Carboxylate complexation by a family of easy-to-make ortho-phenylenediamine based bis-ureas: studies in solution and the solid state†
Simon J. Brooks, Peter R. Edwards, Philip A. Gale* and Mark E. Light
School of Chemistry, University of Southampton, Southampton, UK SO17 1BJ. E-mail: philip.gale@soton.ac.uk; Fax: +44 23 8059 6805; Tel: +44 23 8059 3332
First published on 28th November 2005
Abstract
Simple bis-urea compounds based on ortho-phenylenediamine function as excellent receptors for carboxylate anions in DMSO-d6–0.5% water solution. By varying the functional groups in these compounds, the binding affinity of carboxylate anions can be modulated. Solid-state studies illustrate the cleft-like arrangement of the DDDD hydrogen bond array in the carboxylate complexes of these compounds.
Introduction
The development of simple hydrogen bonding motifs for anion recognition that are easy to make and functionalize has had a profound effect on anion complexation chemistry. Particularly notable are the isophthalamide-based systems reported by Crabtree1 and Smith2 independently in 1997, which have led on to many new anion3 and ion pair4 receptors and anion-templated helices,5 catenanes6 and rotaxanes.7 Another example is the calixpyrroles8 which have developed into a diverse family of selective receptors,9 sensors10 and separation agents11 for anionic species. Hence a continuing theme in our research is the development of new simple receptors12 with the potential to develop into families of anion complexation agents.Receptors for carboxylate anions are important for the recognition of a variety of biomolecules and in particular amino acids.13 Many of the carboxylate binding sites in these systems contain urea or thiourea groups and many receptors containing either one14 or two15 of these moieties have been reported and shown to be excellent carboxylate receptors and sensors. As we have recently reported, compounds 1–3 were synthesized in an attempt to explore the anion-binding properties of 1,2-phenylenediamine derived neutral hydrogen bonding receptors.16 Little work has been done on anion receptors based upon ortho-phenylenediamine or analogous diamines.17 The potential carboxylate complexation properties of these systems has so far yet to be exploited. In 2000, Reinhoudt and co-workers reported the anion-binding ability of cyclic and acyclic receptors containing two ortho-phenylenediamine based bis-urea units.18 This work showed that these receptors were selective for dihydrogen phosphate in DMSO. We initially wished to ‘extract’ the bis-urea unit and investigate the intrinsic anion-binding properties of 1,1′-(1,2-phenylene)bis(3-phenylurea) (3) and those of the structurally related compounds 1 and 2.16
Compound 1 had previously been reported by Rebek and co-workers,19 whilst the anion- (but not carboxylate-) binding properties of 2 were reported by Cheng and co-workers, whilst our initial study was in progress.20 These compounds were studied in order to provide insight into their anion-complexation properties with the aim of designing an optimum hydrogen bond donor arrangement motif containing an ortho-phenylenediamine unit that could be subsequently exploited as an oxo-anion recognition motif. Anion-binding studies were conducted using 1H NMR titration techniques in DMSO-d6–0.5% water and revealed that all three receptors displayed varying degrees of oxo-anion selectivity. Compound 1, which contains two hydrogen bond donor groups, displayed the lowest stability constants (Table 1). Compound 2 shows a higher affinity for oxo-anions than compound 1. X-Ray‡ quality crystals of the complexes of receptor 2 with tetrabutylammonium fluoride,§ chloride¶ and acetate16 were obtained by slow evaporation of acetonitrile solutions of the complexes. The structures of the fluoride and chloride complexes illustrate the pyrrole NH⋯OC amide hydrogen bonded dimer that has been frequently observed in the structure of 2-amidopyrroles.12 The fluoride complex [Fig. 1(a)] forms a dimer wherein each anion is bound by two amide NH groups and a pyrrole NH group [N⋯F 2.639(8)–2.733(7) Å]. The other pyrrole is involved in dimer formation with an adjacent complex. The structure of the chloride complex [Fig. 1(b)] reveals the anion bound by only two hydrogen bonds from the central amide groups [N⋯Cl 3.165(6) and 3.225(5) Å], with the two pyrrole rings oriented outwards and involved in amidopyrrole dimer formation [N⋯O 2.809(7) and 2.825(7) Å], with the result that the chloride complex forms an anionic tape in the solid state. The structure of the acetate complex [Fig. 1(c)] which we have reported previously,16 shows the anion bound by two amide NH groups [N⋯O 2.751(3) and 2.731(3) Å] and one pyrrole NH group [N⋯O 2.781(3) Å]. The other pyrrole NH group is once again oriented out of the cavity, forming an intermolecular hydrogen bond to an adjacent complex [N⋯O 2.874(3) Å].
