 | F | I | (11) 91 | (12) 92 |
| 7 | H | Br | (1) 47 | (2) 63d |
| 8 | H | Cl | (3) <1e | |
| 9 | Me | Br | (4) 10e | (5) 15d,e |
| 10 | CF3 | Br | (6) 75 | (7) 90d |
| 11 | CH3CO | Br | (8) 76 | (9) 86d |
| 12 | F | Br | (10) 69 | (11) 85d |
| 13 | NO2 | Br | (12) 81 | (13) 89d |

In order to continue to evaluate the catalytic system, we used aryl bromides as reactants and found that their electronic nature has a significant effect on the yields of coupled products. The cross-coupling reaction of bromobenzene with n-butyl acrylate at 110 °C for 3 h gave rise to trans-cinnamic acid n-butyl ester in a 47% yield (Table 3, entry 7). Aryl bromides bearing electron-withdrawing substituents, such as 4-bromo(trifluoromethyl)benzene, 4-bromoacetophenone and 4-bromofluorobenzene, reacted with n-butyl acrylate under identical conditions and gave the corresponding coupled products in high isolated yields (Table 3, entries 10–13); prolonged reaction time resulted in the enhancement of the isolated yields. However, a similar reaction using electron-rich 4-bromotoluene in the system gave only a 15% GC yield even after 15 h (Table 3, entry 9). With 4-chlorobenzene, only a trace of the target product was detected after 15 h (Table 3, entry 8).
Palladium(II)-catalyzed Suzuki cross-coupling reactions in ionic liquid 2d
Since our goal was to develop a highly recyclable phosphine-free catalytic system, we next applied this system to Suzuki cross-coupling reactions, another very important carbon-carbon bond formation reaction for the synthesis of biaryl compounds. The cross-coupling reactions of phenylboronic acid with aryl bromides and activated aryl chloride were evaluated using procedures reported previously.26 In the present system, Na2CO3 was used as a base, and water was added as a co-solvent to improve the solubilization of the inorganic base. As shown in Table 4, treatment of phenylboronic acid with bromobenzene and deactivated 4-bromoanisole in the presence of Na2CO3 and 2 mol% catalyst in 2d at 110 °C for 1 h produced the corresponding coupled products in 76 and 53% isolated yields, respectively. The higher isolated yields were obtained when the reaction time was prolonged to 15 h (Table 4, entries 1 and 2). The activated 4-chloroacetophenone also reacted with phenylboronic acid, and a 31% isolated yield was obtained after reaction at 110 °C for 15 h (Table 4, entry 3). The reactions of activated aryl bromides and phenylboronic acid were completed within 1 h, and the corresponding binary compounds were obtained in high isolated yields (Table 4, entries 4–7). These results showed that the palladium-containing pyrazolyl-functionalized ionic liquid is more active for Suzuki than for Heck cross-coupling reactions. It is noteworthy that the palladium(II)-containing catalytic solution was recycled fourteen times without obvious loss of catalytic activity.|
|
||||
|---|---|---|---|---|
| Entry | R | X | (Cycle) Yield (%)b | |
| a Unless otherwise stated, the reactions were carried out using 1.0 mmol of aryl halide, 1.1 mmol phenylboric acid, 2.0 mol Na2CO3, 1 mL H2O, 2 mol% catalyst and 3.0 g 2d at 110 °C for 1 h. b Isolated yield. c Reaction time 15 h. d GC yield. e Reaction time 3 h. | ||||
| 1 | H | Br | (1) 76 | (2) 91c |
| 2 | MeO | Br | (3) 53 | (4) 82c |
| 3 | CH3CO | Cl | (5) 10d | (6) 31c |
| 4 | CH3CO | Br | (7) 91 | (8) 93c |
| 5 | F | Br | (9) 92 | (10) 90e |
| 6 | CF3 | Br | (11) 91 | (12) 95e |
| 7 | NO2 | Br | (13) 89 | (14) 91e |
Palladium(II)-catalyzed Sonogashira cross-coupling reactions in ionic liquid 2l
