A paramagnetic contrast agent with myeloperoxidase-sensing properties†
Manel
Querol
a,
John W.
Chen
b and
Alexei A.
Bogdanov, Jr.
*a
aS2-804, University of Massachusetts Medical School, 55 Lake Avenue, Worcester, Massachusetts 01655, USA. E-mail: Alexei.Bogdanov@umassmed.edu
bCentre for Molecular Imaging Research at Massachusetts General Hospital Bldg 149, 13th Street, Charlestown, Massachusetts 02129, USA
First published on 12th April 2006
Abstract
Myeloperoxidase (MPO) is an essential component of inflammatory response in norm and pathology. With an ultimate goal of non-invasive imaging of MPO we used a gadolinium-chelating bis(5-hydroxytrytamide) derivative of diethylenetetraamine pentaacetic acid (L1–Gd3+salt) as a paramagnetic sensor of enzymatic activity. We tested whether L1–Gd3+ is active in reducing the oxidized form of myeloperoxidase, generated as a result of hydrogen peroxide reduction by the enzyme. We expected that upon activation by MPO/H2O2 L1–Gd3+ would not only oligomerise but also bind to other macromolecules present in the media and that the overall process will give rise to a net T1-weighted MRI signal increase.
Introduction
It is widely anticipated that magnetic resonance imaging (MRI) would benefit from the use of “MR-responsive” contrast agents (CAs) for specific visualization of molecules in vivo.1,2 Imaging at the molecular level would eventually lead to a better understanding of biological processes in intact living systems and, ultimately, would lead to an improvement in diagnostic techniques.To pursue the goal of MR imaging at the molecular level, CAs with specific molecule targeting and/or specific “in situ” activation are ultimately required.3,4 The need of such CAs is dictated by a relatively low sensitivity of MRI to the presence of Gd in tissue. Specific targeting and/or specific “in situ” activation are expected to provide means of site-specific signal enhancement either by CA controlled local accumulation and/or as a result of site-specific relaxivity increase and concomitant site-specific signal amplification. Several applications of the above CAs have already been reported.5–8 Recently, CAs with biologically relevant targeting moieties9–11 as well as “pseudo-biotic” nanoparticles12 were developed to gain MR signal intensity (SI) enhancement due to specific delivery in vivo.
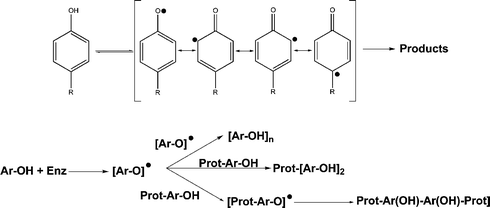
The most well-studied approaches enabling MRI SI enhancement to be achieved involve either a change in CA water coordination (q value), i.e. increasing the number of molecules directly interacting with the paramagnetic center13,14 or decreasing the rotational correlation times (τR) by increasing the size of the CA.11,15 In the current study, we combined the above approaches to obtain an enzyme-specific Gd-based CA enabling MRI SI enhancement due to a synergism of two effects: 1) enzyme-mediated structural changes; 2) specific site accumulation upon enzyme activation. The driving force behind the aforementioned MR SI enhancement is based on the ability of oxidoreductases to generate phenolic radicals that, once formed, can undergo various permutations including: a) oligomerisation16–20 and b) protein attachment via covalent bond formation with aromatic amino acids present in the protein.21–27 This effect is not limited to small molecules (tyrosine, tyramine, serotonin, etc.) but also includes macromolecules (e.g. proteins), either directly, or via intermolecular radical transfer mechanisms (Fig. 1).
 | ||
| Fig. 1 a) Phenyl radical resonance forms; b) phenyl radical reactivity in the presence of other phenols and phenol-containing proteins. | ||
We have previously demonstrated that DOTA-based gadolinium (DOTA = 1,4,7,10-tetra(carboxymethyl)-1,4,7,10-tetraazacyclododecane) CAs bearing a phenolic group (Fig. 2a) can interact “in situ” with oxidoreductases generating oligomers as a result. The oligomerisation process has been shown to produce a net increase in MRI signal. Initially, a Gd–DOTA derivative mono-functionalized with a catechol moiety was shown to reduce oxidized horseradish peroxidase (HRP). In this experiment, MR imaging detected picomolar concentrations of HRP. The same system also allowed imaging of E-selectin on the surface of endothelial cells.28 Later, the catechol unit was substituted by a serotonin moiety (Fig. 2b).29 Serotonin is a naturally occurring neuromediator that also functions as a donor-substrate of inflammation-related enzyme myeloperoxidase (EC 1.11.1.7).30,31 It is also known that monocytes and macrophages present in atherosclerotic lesions in the arterial intima secrete catalytically active myeloperoxidase (MPO) in plaques. Ample clinical data suggest that MPO is a biomarker that can serve as a predictor of acute coronary events.31–33 Since MPO catalyzed a 1.7-fold increase in relaxivity of serotonin–DOTA(Gd), we proposed a concept of vulnerable plaque detection that is based on enzymatic activity. Recently, MPO-mediated chemical bond formation concept has been adapted for a radioactive 67Ga probe for single photon computed tomography (SPECT, Fig. 2c).34 Initially we tested the extent of probe attachment to protein upon MPO activation in order to assess the possibility of site-specific accumulation. Preliminary results confirmed site-specific targeting/accumulation within MPO rich areas.