| Compounds | |||
|---|---|---|---|
| a Errors estimated to be no more than ±10%.b We have repeated the anion-complexation studies with compound 2 under slightly different experimental conditions from those originally reported,20 however our results are broadly similar. | |||
| Anion | 1 | 2b | 3 |
| Cl− | 13 | 12 | 43 |
| Br− | — | — | <10 |
| CH3CO2− | 98 | 251 | 3210 |
| C6H5CO2− | 43 | 113 | 1330 |
| H2PO4− | 149 | 295 | 732 |
| HSO4− | — | — | 10 |
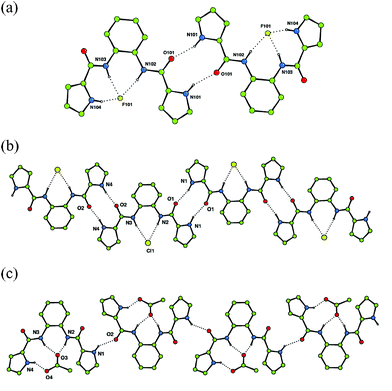 | ||
| Fig. 1 (a) Hydrogen bonded dimer formation in the tetrabutylammonium fluoride complex of receptor 2 and chain formation in the chloride (b) and acetate (c) complexes of receptor 2. Tetrabutylammonium counter cations and non-acidic hydrogen atoms have been omitted for clarity. | ||
The synthesis of compound 3 had previously been reported21 and was synthesized in order to increase the affinity for carboxylates by opening up the binding site. The crystal structure of the benzoate complex of receptor 3 revealed that this compound is capable of binding carboxylates via four hydrogen bonds in an almost symmetrical arrangement (Fig. 2).16 In addition, higher carboxylate affinities in solution suggested that all four of the NH hydrogen bond donor groups are involved in carboxylate complexation in DMSO-d6–0.5% water solution. The shifts of the amide NH protons of 3 upon addition of anions are illustrated in Fig. 3.
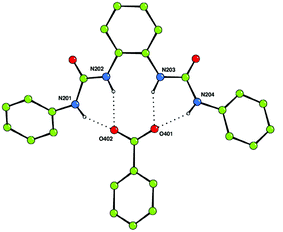 | ||
| Fig. 2 The benzoate complex of receptor 3 showing the formation of four hydrogen bonds between the receptor and carboxylate anion in the solid state. One of the two independent complexes in the asymmetric unit is shown (the other is in essentially the same arrangement). Tetrabutylammonium counter cations and non-acidic hydrogen atoms have been omitted for clarity. | ||
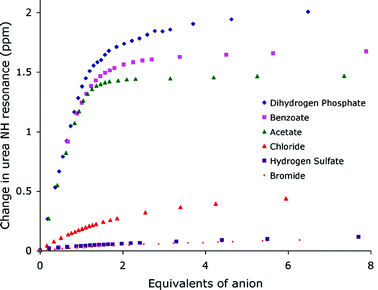 | ||
| Fig. 3 Shifts of the central NH groups in compound 3 upon addition of tetrabutylammonium anion salts in DMSO-d6–0.5% H2O. | ||
Etter has previously shown in a variety of crystal structures of diaryl ureas that interactions between urea carbonyl oxygen atoms and adjacent aryl CH groups can contribute towards an increase in the angle between the aryl groups.22 The conformation adopted by receptor 3 upon binding of anions may also be stabilized by internal interactions that might help to preorganize the hydrogen bonding array. Whilst stabilising hydrogen bond interactions and resonance stabilization favour an in-plane conformation of the aryl group, equilibrium hydrogen bond distances are typically longer than the covalent bonds that connect the two groups and this may deform the binding mode. The crystal structure of the benzoate complex of 316 shows that there are a variety of different CH⋯O hydrogen bond lengths between the aryl groups and the urea oxygen atoms in the solid state [between 2.740(4)–2.939(4) Å], however the pendant aryl groups are not coplanar with the central aryl ring.