Palladium-catalyzed Sonogashira cross-coupling reactions have been extensively used for the synthesis of substituted and conjugated alkynes in natural products chemistry and materials science. Generally, the reaction requires the presence of phosphine ligands and a co-catalyst, such as CuI, in order to achieve high activities. Several improvements have recently been accomplished which eliminate the need for CuI and/or phosphine ligands.27 Encouraged by the fine results from the present metal-containing ionic liquids, we further evaluated Sonogashira cross-coupling reactions. The reaction was carried out using 1.0 mmol aryl iodides, 1.0 mmol phenylacetylene, 1.5 mmol piperidine, and 1.0 mol% 3b dissolved in ionic liquid 2l (Table 5). The corresponding cross-coupled products were obtained in high isolated yields. There was no apparent difference in yields obtained with electron-rich and electron-deficient aryl iodides. The heteroaromatic reactant, 2-iodothiophene, also reacted with phenylacetylene to produce 2-(phenylethynyl)thiophene in an 87% isolated yield in the sixth cycle. It was important to note that Glaser-type homocoupling products were not detected, which suggested a high catalytic activity of the catalytic solution in Sonogashira cross-coupling reactions. The catalytic solution was recycled six times with different reactants without significant loss of catalytic activity. This showed that the catalytic process tolerates heteroaryl iodide and both electron-donating and electron-withdrawing substituents in the aryl iodides.|
|
||||||
|---|---|---|---|---|---|---|
| R | H | Me | MeO | F | NO2 | c |
| a All reactions were carried out using 1.0 mmol of aryl iodide, 1.0 mmol of phenylacetylene, 2.0 mmol piperidine, 1.0 mol% 3b and 3.0 g 2l at 70 °C for 3 h. b Isolated yield. c 2-Iodothiophene acts as a substrate. | ||||||
| Cycle | 1 | 2 | 3 | 4 | 5 | 6 |
| Yield (%)b | 93 | 91 | 94 | 95 | 91 | 87 |

Conclusions
The outstanding stability and activity of the palladium catalysts of the pyrazolyl- and dimethylpyrazolyl-functionalized 2-methylimidazolium-based ionic liquids in the corresponding reaction media result from a highly synergistic effect between the pyrazolyl or dimethylpyrazolyl coordinating group and 2-methylimidazolium. Since our ionic liquids can serve as both solvents and ligands, the palladium catalysts are part of the ionic liquids in the catalytic system, and the decomposition and leaching of catalysts throughout the reaction and product separations can be markedly reduced. Moreover, since the imidazolium group was methylated at C2, the formation of potentially detrimental side-products and strong hydrogen bonding interactions were precluded. When the palladium(II)-catalyzed cross-coupling reactions were carried out using catalysts without a pendant ionic group in the methylimidazolium-based ionic liquids or using pyrazole-palladium(II) complexes in non-functionalized ionic liquids,21 the catalysts showed poor activity with concomitant formation of black precipitates. This indicates that neither the pyrazole nor the methylimidazolium-based ionic liquid alone can effectively activate and stabilize the palladium(II) catalysts.In summary, a family of 2-methylimidazolium-based ionic salts containing pyrazolyl- and 3,5-dimethylpyrazolyl-functional groups was synthesized. The relationship between their structures and melting points as well as thermal stabilities was determined. Their reactions with palladium(II) chloride gave rise to palladium(II) ionic liquid complexes which were employed as catalyst precursors for the Heck, Suzuki and Sonigashira cross-coupling reactions in corresponding ionic liquids. The catalytic solutions showed high stability and extended catalytic activity, and were effectively recovered and recycled in the chemical transformations without significant loss in the catalytic activity. This work has demonstrated that the combination of 2-methylimidazolium and pyrazolyl coordination groups is a promising pathway toward design and synthesis of highly stable and active phosphine-free catalytic systems.
