Synthesis and molecular structures of the first phosphoranylidene complexes of rare earth metals†‡
Konstantin A.
Rufanov
*,
Bernd H.
Müller
,
Anke
Spannenberg
* and
Uwe
Rosenthal
Leibniz-Institut für Organische Katalyse an der Universität Rostock, Albert-Einstein-Straße 29A, 18059, Rostock, Germany. E-mail: k_rufanov@gmx.de; Fax: +49 (0)381 1281 5000
First published on 9th December 2005
Abstract
Metallation of the donor-functionalised ylide ligand Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–(o-CH3OC6H4) with homoleptic alkyl and aryl complexes of yttrium and lutetium furnished unprecedented phosphoranylidene complexes, featuring a central μ2-M2C2 core.
CH–(o-CH3OC6H4) with homoleptic alkyl and aryl complexes of yttrium and lutetium furnished unprecedented phosphoranylidene complexes, featuring a central μ2-M2C2 core.
The organometallic chemistry of rare earth metals, which to date has been dominated by metallocene complexes, has recently received some impetus towards the search for new ligand systems to expand its scope beyond this traditional realm.1–8 In contrast to main-group9–11 and transition metals,12 there are no examples of the activation of the CH protons of ylenes by rare-earth metals. Because the increased Lewis acidity of the lanthanides may lead to reactivity patterns that are different from those for transition metals, we were encouraged to develop new synthetic approaches towards lanthanide complexes of these phosphorus-ylide ligands.
Previously Schumann has found that Ph3PCH2 and Ph3PCHSiMe3 simply substitute THF in the complexes Cp2LuX(THF) (X = But, Cl),13 while (C5Me5)2LuMe reacts with ortho-metallation of one of the phenyl substituents and formation of a 5-membered cyclic ylide complex,14–16 rather than in metallation of the CH2 group (Scheme 1). Similar donor–acceptor adducts were later reported for other rare earth metals.17
 | ||
| Scheme 1 | ||
For our study we have chosen the known chelating phosphoranylidene ligand Ph3PCH-(o-CH3OC6H4) 1,18 whose structure we determined for comparison purposes (Fig. 1).§
![ORTEP drawing of the molecular structure of 1; 50% probability thermal ellipsoids are presented.Data were collected on a STOE IPDS diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined by full-matrix least-squares techniques against F2 using SHELX-97 software package.25Crystal data: for 1: C26H23OP, M = 382.41, triclinic, space group P1̄, a = 9.746(2) Å, b = 10.046(2) Å, c = 10.715(2) Å, α = 73.76(2)°, β = 85.61(2)°, γ = 89.94(2)°, V = 1004.0(3) Å3, Z = 2, Dc = 1.265 g cm−3, μ(Mo-Kα) = 0.151 mm−1. T = −123 °C, crystal dimensions 0.76 × 0.40 × 0.08 mm, 6420 reflections were measured, 3401 were symmetry independent and 2577 were observed [I > 2σ(I)], R1 = 0.0376 and wR2 = 0.0945 (all data). CCDC reference number 245413. For 3: C84H80Lu2O4P2, M = 1565.36, monoclinic, space group P21/c, a = 14.192(5) Å, b = 12.461(4) Å, c = 20.490(5) Å, β = 105.51(3)°, V = 3491.6(18) Å3, Z = 2, Dc = 1.489 g cm−3, μ(Mo-Kα) = 2.908 mm−1. T = −93 °C, crystal dimensions 0.40 × 0.28 × 0.20 mm, 17113 reflections were measured, 4979 were symmetry independent and 2802 were observed [I > 2σ(I)], R1 = 0.0843 and wR2 = 0.2278 (all data). The intensities were corrected for Lorentz polarization and absorption effects using ABSCOR, a modification of DIFABS (Tmin = 0.366, Tmax = 0.558).26 CCDC reference number 245412. For 5a: C68H72O6P2Y2, M = 1225.02, triclinic, space group P1̄, a = 12.513(3) Å, b = 14.455(3) Å, c = 19.023(4) Å, α = 86.89(3)°, β = 75.64(3)°, γ = 70.68(3)°, V = 3144.4(11) Å3, Z = 2, Dc = 1.294 g cm−3, μ(Mo-Kα) = 1.936 mm−1. T = −73 °C, crystal dimensions 0.45 × 0.25 × 0.20 mm, 40601 reflections were measured, 11055 were symmetry independent and 6310 were observed [I > 2σ(I)], R1 = 0.0624 and wR2 = 0.1511 (all data). CCDC reference number 286123. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b514439f.](/image/article/2006/NJ/b514439f/b514439f-f1.gif) | ||
| Fig. 1 ORTEP drawing of the molecular structure of 1; 50% probability thermal ellipsoids are presented.¶ | ||
In the early stages of our investigation we attempted to metallate 1 with rather stable Cp#2Lu(THF)CH3 (Cp# = [1,3-(SiMe3)2C5H3]) and M[N(SiMe3)2]3 (M = Y, Lu) complexes. However, NMR-monitoring of these reactions revealed only the simple substitution of the THF molecule or reversible formation of unstable adducts, similar to those described earlier.14 The metallation of 1, however, was not observed. Although the protonolysis of the more reactive homoleptic alkyl- or aryl-complexes of the types Ln(CH2SiMe3)3(THF)n (n = 2, 3) or Ln(o-Me2NCH2C6H4)3 with different CH acids, such as Cp and acetylenes, is well established,19–21 such reactions have not been reported for phosphoranylidenes.