 | ||
| Fig. 2 Oxidoreductase sensitive chelating compounds: a, b) chelating units for Gd(III),28,29 c) chelating unit for 67Ga.34 | ||
In this research, to achieve further improvement in enzyme-mediated MRI SI enhancement, we linked two groups35 prone to oligomerisation onto each chelate in order to increase the possibility of oligomerisation/protein attachment. For the above purpose, DTPA (diethylenetriaminepentaacetic acid) was chosen as a chelate since its bis(anhydride) derivative is easily available and DTPA–bis(monomethyl)amides were shown to have no toxicity and are approved for clinical use. The goals of this work were: a) to assess the possibility of enzyme-mediated MRI signal amplification; b) to estimate the possibility of site-specific CA protein attachment; c) to test the combined effect of CA oligomerisation/protein attachment for MPO detection and potential atherosclerotic vulnerable plaque imaging.
Results
Preliminary oligomerisation formation and relaxometry
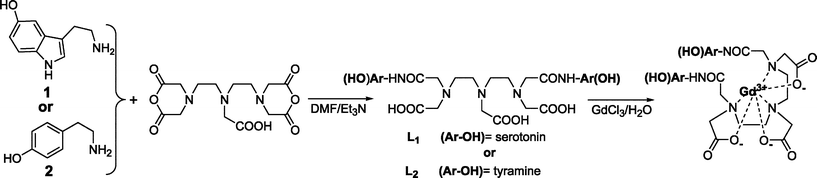 | ||
| Scheme 1 Synthetic scheme for the preparation of the CAs used in the present work: 5-hydroxytrytamide (5-HT, serotonin) or tyramine were reacted with DTPA–bis(anhydride) in the presence of Et3N to afford either L1 (di-5HT–DTPA) or L2 (di-tyramido–DTPA). Reaction of the previous ligands with an excess of GdCl3 afforded the corresponding Gd–CAs. | ||
Further, compound L1–Gd3+ in a concentration of 0.5 mM was incubated with 5 iU of MPO and a large excess of H2O2 for two hours. After incubation, the mixture was analyzed with C18-HPLC. Fig. 3 shows the chromatogram before and after the enzyme addition. The sharp peak assigned to L1–Gd3+ disappeared giving rise to a broad band that can be assigned to oligomerisation products. Intensities, in arbitrary units, for the peaks at 21.6 min (top) and 21.5 min (bottom), were 1 and 0.05, respectively, suggesting a conversion degree that exceeded 90% (Fig. 3).
 | ||
| Fig. 3 Chromatograms of compound L1–Gd3+. Starting material (top), after reaction with MPO in the presence of H2O2 (bottom). | ||
Further, we tested the effect of molecular weight increase over the relaxometric properties of these complexes (Table 1).
| Compound | Buffer components | Enzyme (iUe) | Peroxide source | r 1o d/mM−1 s−1 | r 1o Increase | K f/s−1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Buffer contained 5.5 mM glucose. b Different R1o values taken at different time-points. c Relative to a reference buffer containing HSA in the absence of MPO and GOX. d Atomic relaxivity values were obtained by using linear fitting of observed R1ovs. initial gadolinium concentration, 4 points/fitting. The concentration range 0.07 mM−1 mM. e International units. f Obtained by fitting R1oversus time from samples containing initial concentration of CA in the range: 0.10–0.13 mM. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ | PBS | — | — | 4.3 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L2–Gd3+ | PBS | — | — | 4.3 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ | PBS | +HRP (4) | 10 mM | 10.5 | 2.4-fold | 4.50 × 10−3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L2–Gd3+ | PBS | +HRP (4) | 10 mM | 15.9 | 3.7-fold | 5.00 × 10−3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ | PBS | +MPO (4) | 10 mM | 5.9 | 37% | 0.70 × 10−3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L2–Gd3+ | PBS | +MPO (4) | 10 mM | 5.3b | 23%b | na | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ | PBS | +MPO (0.5) | GOXa | 6.2 | 44% | 0.43 × 10−3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ | PBS | +HRP (0.5) | GOXa | 9.1 | 2.1-fold | 5.60 × 10−3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L2–Gd3+ | PBS | +MPO (0.5) | GOXa | 5.1b | 18%b | na | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L2–Gd3+ | PBS | +HRP (0.5) | GOXa | 11.8 | 2.7-fold | 4.83 × 10−3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ | HSA (40 mg per mL PBS) | — | — | 5.5 | 27% | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ | Lys (40 ng per mL PBS) | — | — | 5.5 | 27% | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ | HSA (40 mg per mL PBS) | +MPO (0.5) | GOXa | 6.6 | 54% (20%c) | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Initially we studied the effect of 0.5iU of MPO at 0.13 mM of compound L1–Gd3+ in PBS buffer containing 5.5 mM of glucose (Table 1). Myeloperoxidase-mediated reactions were studied under four conditions by keeping MPO and L1–Gd3+ concentrations constant and testing sources of hydrogen peroxide: a) 10 mM H2O2, b) 10 µg GOX, c) 1 µg GOX, d) 50 µg GOX. The corresponding T1 values of these test reactions were: 1150 ms, 832 ms, 815 ms and 825 ms, respectively. A value of 831 ms was also obtained when a solution 0.13 mM L1–Gd3+ in 5.5 mM glucose, 10 mM of H2O2 was treated with a 10-fold amount of MPO (5 iU). To further validate the experiment, the same conditions were chosen if HRP was used instead of MPO and this resulted in a similar T1 value around 664 ± 20 ms in all four cases. When 1.1 mM L1–Gd3+ solutions were combined with GOX in the presence of glucose with no HRP or MPO present, no changes in T1 were seen. These results suggest that the use of the GOX/glucose H2O2 generating system results in higher levels of relaxivity enhancement. When the effect of this system was measured at several different concentrations of L1–Gd3+, the relaxivity at 0.47 T (40 °C) was 6.2 mM−1 s−1 that corresponds to a relaxivity increase of 44% over that of the control. Glucose alone has no effect on relaxivity. Furthermore, the GOX/glucose system resulted in stable reaction products for at least 24 h as opposed to H2O2 and 5 iU of MPO that resulted in the early formation of insoluble reaction products. By replacing MPO with HRP, a relaxivity of 9.1 mM−1 s−1 was obtained suggesting that HRP is not affected by H2O2 concentration. When the MPO–GOX/glucose system was assayed using L2–Gd3+, partial insolubility of the product did not allow further characterization. As before, T1 changes at various concentrations were continuously monitored until a T1 descending trend was reversed. The final relaxivity value of 5.1 mM−1 s−1 has to be interpreted with caution because of inhomogeneity of the system.