Experimental
General
The compounds, 1-(chloromethyl)pyrazole hydrochloride, 1-(chloromethyl)-3,5-dimethylpyrazole hydrochloride,28 and 1-butyl-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)amide18,29 were prepared as previously reported. DMF was distilled prior to use. The other chemicals were obtained commercially and were used as purchased. A standard Schlenk line system was used for handling the air- and moisture-sensitive reactions under nitrogen. 1H, 13C and 19F NMR spectra were recorded on a 300 MHz spectrometer operating at 300, 75 and 282 MHz, respectively, by using acetone-d6 as locking solvent except where otherwise indicated. Chemical shifts were reported in ppm relative to the appropriate standard: CFCl3 for 19F and TMS for 1H and 13C NMR spectra. GC-MS spectra were determined using an appropriate instrument. Differential scanning calorimetry (DSC) measurements were performed using a calorimeter equipped with an auto-cool accessory and calibrated using indium. The following procedure was used for each sample: cooling from 40 °C to −80 °C and heating to 400 or 500 °C at 10 °C min−1. The transition temperature, Tm, was taken as peak maximum. Onset of decomposition was taken as when the abnormal section of the plot began. Thermogravimetric analysis (TGA) measurements were carried out by heating samples at 10 °C min−1 from room temperature to 500 °C in a dynamic nitrogen atmosphere (flow rate = 70 mL min−1). Thin-layer chromatography (TLC) analysis was performed with Al-backed plates pre-coated with silica gel and examined under UV (254 nm). Flash column chromatography was executed on silica gel (60–200 µm, 60 A). Elemental analyses were obtained on a CE-440 Elemental Analyzer.General procedure for the preparation of 2a–2q
Compound 1 (1.0 mmol) and alkyl polyfluoroalkyl iodides or 1-bromohexane (1.5 mmol) were placed in a Pyrex glass tube. After cooling to −195 °C, the tube was evacuated and sealed. The neat reaction mixture was heated at 80 °C for 12 h. After cooling and carefully opening the tube, the volatile materials were removed at 80 °C under reduced pressure. The residue was dissolved in a mixture of water (5 mL) and acetone (5 mL), and then lithium bis(trifluoromethanesulfonyl)amide or potassium haxafluorophosphate (1.5 mmol) was added. The reaction mixture was stirred at 25 °C for 5 h. The acetone was evaporated under reduced pressure, and the water layer was extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed with water (3 × 10 mL), once with saturated Na2S2O3 solution (5 mL), and then dried over anhydrous Na2SO4. After the solvent was removed in vacuo, the products were washed with CH2Cl2 (3 mL) and were filtered to remove inorganic salts. The solvent was removed again in vacuo to give pure products 2a–2q.General procedure for the preparation of palladium(II) complexes 3a and 3b
PdCl2 (0.035 g, 0.2 mmol) was added to a stirred solution of ionic liquid 2d or 2l (0.4 mmol) in methanol (10 mL). The mixture was stirred for 12 h at 25 °C. Slow evaporation of the resulting solution gave rise to pale-yellow solid which was washed three times with ethyl ether (10 mL).General procedure for the Heck reactions in ionic liquid
Palladium(II) complex 3a (23.4 mg, 0.02 mmol) was dissolved in ionic liquid 2d (3.0 g), and then the solvent was degassed under reduced pressure at 60 °C for 1 h before dry nitrogen was introduced. The aryl halide (1.0 mmol), n-butyl acrylate (1.25 mmol) and Et3N (1.5 mmol) were subsequently added under nitrogen at 25 °C. The resulting mixture was stirred for an appropriate time at 110 °C. The product was extracted from the reaction mixture by addition of ethyl ether (3 mL), followed by decanting off the ethyl ether solution of the product. This process was repeated (3 × 3 mL). The combined organic layer was concentrated by rotary evaporation. The residue was purified by flash chromatography on silica gel to give the desired product. The ionic liquid containing Pd(II) catalyst was washed with water (3 × 10 mL) to remove excess base and its salt, then dried under reduced pressure at 60 °C for 4 h to remove traces of ethyl ether and water and employed for the next cycle.General procedure for the Suzuki reactions in ionic liquid