Initially we discovered that reaction of Lu(CH2SiMe3)3(THF)2 with 1 leads to exhaustive protonolysis. Indeed, NMR monitoring of the reaction mixture revealed the gradual growth of a new broad 31P signal at 12.2 accompanied by the decline of the sharp signal of 1 at 10.8. The reaction, which is complete after 2 d, furnished yellow, microcrystalline material in 46% yield. Single crystals were obtained from a saturated benzene solution that had been standing at 10 °C for several weeks. The broad 1H resonances for the aromatic protons and the methoxy group suggest that 3 displays a dynamic behaviour in solution and that the ethereal ligands are hemi-labile.22
We also studied the metallation of 1 with the thermally more stable and less reactive yttrium complex Y(o-Me2NCH2C6H4)3, intending to synthesize heteroleptic mono-ylide complex 4 (Scheme 2). A reaction of equimolar amounts of 1 and the metal complex furnished, after heating at 65 °C for 5 h, unexpectedly a similar product of the exhaustive protonolysis of 1, namely 5. This yellow microcrystalline material has very low solubility in common solvents and was characterized by 1H-, 31P-NMR and elemental analysis and shown to be an analogue of binuclear complex 3. Recrystallization of 5 from boiling THF furnished, upon slow cooling of the solution, the light yellow complex 5a. The molecular structures of 3 and 5a differ only in the number of coordinated THF molecules. Each lutetium atom in 3 is coordinated by only one THF donor ligand and the molecule has a center of inversion, with both lutetium atoms having distorted octahedral coordination geometry. In compound 5a, by contrast, one yttrium is six-coordinate, bearing one THF ligand, while the other is seven-coordinate, bearing two THF ligands (Fig. 2).23
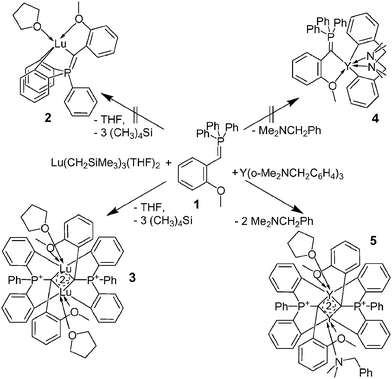 | ||
| Scheme 2 Synthesis of binuclear phosphoranylidene complexes 3 and 5. | ||
 | ||
| Fig. 2 ORTEP drawing of the molecular structure of 5a. All hydrogen atoms are omitted; for non-metallated Ph-rings and co-ordinated THF ligands only ipso-C and oxygen atoms are shown for clarity. 50% probability thermal ellipsoids are presented only for the labelled atoms. | ||
The difference in bond lengths within the central μ2-M2C2 core is larger for 5a (Δ = 0.18 Å) than for 3 (Δ = 0.14 Å), but the poor crystal quality of 3 (R1 = 0.0843) prohibits detailed comparisons of the bond lengths and angles of 3 with those of 1 and 5a. Selected structural parameters of the structures of the free ligand 1 and of 5a are given in Table 1.