In order to estimate the pseudo-first order kinetics constant of reactions in the presence of GOX/glucose, T1 kinetics of 0.13 mM of compound L1–Gd3+ using MPO and 0.10 mM L2–Gd3+ in the presence of HRP were continuously monitored (Fig. 4). The apparent kinetic constants were 0.43 × 10−3 s−1, 5 × 10−3 s−1 and 4.83 × 10−3 s−1, respectively. HRP catalysis resulted in kinetics that were10-fold faster than MPO-mediated catalysis. This allowed 90% conversion after approximately 90 min. The catalytic conversion of the substrate in the presence of MPO took up to 4 hours with precipitation of oligomeric products.
 | ||
| Fig. 4 Kinetics of T1 changes. a: 1) L1–Gd3+ + MPO–GOX, 2) L1–Gd3+ + HRP–GOX, 3) L2–Gd3+ + HRP–GOX. b: 1) L1–Gd3+ + MPO–GOX. Graph a shows a time-span of 120 minutes, graph b shows a time-span of 1000 minutes for situation 1. | ||
Changes in composition were detected by using spectrophotometry when solutions of both L1–Gd3+ and L2–Gd3+ were reacted with enzymes under various conditions at 40 °C. Fig. 5 shows several representative spectra obtained using L1-Gd3+. In the case of L2–Gd3+ all spectra showed the presence of insoluble products after ∼5 min. Insolubility resulted in spectra showing a continuous increase of absorbance in the far-red range (800 nm) due to insoluble particles of polymerized product. In general, the UV/Vis spectrum of compound L1–Gd3+ reacted in the presence of either MPO or HRP, showed a shift of the bands into the visible range. While in the case of MPO–H2O2 the spectral changes were comparatively slow and continuous, the rest of the reaction mixtures showed a new band at 440 nm, which emerged within 1 min of enzyme addition. While the overall absorbance kept increasing for a period of at least 4 h, the major change took place within the first hour. In general, the band corresponding to the serotonin aromatic ring that shows a maximum at 275 nm with a shoulder at 315 nm in the absence of enzyme transformed into a broader band with two maxima if hydrogen peroxide was provided by the GOX/glucose system.
 | ||
| Fig. 5 UV/Vis spectra of L1–Gd3+ in the presence of: a) MPO–GOX, b) HRP–GOX, c) MPO–H2O2, d) HRP–H2O2. Insets show the time evolution of the newly formed band at 440 nm. | ||
Interaction with proteins
It is widely accepted that in the presence of peroxidases serotonin or tyrosine do not exclusively oligomerise but either transfer the radical to other aromatic groups present in the media, or get covalently attached to other macromolecules or molecular aggregates. If the above holds true for the systems described in this paper, two main effects are to be expected: 1) attachment to other macromolecules, i.e. proteins, should result in a relaxivity increase due to the increase in the overall size of the paramagnetic system with a concomitant change of the tumbling rate; 2) the formation of large aggregates should change the pharmacokinetics of the compounds transforming them into products with longer retention times. These points are further exemplified below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) or lysozyme (Mw = 15000 Da). T1 in both cases increased from 0.83 s−1 in the absence of protein to 1.25 s−1 and 1.17 s−1 at 40 mg mL−1 of HSA or lysozyme (0.6 mM HSA; 2.6 mM lysozyme), respectively. These values suggest the absence of non-covalent binding between L1–Gd3+ and proteins used (effective KD < 200 M−1; binding efficiency <1%).41–43 Further, we titrated solutions containing 40 mg protein mL−1 with various amounts of L1–Gd3+. The relaxivity of compound L1–Gd3+ was estimated as 5.5 mM−1 s−1 in both cases. Identical relaxivities suggested no true interaction with the proteins but rather could be attributed to the differences in relaxivity caused by high protein concentration in the solutions.