Palladium(II) complex 3a (23.4 mg, 0.02 mmol) was dissolved in ionic liquid 2d (3.0 g), and the mixture was degassed under reduced pressure at 60 °C for 1 h before nitrogen was introduced. The aryl halide (1.0 mmol), phenylboric acid (1.34 g, 1.1 mmol), Na2CO3 (0.21 g, 2.0 mmol) and H2O (1 mL) were added to the ionic liquid solution containing the Pd(II) catalyst under nitrogen. The resulting mixture was stirred for an appropriate time at 110 °C. The product was extracted from the reaction mixture by addition of ethyl ether (3 mL), followed by decanting the ethyl ether solution of the product. This process was repeated (3 × 3 mL). The combined organic layer was concentrated by rotary evaporation. The residue was purified by flash chromatography on silica gel to give the desired product. The ionic liquid containing the Pd(II) catalyst was washed with water (3 × 10 mL) to remove excess base and its salt, then dried under reduced pressure at 60 °C for 4 h to remove traces of ethyl ether and water and employed for the next cycle.General procedure for the Sonogashira reactions in ionic liquid
Palladium(II) complex 3b (12.3 mg, 0.01 mmol) was dissolved in ionic liquid 2l (3.0 g), and then the solvent was degassed under reduced pressure at 60 °C for 1 h before nitrogen was introduced. The aryl iodide (1.0 mmol), phenylacetylene (0.102 g, 1.0 mmol) and piperidine (0.128 g, 1.5 mmol) were added to the ionic liquid solution containing the Pd(II) catalyst under nitrogen. The resulting mixture was stirred at 70 °C for 3 h. The product was extracted from the reaction mixture by addition of ethyl ether (3 mL), followed by decanting off the ethyl ether solution of the product. This process was repeated (3 × 3 mL). The combined organic layer was concentrated by rotary evaporation. The residue was purified by flash chromatography on silica gel to give the desired product. The palladium-containing ionic liquid was washed with water (3 × 10 mL) to remove excess base and its salt, then dried under reduced pressure at 60 °C for 4 h to remove traces of ethyl ether and water and employed for the next cycle.Acknowledgements
We gratefully acknowledge the support of the National Science Foundation (Grant CHE-0315275), AFOSR (Grant F49620-03-1-0209) and ONR (Grant N00014-02-1-0600).References
- (a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS; (b) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS; (c) V. Farina, Adv. Synth. Catal., 2004, 346, 1553–1582 CrossRef CAS; (d) R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283–2321 CrossRef CAS.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) J. E. Milne and S. L. Buchwald, J. Am. Chem. Soc., 2004, 126, 13028–13032 CrossRef CAS; (c) A. Cwik, Z. Hell and F. Figueras, Org. Biomol. Chem., 2005, 3, 4307–4309 RSC.
- (a) T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS; (b) J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 12527–12530 CrossRef CAS; (c) A. Zapf, A. Ehrentraut and M. Beller, Angew. Chem., Int. Ed., 2000, 39, 4153–4155 CrossRef CAS.
- (a) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–907 CrossRef CAS; (b) K. Selmeczi, M. Réglier, M. Giorgi and G. Speier, Coord. Chem. Rev., 2003, 245, 191–201 CrossRef CAS.
- (a) G. Chelucci and R. P. Thummel, Chem. Rev., 2002, 102, 3129–3170 CrossRef CAS; (b) Y. Yamamoto, A. Nagata, H. Nagata, Y. Ando, Y. Arikawa, K. Tatsumi and K. Itoh, Chem.–Eur. J., 2003, 9, 2469–2483 CrossRef CAS.