| 1 | 5a | |
|---|---|---|
| Y–C(shortest) | 2.396(6) | |
| Y–C(longest) | 2.576(6) | |
| Y–CPh | 2.480(7)–2.572(6) | |
| Y–OTHF | 2.408(5), 2.450(5), 2.471(4) | |
| Y–OOMe | 2.431(4), 2.577(4) | |
| P–CYlide | 1.693(2) | 1.701(6), 1.714(6) |
| P–CPh | 1.818(2)–1.831(2) | 1.807(7)–1.851(6) |
| CYlide–CAryl | 1.452(2) | 1.449(9), 1.455(8) |
| CAryl–OOMe | 1.382(2) | 1.398(7), 1.404(8) |
| C–P–C | 103.4(1)–120.2(1) | 104.4(3)–115.4(3) |
| P–CYlide–CAryl | 132.0(1) | 117.8(4), 119.9(4) |
| CYlide–Y–CYlide | 85.4(2), 87.5(2) | |
| Y–C–Y | 93.2(2), 93.7(2) |
The shortest Y–C bond in the μ2-Y2C2 core is of 2.396(6) Å long, and it lies in a range typical for neutral, low-coordinated, non-Cp complexes of yttrium with at least two hard-donor ligands.24a The THF molecules form strong bonds with the yttrium atoms with a maximum bond length of 2.47 Å, which is a similar value to those found in other Y–THF adducts.24 One of the Y–O bonds to the methoxy groups is much longer and indicates weaker bonding. The phosphonium centers are nearly tetrahedral with a narrower range of C–P–C angles 110 (±5)° and significantly smaller P–CYlide−CAryl angle 119(±1)° than that in the free ligand 1—112(±8)° and 132°, respectively.
These new phosphoranylidene complexes of lutetium and yttrium should be important both for our understanding of fundamental bonding concepts of organometallic chemistry and as catalyst precursors. Full details of our results on rare earth complexes in this series will be discussed in a forthcoming paper.
Acknowledgements
The authors thank Prof. H. Schumann and Dr S. I. Troyanov for fruitful discussions, Dr B. Ziemer (Humboldt Universität zu Berlin) for crystallographic studies of 1 and 3 and Dipl.-Ing. S. Schutte (Technische Universität Berlin) and Petra Bartles for technical assistance. Financial support through the DFG programmes (SPP 1166, GrK 352 and GrK 1213) is gratefully acknowledged.References
- W. J. Evans, Polyhedron, 1987, 6, 803 CrossRef CAS.
- C. J. Schaverein, Adv. Organomet. Chem., 1994, 36, 283 CAS.
- F. T. Edelmann, in Comprehensive Organometallic Chemistry, ed. E.W. Abel, F. G. A. Stone, G. Wilkinson and C. E. Housecroft, Pergamon Press, 1995, vol. 4, p. 11 Search PubMed.
- H. Schumann, J. A. Meese-Marktscheffel and L. Esser, Chem. Rev., 1995, 95, 865 CrossRef CAS.
- F. T. Edelmann, D. M. M. Freckmann and H. Schumann, Chem. Rev., 2002, 102, 1851 CrossRef CAS.
- R. Anwander, in Topics in Organometallic Chemistry, vol. 2, ed. S. Kobayashi, Springer-Verlag, Berlin, 1999, p. 1 Search PubMed.
- Z. Hou and Y. Wakatsuki, in Science of Synthesis, ed. R. Noyori and T. Imamoto, Thieme, Stuttgart, 2001, ch. 1, p. 2 Search PubMed.
- W. E. Piers and D. J. H. Emslie, Coord. Chem. Rev., 2002, 233–234, 131 CrossRef CAS.
- H. Schmidbaur and W. Malisch, Angew. Chem., 1970, 82, 84 ( Angew. Chem., Int. Ed. Engl. , 1970 , 9 , 77 ) CrossRef.
- H. Schmidbaur and W. Malisch, Chem. Ber., 1971, 104, 150 CrossRef CAS.
- K. Korth and J. Sundermeyer, Tetrahedron Lett., 2000, 41, 5461 CrossRef CAS.
- W. C. Kaska and K. A. Ostoja Starzewski, in Transition metal complexes with ylides, Ylides and Imines of Phosphorus, ed. A.W. Johnson, John Wiley & Sons, NY, 1993, p. 485, and references cited therein Search PubMed.
- H. Schumann, I. Albrecht, F. W. Reier and E. Hahn, Angew. Chem., 1984, 23, 522.
- P. L. Watson, J. Chem. Soc., Chem. Commun., 1983, 276 RSC.
- H. Schumann, F. W. Reier and E. Palamidis, J. Organomet. Chem., 1985, 297, C30 CrossRef CAS.
- H. Schumann and F. W. Reier, J. Organomet. Chem., 1984, 269, 21 CrossRef CAS.