000 Da) or lysozyme (Mw = 15000 Da). T1 in both cases increased from 0.83 s−1 in the absence of protein to 1.25 s−1 and 1.17 s−1 at 40 mg mL−1 of HSA or lysozyme (0.6 mM HSA; 2.6 mM lysozyme), respectively. These values suggest the absence of non-covalent binding between L1–Gd3+ and proteins used (effective KD < 200 M−1; binding efficiency <1%).41–43 Further, we titrated solutions containing 40 mg protein mL−1 with various amounts of L1–Gd3+. The relaxivity of compound L1–Gd3+ was estimated as 5.5 mM−1 s−1 in both cases. Identical relaxivities suggested no true interaction with the proteins but rather could be attributed to the differences in relaxivity caused by high protein concentration in the solutions.
Further, HSA titration experiments were conducted in the presence of 0.5 iU MPO using the GOX/glucose system as a source of H2O2 that resulted in relaxivity of 6.6 mM−1 s−1. This value reflects a 54% relaxivity increase if compared with the relaxivity of compound L1–Gd3+ in PBS alone, or a 20% increase for compound L1–Gd3+ in the presence of 40 mg HSA per mL. The presence of HSA (r1o = 6.6 mM−1 s−1) also resulted in a further increase of 8% when compared with MPO effects in the absence of HSA (r1o = 6.2 mM−1 s−1). It should be noted that in the absence of HSA and in the presence of MPO–GOX/glucose, R1o keeps increasing over a period of approximately 3 hours, whereas the presence of albumin results in maximum relaxivity in 15–20 min.
In another experiment we set a goal to determine whether pre-formed oligomers bind HSA and also if those preformed oligomers are adequate substrates for MPO. We used 5.6 mM L1–Gd3+ solution and incubated it with 2 iU of MPO in the presence of GOX/glucose. After 40 °C incubation for 18 hours, the solution was sonicated to produce a clear solution of “oligomerised L1–Gd3+” with relaxivity r1o = 10.1 mM−1 s−1. This product was used to titrate HSA solutions either in the absence, or in the presence of MPO–GOX/glucose. The relaxivity values obtained where similar in both experiments −13.1 and 12.4 mM−1 s−1, respectively. The obtained results suggested that although the pre-formed oligomers could have some affinity for albumin, further addition of the enzyme does not play any significant role, which means that oligomers are poor substrates of MPO at best. The observed high relaxivity values are associated with the use of larger enzyme activities (i.e. 10-fold) during longer periods of time (18 h). Finally, we estimated the overall size of the oligomerisation products. Solutions of L1–Gd3+ were prepared as follows: solution A contained MPO–GOX/glucose, solution B contained 40 mg mL−1 HSA and solution C had all components above present (i.e. HSA–MPO–GOX/glucose). The solutions were incubated at 40 °C overnight followed by exhaustive dialysis using membranes with a 10000 Da cut-off.
The dialysis experiment (see Table 2) suggested that compound L1–Gd3+ does not interact with HSA in the absence of MPO. Furthermore, the presence of the MPO–GOX system lead to a population of oligomers that contained 40% of molecules with masses exceeding 10000 Da. If HSA was additionally present, the amount of retained oligomers post dialysis increased to 65%. The observed differences can be assigned to the covalent binding of L1–Gd3+ to HSA.
| Composition | [Gd], pre-dialysis/mM | [Gd], post-dialysis/mM | Gd loss (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Values are average of two independent experiments. Error, defined as the observed difference/average, was below 5% in all cases. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+–HSA + MPO + GOX | 0.18 | 0.07 | 60 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ + HSA | 0.35 | 0.02 | 95 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L1–Gd3+ + HSA + MPO + GOX | 0.29 | 0.19 | 35 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 | ||
| Fig. 6 L1–111In chromatograms: Labeling (top), after incubation with MPO–H2O2 (bottom). Chromatograms show both radioactivity (upper) and UV (lower) traces. | ||
In vivo model studies
To further evaluate the combined effect of oligomerisation and protein attachment of CA we devised an in vivo experiment that included mice harbouring implants containing human MPO. The animal model used for MPO imaging was obtained by injecting subcutaneously 0.4 mL of Matrigel™ (Beckton–Dickinson) in both right and left thighs of a mouse. Matrigel™ when injected subcutaneously undergoes a gelation thereby creating a permeable and dynamic reservoir within the interstitium allowing extravasation of CA while keeping the enzyme trapped in the implant due to the higher molecular mass. The experimental implant contained 6 iU of human MPO (final concentration 1.5 iU mg−1 protein; for comparison, MPO concentration in human atherosclerotic plaques ranges between 71 and 511 U mg−1 protein44) and 10 µL GOX, whereas the left thigh was injected with Matrigel™ only. We subsequently performed T1-weighted MR imaging after intravenous injection of L1–Gd3+ at a dose of 0.3 mmol kg−1 (Fig. 7a). The comparison of contrast-to-noise ratio MR signal intensities measured in the right (MPO-containing) and the left (control) extremities of animals increased 1.6–1.9-times during the 3 h of in vivo monitoring (Fig 7b). The same kinetic experiment performed using Gd–DTPA–dimeglumine (Magnevist™, Schering) did not result in any appreciable signal difference between MPO-containing and control Matrigel™ implants. | ||
| Fig. 7 a) MR image of Matrigel™ implants obtained at 2 hours after the addition of CA. The right extremity contains exogenous-MPO, the left one contains only Matrigel™ and works as an internal control. b) MRI signal evolution along the time-span of the experiment. | ||
The possibility of in vivo accumulation due to the ability of the MPO reaction product to bind to the proteins was assessed by injecting L1–111In at a tracer dose (80–100 µCi) into a mouse using the above Matrigel™ implant model and performing SPECT/CT imaging (Fig. 8). Monitoring of the radioactivity levels for 3 hours after injection revealed a 2.3-fold difference between the right and the left extremities of the animals. Excision of the implants after 6 hours followed by radioactivity measurements showed a 4-times higher radioactivity for the MPO-containing implant.