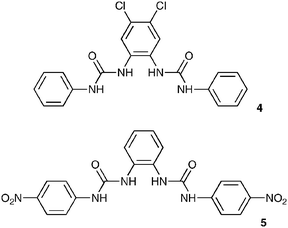
The high affinity of compound 3 for carboxylates merited further investigation. We initially decided to study how the introduction of electron-withdrawing substituents would affect the observed anion association constants and therefore synthesized compounds 4 and 5, in which chloro- and nitro-substituents were located on the central and pendant aryl groups, respectively. Compound 4 displayed significantly enhanced anion affinities relative to compound 3, with the largest relative change in association constant observed for dihydrogen phosphate which increased by a factor of 6.4 compared to acetate which increased by a factor of 2.5 (Table 2). This observation is consistent with the hypothesis that dihydrogen phosphate is interacting strongly with the central urea hydrogen atoms. The increase of acidity of these hydrogen atoms appears to have the most significant effect upon dihydrogen phosphate association. The bond angles in H2PO4−, being slightly smaller than those in benzoate, may cause a donor–acceptor mismatch if the dihydrogen phosphate were bound in a similar fashion to that observed for benzoate in the solid state, which in turn may lead to a different binding mode predominating in solution. Interestingly, significant improvements in the observed binding constants for both acetate and benzoate were also observed for compound 4 relative to 3, with a value of 8079 M−1 observed for acetate, an exceptionally high value for an acyclic receptor considering the competitive nature of DMSO–water solvent mixtures. The anion stability constant values obtained for compound 5 however were only slightly higher than those observed for compound 3. This may be due to an increase in the preorganization within compound 4 relative to 5 arising from the localization of acidity within the molecule. Positioning of chloro substituents in the 4- and 5-positions of the central aromatic ring has the effect of increasing the acidity of hydrogen atoms attached to that ring with 1H NMR experiments revealing a greater difference in the chemical shift of the aromatic protons on the central ring than either of the NH protons when comparing 3 and 4 (ΔδXH = δXH(4) − δXH(3)) (ΔδNHINT = 0.14 ppm, ΔδNHEXT = 0.08 ppm, ΔδCHINT = 0.32 ppm, ΔδCHEXT = 0.003 ppm). The electron withdrawing groups on the central ring may make the formation of intramolecular CH⋯O hydrogen bonds with the urea carbonyl oxygen more favourable, with the effect that the molecule adopts a more preorganized conformation. When comparing the chemical shifts of the NH protons of compound 5 with 4, the chemical shifts of the NH hydrogen atoms in the nitro-derivative are shifted downfield relative to the chloro compound (ΔδXH = δXH(5) − δXH(3)) (ΔδNHINT = 0.23 ppm, ΔδNHEXT = 0.75 ppm, ΔδCHINT = 0.012 ppm, ΔδCHEXT = 0.71 ppm), an indication of their higher degree of acidity. However compound 4 has a higher affinity for anions than 5. It therefore seems that the increasing acidity of the urea protons alone does not automatically result in higher association constants. In this case the 1H NMR evidence suggests that stabilizing internal hydrogen bonds are formed in compound 4, presumably resulting in a more preorganized conformation and hence significantly increasing the affinity of the receptor for anionic guests. This is illustrated in Fig. 4.