- M. Booij, J. Berth, R. Duchateau, D. S. Postma, A. Meetsma and J. H. Teuben, Organometallics, 1993, 12(9), 3531 CrossRef CAS.
- L. K. Johnson, S. C. Virgil, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1990, 112, 5384 CrossRef CAS.
- K. C. Hultzsch, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 1999, 38, 227 CrossRef CAS.
- O. Tardif, M. Nishiura and Z. Hou, Organometallics, 2003, 22, 1171 CrossRef CAS.
- T. M. Cameron, J. C. Gordon and B. L. Scott, Organometallics, 2004, 23, 2995 CrossRef CAS.
- Poor solubility of 3 at lower temperatures prohibits study of its dynamic behaviour with NMR in more detail.
- Two solvate C6H6 molecules per asymmetric unit of 3 and one solvate THF molecule per asymmetric unit of 5a have been found.
- See for example: (a) W. J. Evans, J. C. Brady and J. W. Ziller, J. Am. Chem. Soc., 2001, 123, 7711 CrossRef CAS; (b) S. Arndt, P. M. Zeimentz, T. P. Spaniol, J. Okuda, M. Honda and K. Tatsumi, Dalton Trans., 2003, 3622 RSC; (c) P. G. Hayes, G. C. Weich, D. J. H. Emslei, C. L. Noack and W. E Piers, Organometallics, 2003, 22, 1577 CrossRef CAS; (d) S. Bambirra, D. van Leusen, A. Meetsma, B. Hessen and J. H. Teuben, Chem. Commun., 2003, 522 RSC.
- (a) G. M. Sheldrick, Programm for solution of crystal structures, SHELXL-97, Universität Göttingen, 1997, 22 Search PubMed; (b) G. M. Sheldrick, Programm for the refinement of crystal structures, SHELXL-97, Universität Göttingen, 1997, 22 Search PubMed.
- N. Walker and D. Stuart, Acta Crystallogr., Sect. A, 1983, A39, 158 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/b514439f |
| ‡ Dedicated to Prof. Dmitry A. Lemenovskii on occasion of his 60th birthday |
| § Synthetic and analytical details for the starting reagents are given in the ESI. Synthesis of3: a 0.5 M benzene solution of Lu(CH2SiMe3)3(THF)2 (5 mL, 2.5 mmol) was slowly added to a stirred solution of 1 (956 mg, 2.5 mmol) in benzene (5 mL) at room temperature. The reaction mixture was then stirred and heated at 45 °C. It gradually changed in colour from orange-red to dark yellow. After 2 d a small amount of precipitate formed that was filtered off and the resulting solution was reduced in volume to approx. 5 mL and very slowly cooled down to 10 °C for crystallization. After several weeks light yellow crystals formed. The product was filtered off, washed with pre-cooled benzene (2 mL) and dried under vacuum. Yield 0.72 g (46%). 1H NMR (300 MHz, C6D6, +23 °C): δ 7.89–6.95 (br m, Aryl-groups and solvated C6H6), 3.60 (br s, CH2–O, THF), 2.75 (br s, CH3–O), 1.70 (br t, CH2–CH2–O, THF). 31P NMR (121.5 MHz, C6D6, +23 °C): δ 12.2 (br s). Elemental analysis was calculated for C60H56Lu2O4P2(C6H6)4, C 64.45%, H 5.15%, Lu 22.35%, observed C 63.92%, H 5.28%, Lu 22.17%. Synthesis of 5: a 0.5 M THF solution of Y(o-Me2NCH2C6H4)3 (5 mL, 2.5 mmol) was added to a stirred solution of 1 (956 mg, 2.5 mmol) in toluene (30 mL). The orange solution was slowly heated to 65 °C and allowed to stir at this temperature for 5 h, while it gradually deepened in colour and a bright yellow precipitate deposited. After slow cooling to room temperature, the dark red solution was decanted and the yellow solid was washed with toluene (2 × 5 mL) and dried under high vacuum. Yield 0.60 g (42%) of C52H40O2P2Y2(C4H8O)(C9H13N). 1H NMR (300 MHz, C5D5N, +23 °C): δ 7.89–7.15 (br m, Aryl-groups), 3.65 (br s, CH2–O, THF), 3.54 (s, Me2NCH2), 3.45 (br s, CH3–O), 2.19 (s, Me2N), 1.59 (br t, CH2–CH2–O, THF). 31P NMR (121.5 MHz, C5D5N, +23 °C): δ 36.8. Elemental analysis was calculated for C65H61NO3P2Y2, C 68.25%, H 5.37%, N 1.22%, observed C 68.96%, H 5.31%, N 1.22%. |