 | ||
| Fig. 8 Fused SPECT-CT image of Matrigel™ implants 3 hours after the injection of L1–111In imaging agent. Red colour indicates the hot-spot of radioactivity. | ||
Discussion
Mammalian oxidoreductases are multifunctional enzymes, involved in the biosynthesis of thyroid hormones (thyroid peroxidase) and in antimicrobial defence (myeloperoxidase). The mature form of myeloperoxidase is found in secretory azurophilic granules of polymorphonuclear leukocytes and monocytes. Usually, MPO secretion is associated with oxidative burst where NADPH-oxidase generates superoxide and hydrogen peroxide. MPO amplifies the oxidative power of hydrogen peroxide by using it as a substrate for hypochlorous acid synthesis from chloride anion. Activated MPO is also capable of further reacting with various organic substrates producing undesirable chemical reactions with proteins that contribute to atherosclerosis plaque formation and rupture, i.e. LDL oxidation,45,46 HDL inactivation47 and metalloproteinase activation.48,49To develop MPO activity sensing probes, we investigated the ability of peroxidases to generate reactive organic species. The concept of such probes is based on tyrosine reactivity patterns in the presence of oxidoreductases, especially MPO (see Fig. 1). We synthesized paramagnetic prototype probes L1–Gd3+ and L2–Gd3+ that in the presence of either HRP or MPO and peroxide showed a formation of complex mixtures containing oligomers of various lengths. MALDI-TOF of these oligomers showed the formation of 9-mer molecules. This qualitative preliminary evidence suggested a complex MPO-catalyzed reaction. Importantly, striking differences between relaxivities of reaction products were observed when MPO was compared to HRP. While HRP resulted in 2.4- to 3.7-fold relaxivities of paramagnetic products compared to the initial monomers, MPO yielded a net relaxivity increase close to 23–37%. Furthermore, pseudo-first order kinetic constants used to compare bulk oligomerisation were 10-times lower in the case of L1–Gd3+ in the presence of MPO if compared with the corresponding constants of HRP-mediated reactions. The above differences suggested fundamental differences between MPO and HRP. Percent homology analyses and X-ray data suggested two superfamilies of peroxidases: superfamily I found in prokaryotes, fungi and higher plants, and superfamily II found in mammals.50,51 In general, the peroxidase catalytic cycle is a feature shared by both superfamilies (shown in Fig. 9).40,52–55 The rate of formation of enzyme intermediates I and II with the different substrates and, hence, native enzyme regeneration, depends on several characteristics of electron donors51 including not only redox potentials (E), but also molecular structure. HRP has very similar E values for compounds I and II (0.9 V). The existing literature data suggest that MPO theoretically is better suited for radical formation than HRP (compound I has E > 1.015 V, and compound II has E = 0.97 V).51,56–59 However, the main feature determining selectivity to a substrate is the accessibility to the active site of the enzyme. While HRP shows very similar and exposed geometries for compounds I and II in a single 40 kDa subunit, the MPO active site is located in a narrow hydrophobic cleft within a larger 60 kDa subunit. This hinders MPO reactivity towards bulky and/or hydrophilic compounds and could eventually restrict the enzyme regeneration cycle.27,50,60 It is now accepted that HRP can interact directly with phenolic compounds in proteins. Some data suggest that this is also a possibility with MPO although at much lower rates and probably with surface-exposed residues only. MPO mediated protein cross-linking is postulated to occur via radical transfer catalysts, e.g. tyrosine in the range 100–200 µM in plasma.22 Therefore, though MPO is electrochemically better suited for the formation of radicals and covalent protein modification, steric restriction in its active site shortens the catalytic cycle turn-over as higher molecular weight substrates replace small, more suitable, substrates from the catalytic cycle. Consequently, the step-wise formation of oligomers exhausts MPO regeneration far more effectively than HRP regeneration sources since oligomers can still react efficiently with HRP-II but not with MPO-II.

In view of the above, our preliminary experiments showed that: a) enzyme mediated oligomerisation of reducing substrates results in a net increase in MRI signal, and b) L1–Gd3+ is a better substrate for MPO than L2–Gd3+ (see also reference 29). Under physiological conditions the following factors are of prime importance: 1) the presence of macromolecules (e.g. serum albumin) due to covalent and non-covalent binding of the substrate and the reaction products; 2) enzyme concentration (0.5 iU MPO was used, which is lower than 71–511 iU mg−1 of human atherosclerotic plaques44); 3) the amount of hydrogen peroxide: the production of peroxide in vivo from O2via NADPH-oxidase in humans proceeds at the rate of 0.2 µM s−1 (although leukocytes can yield micromolar concentrations in inflammation).61 Importantly, the excess of peroxide can render MPO inactive via two pathways (Fig. 9).