| Compounds | |||||
|---|---|---|---|---|---|
| a Slightly sigmoidal titration curves were obtained in these cases. This data was fitted to a 1∶1 binding model (attempts to fit to a 1∶2 receptor∶anion model failed). These values should be treated with caution as there may be complexes of stoichiometries other than 1∶1 present in solution.b Deprotonation. | |||||
| Anion | 4 | 5 | 6 | 7 | 8 |
| Cl− | 67 | 78 | — | 18 | 18 |
| Br− | <10 | <10 | — | — | — |
| CH3CO2− | 8079 | 4018 | 1739 | b | 188a |
| C6H5CO2− | 2248 | 1399 | 541 | 1249 | 233 |
| H2PO4− | 4724 | 666a | 349 | 1637 | 1490a |
| HSO4− | <10 | <10 | — | — | — |
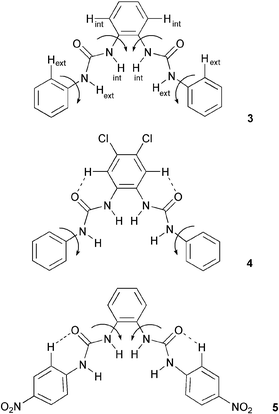 | ||
| Fig. 4 An increase in acidity on the central ring in compound 4 leads to higher stability constants, possibly due to a higher degree of pre-organization occurring in this system as compared to compounds 3 and 5. | ||
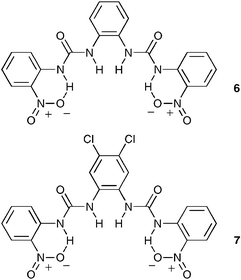
We decided to investigate the effect of functionalizing the pendant aryl rings with nitro groups in the 2-positions so as to reduce the hydrogen-bonding ability of the outer urea NH groups by involving them in intramolecular hydrogen bonding interactions. The nitro groups may also introduce a steric constraint on the binding site. Therefore compound 6 was synthesized and, as expected, a reduction in anion association constants was observed. The relative reduction in the affinities for the observed anions remained fairly constant, indicating that the presence of the nitro groups was not altering the selectivity of the bis-urea skeleton. In order to try and increase the observed association constants in the intramolecularly hydrogen bonded cavity, we combined both the 2-positioned nitro group on the pendant aryl rings, with the 4,5-dichloro central ring to afford compound 7. This led to an increase in stability constants, especially with dihydrogen phosphate. Again this may be an indication that dihydrogen phosphate is interacting strongly with the inner urea hydrogens. In this case, the acidity of the urea protons was increased to such a degree that behaviour that may be indicative of deprotonation was observed in the 1H NMR titration with acetate,23 a process that was accompanied by a colour change from yellow to red.
Finally we decided to investigate whether a bis-thiourea 8 would exhibit similar behaviour to that displayed by the bis-urea 4, considering that stability constants of thioureas with anions are generally higher due to the greater acidity of the NH protons.24 The synthesis of this compound has previously been reported.25 Interestingly, the measured stability constants with carboxylates were almost an order of magnitude lower than those obtained with 3 (the urea analogue). This may be due to the larger sulfur atom distorting the shape of the binding site (Fig. 5), with the outer thiourea NH groups no longer capable of simultaneously coordinating to the anion.
The improvement in the stability of the dihydrogen phosphate complex could therefore be exclusively the result of the inner NH protons still being approximately in the correct geometry to coordinate the anion.
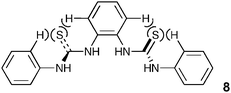 | ||
| Fig. 5 Steric interactions in 8 may twist the binding site in solution, so lowering the affinity of the receptor for carboxylates. | ||
Crystals of compound 8 were obtained by slow evaporation of an acetonitrile solution of the compound.|| Interestingly, in the solid state the bis-thiourea adopts a twisted conformation with neither sulfur atom being found in the plane of the central aromatic ring (Fig. 6). Hydrogen bonding interactions [N⋯S hydrogen bond lengths in the range 3.118(2)–3.388(2) Å] result in the formation of sheets of molecules in the ac plane.
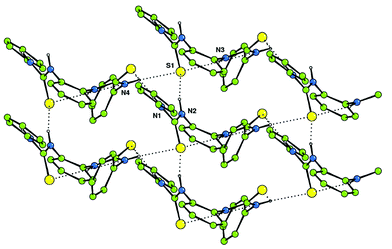 | ||
| Fig. 6 Hydrogen bonding interactions form sheets of bis-thiourea 8 that extend in the ac plane in the solid state. | ||
The new compounds reported here, and in particular compound 4, demonstrate that the bis-urea compounds are excellent carboxylate receptors under competitive conditions. Carboxylate/dihydrogen phosphate selectivity can be tuned by functionalising the bis-urea skeleton or by converting the urea groups to thioureas. We are currently investigating this hydrogen bonding motif as a carboxylate binding site in receptors for amino acids and as a hydrogen bond donor in macrocyclic systems. The results of these studies will be reported in due course.
We would like to thank the EPSRC for a DTA studentship (to SJB) and the EPSRC together with Professor Mike Hursthouse for use of the crystallographic facilities at the University of Southampton. PAG would like to thank the Royal Society for a University Research Fellowship.