¶ Data were collected on a STOE IPDS diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined by full-matrix least-squares techniques against F2 using SHELX-97 software package.25Crystal data: for 1: C26H23OP, M = 382.41, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.746(2) Å, b = 10.046(2) Å, c = 10.715(2) Å, α = 73.76(2)°, β = 85.61(2)°, γ = 89.94(2)°, V = 1004.0(3) Å3, Z = 2, Dc = 1.265 g cm−3, μ(Mo-Kα) = 0.151 mm−1. T = −123 °C, crystal dimensions 0.76 × 0.40 × 0.08 mm, 6420 reflections were measured, 3401 were symmetry independent and 2577 were observed [I > 2σ(I)], R1 = 0.0376 and wR2 = 0.0945 (all data). CCDC reference number 245413. For 3: C84H80Lu2O4P2, M = 1565.36, monoclinic, space group P21/c, a = 14.192(5) Å, b = 12.461(4) Å, c = 20.490(5) Å, β = 105.51(3)°, V = 3491.6(18) Å3, Z = 2, Dc = 1.489 g cm−3, μ(Mo-Kα) = 2.908 mm−1. T = −93 °C, crystal dimensions 0.40 × 0.28 × 0.20 mm, 17113 reflections were measured, 4979 were symmetry independent and 2802 were observed [I > 2σ(I)], R1 = 0.0843 and wR2 = 0.2278 (all data). The intensities were corrected for Lorentz polarization and absorption effects using ABSCOR, a modification of DIFABS (Tmin = 0.366, Tmax = 0.558).26 CCDC reference number 245412. For 5a: C68H72O6P2Y2, M = 1225.02, triclinic, space group P, a = 12.513(3) Å, b = 14.455(3) Å, c = 19.023(4) Å, α = 86.89(3)°, β = 75.64(3)°, γ = 70.68(3)°, V = 3144.4(11) Å3, Z = 2, Dc = 1.294 g cm−3, μ(Mo-Kα) = 1.936 mm−1. T = −73 °C, crystal dimensions 0.45 × 0.25 × 0.20 mm, 40601 reflections were measured, 11055 were symmetry independent and 6310 were observed [I > 2σ(I)], R1 = 0.0624 and wR2 = 0.1511 (all data). CCDC reference number 286123. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b514439f. , a = 9.746(2) Å, b = 10.046(2) Å, c = 10.715(2) Å, α = 73.76(2)°, β = 85.61(2)°, γ = 89.94(2)°, V = 1004.0(3) Å3, Z = 2, Dc = 1.265 g cm−3, μ(Mo-Kα) = 0.151 mm−1. T = −123 °C, crystal dimensions 0.76 × 0.40 × 0.08 mm, 6420 reflections were measured, 3401 were symmetry independent and 2577 were observed [I > 2σ(I)], R1 = 0.0376 and wR2 = 0.0945 (all data). CCDC reference number 245413. For 3: C84H80Lu2O4P2, M = 1565.36, monoclinic, space group P21/c, a = 14.192(5) Å, b = 12.461(4) Å, c = 20.490(5) Å, β = 105.51(3)°, V = 3491.6(18) Å3, Z = 2, Dc = 1.489 g cm−3, μ(Mo-Kα) = 2.908 mm−1. T = −93 °C, crystal dimensions 0.40 × 0.28 × 0.20 mm, 17113 reflections were measured, 4979 were symmetry independent and 2802 were observed [I > 2σ(I)], R1 = 0.0843 and wR2 = 0.2278 (all data). The intensities were corrected for Lorentz polarization and absorption effects using ABSCOR, a modification of DIFABS (Tmin = 0.366, Tmax = 0.558).26 CCDC reference number 245412. For 5a: C68H72O6P2Y2, M = 1225.02, triclinic, space group P, a = 12.513(3) Å, b = 14.455(3) Å, c = 19.023(4) Å, α = 86.89(3)°, β = 75.64(3)°, γ = 70.68(3)°, V = 3144.4(11) Å3, Z = 2, Dc = 1.294 g cm−3, μ(Mo-Kα) = 1.936 mm−1. T = −73 °C, crystal dimensions 0.45 × 0.25 × 0.20 mm, 40601 reflections were measured, 11055 were symmetry independent and 6310 were observed [I > 2σ(I)], R1 = 0.0624 and wR2 = 0.1511 (all data). CCDC reference number 286123. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b514439f. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2006 |