An excess of peroxide results in a transformation of native enzyme into MPO-II that reacts very slowly with small organic substrates and is nearly inactive in the presence of medium or large organic substrates. A large excess of hydrogen peroxide transforms MPO-I and MPO-II into inactive MPO-III,40 attainable also in the presence of considerable amounts of superoxide (Fig. 9). Unlike the glucose oxidase/glucose system (see Results section), manual addition of hydrogen peroxide always resulted in lower R1o values. Conversely, HRP catalysis did not show differences associated with H2O2 administration confirming that compound II reactivity differs in the case of MPO if compared to HRP. The use of enzymatic hydrogen peroxide production vs. peroxide administration resulted in higher relaxivities although the levels achieved with the use of HRP where still unattainable.
As mentioned previously, tyrosyl radicals interact with proteins resulting in protein cross-linking and covalent attachment of tyrosine to proteins. Experiments performed using MPO, L1–Gd3+, HSA or lysozyme (a protein with no known affinity to indole) suggested that the presence of the enzyme always afforded a net increase in relaxivity. Although relaxivity increased in the presence of proteins and in the absence of MPO, the magnitude of this increase was not sufficient to cause high levels of non-covalent binding of L1–Gd3+ to proteins. The lack of changes in R1o upon titration of the substrate with proteins and similarity to GdDTPA titrations suggested that the degree of non-covalent binding was negligible (i.e. below 1% at physiological levels of albumin concentrations). Overall, albumin not only produced an increase in relaxivity but also resulted in substantial stabilization of the oligomerisation products in solution. The presence of protein molecules in solution could result in either diffusion-limited quenching of radicals, and, therefore, hindering of the oligomerisation process, or, in accelerating competing reactions, i.e. protein cross-linking or an increase in the rate and patterns of oligomerization and protein attachment. Furthermore, we assessed whether: 1) the pre-formed oligomers could interact non-covalently with HSA, and 2) if oligomers could host radicals and therefore covalently react with HSA. We found that covalent binding of preformed oligomers to HSA can be ruled out. The above results suggest that enzyme mediated relaxivity increase in the presence of HSA is mainly associated with the covalent attachment of monomers to proteins.
Dialysis experiments were aimed not only at assessing oligomer size but also as a final probe of covalent attachment to proteins. The results showed that a large proportion of the monomeric starting material (i.e. 40% and 65% in the presence of HSA) is incorporated into the aggregates with the masses exceeding 10000 Da. The above result has been confirmed using MALDI, which often shows under-representation of high-mass components.62
In the absence of HSA and MPO HSA solutions did not retain any gadolinium. In the presence of MPO higher amounts of gadolinium were associated with HSA, suggesting enzyme-mediated covalent attachment which agrees with the gel-permeation chromatography study that used 111In-labeled substrate. The assessment of the above combined effect on signal enhancement in vivo showed that systemic administration of L1–Gd3+ and Gd–DTPA demonstrated similar initial signal increases as could be expected due to similarities in molecular mass. However, the use of non-reactive Gd–DTPA in conjunction with MPO did not show any lasting signal intensity after the initial period. The same was true when L1–Gd3+ enhancement of MPO-free implants was studied.
We believe that the above mechanistic study could potentially result in methods enabling early detection and staging of vulnerable atherosclerotic plaque.
Conclusions
1. The MRI signal amplification/accumulation effects observed after catalytic conversion of reducing 5-HT-containing substrates of peroxidase is a combined result of: a) enzyme-mediated increase in contrast agent size; b) covalent attachment of the contrast agent to macromolecular species present in MPO rich areas.2. Imaging at clinical magnetic field strength can identify MPO rich zones even when the MPO activities are far below the actual values reported in the literature for MPO vulnerable plaque contents.
Further experiments that use realistic animal models i.e. lipopolysaccharide-induced inflammation in mice and direct imaging of atherosclerotic aorta in rabbits, are currently being performed and the obtained animal imaging results will be submitted for publication in the future.
Acknowledgements
This study was supported in part by a National Institute of Biomedical Imaging and BioEngineering grant (AAB) and the Radiological Society of North America Research and Education Foundation (JWC).References
- A. Y. Louie and T. J. Meade, New Technol. Life Sci.: Trends Guide, 2000, 7–11 Search PubMed.
- J. Meade Thomas, K. Taylor Alisha and R. Bull Steven, Curr. Opin. Neurobiol., 2003, 13, 597–602 CrossRef CAS.
- S. Aime, C. Cabella, S. Colombatto, S. Geninatti Crich, E. Gianolio and F. Maggioni, J. Magn. Reson. Imaging, 2002, 16, 394–406 CrossRef.
- S. Aime, M. Fasano and E. Terreno, Chem. Soc. Rev., 1998, 27, 19–29 RSC.
- M. P. Lowe, Curr. Pharm. Biotechnol., 2004, 5, 519–528 CAS.
- S. Aime, A. Barge, C. Cabella, S. G. Crich and E. Gianolio, Curr. Pharm. Biotechnol., 2004, 5, 509–518 CAS.