- (a) M. Ohff, A. Ohff and D. Milstein, Chem. Commun., 1999, 357–358 RSC; (b) H. Weissman and D. Milstein, Chem. Commun., 1999, 1901–1902 RSC.
- (a) L. Botella and C. Nájera, Angew. Chem., Int. Ed., 2002, 41, 179–181 CrossRef CAS; (b) D. A. Alonso, C. Nájera and M. C. Pacheco, Org. Lett., 2000, 2, 1823–1826 CrossRef CAS.
- (a) H. M. Lee, P. L. Chiu, C. H. Hu, C. L. Lai and Y. C. Chou, J. Organomet. Chem., 2005, 690, 403–414 CrossRef CAS; (b) R. Tribo, S. Munoz, J. Pons, R. Yanez, A. Alvarez-Larena, J. F. Piniella and J. Ros, J. Organomet. Chem., 2005, 690, 4072–4079 CrossRef CAS and references therein.
- (a) D. B. Zhao, Z. F. Fei, T. J. Geldbach, R. Scopelliti and P. J. Dyson, J. Am. Chem. Soc., 2004, 126, 15876–15882 CrossRef CAS; (b) D. B. Zhao, Z. F. Fei, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2004, 43, 2197–2205 CrossRef CAS.
- C. J. Mathews, P. J. Smith and T. Welton, J. Mol. Catal. A: Chem., 2003, 206, 77–82 CrossRef CAS.
- (a) J. C. Xiao, B. Twamley and J. M. Shreeve, Org. Lett., 2004, 6, 3845–3847 CrossRef CAS; (b) J. C. Xiao and J. M. Shreeve, J. Org. Chem., 2005, 70, 3072–3078 CrossRef CAS; (c) J. C. Xiao, C. F. Ye and J. M. Shreeve, Org. Lett., 2005, 7, 1963–1965 CrossRef CAS; (d) R. H. Wang, B. Twamley and J. M. Shreeve, J. Org. Chem., 2006, 71, 426–429 CrossRef CAS; (e) J. Kim and J. M. Shreeve, Org. Biomol. Chem., 2004, 2, 2728–2734 RSC.
- (a) S. B. Park and H. Alper, Chem. Commun., 2004, 1306–1307 RSC; (b) S. B. Park and H. Alper, Org. Lett., 2003, 5, 3209–3212 CrossRef CAS.
- G. A. Grasa, A. C. Hillier and S. P. Nolan, Org. Lett., 2001, 3, 1077–1080 CrossRef CAS.
- (a) F. Sinner, M. R. Buchmeiser, R. Tessadri, M. Mupa, K. Wurst and G. K. Bonn, J. Am. Chem. Soc., 1998, 120, 2790–2797 CrossRef CAS; (b) M. R. Buchmeiser and K. Wurst, J. Am. Chem. Soc., 1999, 121, 11101–11107 CrossRef CAS; (c) V. Gallo, P. Mastrorilli, C. F. Nobile, R. Paolillo and N. Taccardi, Eur. J. Inorg. Chem., 2005, 582–588 CrossRef CAS.
- (a) B. C. Ranu and S. Banerjee, J. Org. Chem., 2005, 70, 4517–4519 CrossRef CAS; (b) V. Calò, A. Nacci and A. Monopoli, J. Mol. Catal. A: Chem., 2004, 214, 45–56 CrossRef CAS; (c) L. Xu, W. Chen, J. Ross and J. Xiao, Org. Lett., 2001, 3, 295–297 CrossRef CAS; (d) A. Perosa, P. Tundo, M. Selva, S. Zinovyev and A. Testa, Org. Biomol. Chem., 2004, 2, 2249–2252 RSC.
- (a) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3691 CrossRef CAS; (b) P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3773–3789 CrossRef; (c) N. Jain, A. Kumar, S. Chauhan and S. M. S. Chauhan, Tetrahedron, 2005, 61, 1015–1060 CrossRef CAS; (d) S. Rogers, Chem. Commun., 2001, 2399–2380 RSC.
- (a) V. K. Aggarwal, I. Emme and A. Mereu, Chem. Commun., 2002, 1612–1613 RSC; (b) F. McLachlan, C. J. Mathews, P. J. Smith and T. Welton, Organometallics, 2003, 22, 5350–5357 CrossRef CAS; (c) N. D. Clement, K. J. Cavell, C. Jones and C. J. Elsevier, Angew. Chem., Int. Ed., 2004, 43, 1277–1279 CAS; (d) A. Corma, H. García and A. Leyva, Tetrahedron, 2005, 61, 9848–9854 CrossRef CAS.
- L. Magna, Y. Chauvin, G. P. Niccolai and J. M. Basset, Organometallics, 2003, 22, 4418–4425 CrossRef CAS.
- (a) A. Mele, C. D. Tran and S. H. De Paoli Lacerda, Angew. Chem., Int. Ed., 2003, 42, 4364–4366 CrossRef CAS; (b) J. Ross and J. Xiao, Chem.–Eur. J., 2003, 9, 4900–4906 CrossRef CAS; (c) A. Noda, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2001, 105, 4603–4610 CrossRef CAS.
- J. C. Hsu, Y. H. Yen and Y. H. Chu, Tetrahedron Lett., 2004, 45, 4673–4676 CrossRef CAS.
- C. J. Mathews, P. J. Smith and T. Welton, J. Mol. Catal. A: Chem., 2004, 214, 27–32 CrossRef CAS.
- (a) I. J. B. Lin and C. S. Vasam, J. Organomet. Chem., 2005, 690, 3498–3512 CrossRef CAS and references therein; (b) T. Sasaki, C. Zhong, M. Tada and Y. Iwasawa, Chem. Commun., 2005, 2506–2608 RSC.
- K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS.
- (a) P. Bonhöte, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef CAS; (b) E. A. Turner, C. C. Pye and R. D. Singer, J. Phys. Chem. A, 2003, 107, 2277–2288 CrossRef CAS and references therein; (c) A. Bagno, C. Butts, C. Chiappe, F. D'Amico, J. C. D. Lord, D. Pieraccini and F. Rastrelli, Org. Biomol. Chem., 2005, 3, 1624–1630 RSC.
- (a) T. J. Geldbach and P. J. Dyson, J. Am. Chem. Soc., 2004, 126, 8114–8115 CrossRef CAS; (b) N. Audic, H. Clavéier, M. Mauduit and J. C. Guillemin, J. Am. Chem. Soc., 2003, 125, 9248–9249 CrossRef CAS; (c) Q. Yao and Y. Zhang, Angew. Chem., Int. Ed., 2003, 42, 3395–3398 CrossRef CAS; (d) Y. Peng, Y. Cai, G. Song and J. Chen, Synlett, 2005, 2147–2150 CAS.
- (a) S. Liu, T. Fukuyama, M. Sato and I. Ryu, Synlett, 2004, 1814–1816 CAS; (b) J. McNulty, A. Capretta, J. Wilson, J. Dyck, G. Adjabeng and A. Robertson, Chem. Commun., 2002, 1986–1987 RSC.
- (a) B. Liang, M. Dai, J. Chen and Z. Yang, J. Org. Chem., 2005, 70, 391–393 CrossRef CAS; (b) L. Djakovitch and P. Rollet, Adv. Synth. Catal., 2004, 346, 1782–1792 CrossRef CAS.
- S. Juliá and C. Martínez-Martorell, Heterocycles, 1986, 24, 2233–2237 CrossRef CAS.
- A. R. Gholap, K. Venkatesan, T. Daniel, R. J. Lahoti and K. V. Srinivasan, Green Chem., 2003, 5, 693–696 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data for coupled products for Heck, Suzuki and Sonogashira cross-coupling reactions. See DOI: 10.1039/b604008j |
| This journal is © The Royal Society of Chemistry 2006 |