- G. M. Lanza, P. Winter, S. Caruthers, A. Schmeider, K. Crowder, A. Morawski, H. Zhang, M. J. Scott and S. A. Wickline, Curr. Pharm. Biotechnol., 2004, 5, 495–507 CAS.
- D. Artemov, Z. M. Bhujwalla and J. W. Bulte, Curr. Pharm. Biotechnol., 2004, 5, 485–494 CAS.
- S. Boutry, C. Burtea, S. Laurent, G. Toubeau, L. Vander Elst and R. N. Muller, Magn. Reson. Med., 2005, 53, 800–807 CrossRef CAS.
- S. Laurent, L. Vander Elst, Y. Fu and R. N. Muller, Bioconjugate Chem., 2004, 15, 99–103 CrossRef CAS.
- P. Caravan, N. J. Cloutier, M. T. Greenfield, S. A. McDermid, S. U. Dunham, J. W. M. Bulte, J. C. Amedio, Jr., R. J. Looby, R. M. Supkowski, W. D. Horrocks, Jr., T. J. McMurry and R. B. Lauffer, J. Am. Chem. Soc., 2002, 124, 3152–3162 CrossRef CAS.
- J. C. Frias, K. J. Williams, E. A. Fisher and Z. A. Fayad, J. Am. Chem. Soc., 2004, 126, 16316–16317 CrossRef CAS.
- A. Y. Louie, M. M. Huber, E. T. Ahrens, U. Rothbacher, R. Moats, R. E. Jacobs, S. E. Fraser and T. J. Meade, Nat. Biotechnol., 2000, 18, 321–325 CrossRef CAS.
- R. A. Moats, S. E. Fraser and T. J. Meade, Angew. Chem., Int. Ed. Engl., 1997, 36, 726–728 CrossRef CAS.
- A. E. Merbach and E. Toth, The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, John Wiley & Sons, Ltd, Chichester, 1st ed., 2001 Search PubMed.
- D. Favretto, A. Bertazzo, C. Costa, G. Allegri, N. Donato and P. Traldi, Rapid Commun. Mass Spectrom., 1998, 12, 193–197 CrossRef CAS.
- A. Bertazzo, C. Costa, G. Allegri, D. Favretto and P. Traldi, Rapid Commun. Mass Spectrom., 1996, 10, 1299–1303 CrossRef CAS.
- D Favretto, A. Bertazzo, C. Costa, G. Allegri and P. Traldi, Rapid Commun. Mass Spectrom., 1997, 11, 1038–1042 CrossRef CAS.
- A. Bertazzo, D. Favretto, C. Costa, G. Allegri and P. Traldi, Rapid Commun. Mass Spectrom., 1998, 12, 767–772 CrossRef CAS.
- A. Bertazzo, C. Costa, G. Allegri, M. Schiavolin, D. Favretto and P. Traldi, Rapid Commun. Mass Spectrom., 1999, 13, 542–547 CrossRef CAS.
- G. Heuther, A. Reimer, F. Schmidt, P. Schuff-Werner and M. M. Brudny, J. Neural Transm. Suppl., 1990, 32, 249–257 Search PubMed.
- J. W. Heinecke, W. Li, G. A. Francis and J. A. Goldstein, J. Clin. Invest., 1993, 91, 2866–2872 CrossRef CAS.
- J. S. Jacob, D. P. Cistola, F. F. Hsu, S. Muzaffar, D. M. Mueller, S. L. Hazen and J. W. Heinecke, J. Biol. Chem., 1996, 271, 19950–19956 CrossRef CAS.
- M. L. McCormick, J. P. Gaut, T. S. Lin, B. E. Britigan, G. R. Buettner and J. W. Heinecke, J. Biol. Chem., 1998, 273, 32030–32037 CrossRef CAS.
- S. L. Hazen, F. F. Hsu, J. P. Gaut, J. R. Crowley and J. W. Heinecke, Methods Enzymol., 1999, 300, 88–105 CAS.
- J. W. Heinecke, Toxicology, 2002, 177, 11–22 CrossRef CAS.
- M. Tien, Arch. Biochem. Biophys., 1999, 367, 61–66 CrossRef CAS.
- A. Bogdanov, Jr., L. Matuszewski, C. Bremer, A. Petrovsky and R. Weissleder, Mol. Imaging, 2002, 1, 16–23 Search PubMed.
- J. W. Chen, W. Pham, R. Weissleder and A. Bogdanov, Jr., Magn. Reson. Med., 2004, 52, 1021–1028 CrossRef CAS.
- S. J. Klebanoff, Proc. Assoc. Am. Physicians, 1999, 111, 383–389 Search PubMed.
- S. J. Klebanoff, J. Leukocyte Biol., 2005, 77, 598–625 Search PubMed.
- J. Arnhold, Biochemistry (Moscow), 2004, 69, 4–9 CrossRef CAS.
- R. Zhang, M. L. Brennan, X. Fu, R. J. Aviles, G. L. Pearce, M. S. Penn, E. J. Topol, D. L. Sprecher and S. L. Hazen, JAMA, 2001, 286, 2136–2142 Search PubMed.
- M. Querol Sans, J. Chen, R. Weissleder and A. Bogdanov, Mol. Imaging Biol., 2005, 7, 403–410 Search PubMed.
- M. Querol, J. W. Chen, R. Weissleder and A. Bogdanov, Jr., Org. Lett., 2005, 7, 1719–1722 CrossRef CAS.
- S. Mutze, U. Hebling, W. Stremmel, J. Wang, J. Arnhold, K. Pantopoulos and S. Mueller, J. Biol. Chem., 2003, 278, 40542–40549 CrossRef.
- B. A. Wagner, G. R. Buettner, L. W. Oberley, C. J. Darby and C. P. Burns, J. Biol. Chem., 2000, 275, 22461–22469 CrossRef CAS.
- G. F. Vile, L. A. Rothwell and A. J. Kettle, Arch. Biochem. Biophys., 1998, 359, 51–56 CrossRef CAS.
- R. G. Midwinter, M. C. Vissers and C. C. Winterbourn, Arch. Biochem. Biophys., 2001, 394, 13–20 CrossRef CAS.
- E. Ghibaudi and E. Laurenti, Eur. J. Biochem., 2003, 270, 4403–4412 CrossRef CAS.
- G. E. Jackson and L. L. Du Toit, Polyhedron, 1992, 11, 1315–1318 CrossRef CAS.
- R. Hovland, A. J. Aasen and J. Klaveness, Org. Biomol. Chem., 2003, 1, 1707–1710 RSC.
- S. Aime, E. Gianolio, D. Longo, R. Pagliarin, C. Lovazzano and M. Sisti, ChemBioChem, 2005, 6, 818–820 CrossRef CAS.
- A. Daugherty, J. L. Dunn, D. L. Rateri and J. W. Heinecke, J. Clin. Invest., 1994, 94, 437–444 CrossRef CAS.
- J. W. Heinecke, Curr. Opin. Lipidol., 1997, 8, 268–274 CrossRef CAS.
- J. W. Heinecke, BioFactors, 1997, 6, 145–155 Search PubMed.
- C. Zhang, J. Yang, J. D. Jacobs and L. K. Jennings, Am. J. Physiol.: Heart. Circ. Physiol., 2003, 285, H2563–2572 CAS.
- C. Bergt, S. Pennathur, X. Fu, J. Byun, K. O'Brien, T. O. McDonald, P. Singh, G. M. Anantharamaiah, A. Chait, J. Brunzell, R. L. Geary, J. F. Oram and J. W. Heinecke, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13032–13037 CrossRef CAS.
- S. Pennathur, C. Bergt, B. Shao, J. Byun, S. Y. Kassim, P. Singh, P. S. Green, T. O. McDonald, J. Brunzell, A. Chait, J. F. Oram, K. O'Brien, R. L. Geary and J. W. Heinecke, J. Biol. Chem., 2004, 279, 42977–42983 CrossRef CAS.
- P. G. Furtmuller, M. Zederbauer, W. Jantschko, J. Helm, M. Bogner, C. Jakopitsch and C. Obinger, Arch. Biochem. Biophys., 2005 Search PubMed.
- W. Jantschko, P. G. Furtmuller, M. Allegra, M. A. Livrea, C. Jakopitsch, G. Regelsberger and C. Obinger, Arch. Biochem. Biophys., 2002, 398, 12–22 CrossRef CAS.
- C. C. Winterbourn and A. J. Kettle, Jpn. J. Infect. Dis., 2004, 57, S31–33 Search PubMed.
- A. J. Kettle and C. C. Winterbourn, Biochemistry, 2001, 40, 10204–10212 CrossRef CAS.
- C. C. Winterbourn and A. J. Kettle, Basic Life Sci., 1988, 49, 823–827 Search PubMed.
- L. A. Marquez, J. T. Huang and H. B. Dunford, Biochemistry, 1994, 33, 1447–1454 CrossRef CAS.
- J. Arnhold, P. G. Furtmuller, G. Regelsberger and C. Obinger, Eur. J. Biochem., 2001, 268, 5142–5148 CrossRef CAS.
- J. Arnhold, P. G. Furtmuller and C. Obinger, Redox. Rep., 2003, 8, 179–186 Search PubMed.
- P. G. Furtmuller, J. Arnhold, W. Jantschko, H. Pichler and C. Obinger, Biochem. Biophys. Res. Commun., 2003, 301, 551–557 CrossRef CAS.
- P. G. Furtmuller, W. Jantschko, M. Zederbauer, C. Jakopitsch, J. Arnhold and C. Obinger, Jpn. J. Infect. Dis., 2004, 57, S30–31 Search PubMed.
- E. Ghibaudi, E. Laurenti, C. Pacchiardo, G. Suriano, N. Moguilevsky and R. Pia Ferrari, J. Inorg. Biochem., 2003, 94, 146–154 CrossRef CAS.
- S. Mueller, K. Pantopoulos, C. A. Hubner, W. Stremmel and M. W. Hentze, J. Biol. Chem., 2001, 276, 23192–23196 CrossRef CAS.
- K. J. Wu and R. W. Odom, Anal. Chem., 1998, 70, 456A–461A CAS.
Footnote |
| † Electronic supplementary information (ESI) available: MALDI-TOF of oligomeric material, synthetic methods, MRI and SPECT imaging details, radiolabelling details. See DOI: 10.1039/b601540a |
| This journal is © The Royal Society of Chemistry 2006 |