Experimental
General methods
Reagents were purchased from the Aldrich Chemical Co. Deuterated solvents were purchased from Apollo Ltd. Chemical shifts are reported in ppm and are referenced to the solvent. Proton and 13C NMR spectra were recorded on a Bruker AV-300 NMR spectrometer. Elemental analyses were conducted by Medac Ltd.References
- K. Kavallieratos, S. R. de Gala, D. J. Austin and R. H. Crabtree, J. Am. Chem. Soc., 1997, 119, 2325 CrossRef CAS.
- M. P. Hughes and B. D. Smith, J. Org. Chem., 1997, 62, 4492 CrossRef CAS.
- M. A. Hossain, J. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2001, 40, 2936 CrossRef CAS.
- (a) M. J. Deetz, M. Shang and B. D. Smith, J. Am. Chem. Soc., 2000, 122, 6201 CrossRef CAS; (b) J. M. Mahoney, A. Beatty and B. D. Smith, J. Am. Chem. Soc., 2001, 123, 5847 CrossRef CAS.
- (a) S. J. Coles, J. G. Frey, P. A. Gale, M. B. Hursthouse, M. E. Light, K. Navakhun and G. L. Thomas, Chem. Commun., 2003, 568 RSC; (b) S. J. Brooks, L. S. Evans, P. A. Gale, M. B. Hursthouse and M. E. Light, Chem. Commun., 2005, 734 RSC.
- M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul and A. R. Cowley, J. Am. Chem. Soc., 2004, 126, 15364 CrossRef CAS.
- (a) J. A. Wisner, P. D. Beer and M. G. B. Drew, Angew. Chem., Int. Ed., 2001, 40, 3606 CrossRef CAS; (b) J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469 CrossRef CAS; (c) J. A. Wisner, P. D. Beer, N. G. Berry and B. Tomapatanaget, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4983 CrossRef CAS; (d) B. Tomapatanaget, T. Tuntulani, J. A. Wisner and P. D. Beer, Tetrahedron Lett., 2004, 45, 663 CrossRef CAS; (e) M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul, A. R. Cowley, F. Szemes and M. G. B. Drew, J. Am. Chem. Soc., 2005, 127, 2292 CrossRef CAS.
- P. A. Gale, J. L. Sessler and V. Král, Chem. Commun., 1998, 1 RSC.
- (a) R. Custelcean, L. H. Delmau, B. A. Moyer, J. L. Sessler, W.-S. Cho, D. Gross, G. W. Bates, S. J. Brooks, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2005, 44, 2537 CrossRef CAS; (b) C. J. Woods, S. Camiolo, M. E. Light, S. J. Coles, M. B. Hursthouse, M. A. King, P. A. Gale and J. W. Essex, J. Am. Chem. Soc., 2002, 124, 8644 CrossRef CAS; (c) C. N. Warriner, P. A. Gale, M. E. Light and M. B. Hursthouse, Chem. Commun., 2003, 1810 RSC.
- (a) H. Miyaji, P. Anzenbacher Jr, J. L. Sessler, E. R. Bleasdale and P. A. Gale, Chem. Commun., 1999, 1723 RSC; (b) P. A. Gale, L. J. Twyman, C. I. Handlin and J. L. Sessler, Chem. Commun., 1999, 1851 RSC; (c) P. A. Gale, M. B. Hursthouse, M. E. Light, J. L. Sessler, C. N. Warriner and R. Zimmerman, Tetrahedron Lett., 2001, 42, 6759 CrossRef CAS.
- J. L. Sessler, P. A. Gale and J. W. Genge, Chem.–Eur. J., 1998, 4, 1095 CrossRef CAS.
- (a) P. A. Gale, Chem. Commun., 2005, 3761 RSC; (b) I. E. D. Vega, P. A. Gale, M. E. Light and S. J. Loeb, Chem. Commun., 2005, 4913 RSC; (c) C. R. Bondy, P. A. Gale and S. J. Loeb, Chem. Commun., 2001, 729 RSC.
- For an excellent comprehensive review see: R. J. Fitzmaurice, G. M. Kyne, D. Douheret and J. D. Kilburn, J. Chem. Soc., Perkin Trans. 1, 2002, 841 Search PubMed.
- (a) P. J. Smith, M. V. Reddington and C. S. Wilcox, Tetrahedron Lett., 1992, 41, 6085 CrossRef; (b) E. Fan, S. A. van Arman, S. Kincaid and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 369 CrossRef CAS; (c) T. R. Kelly and M. H. Kim, J. Am. Chem. Soc., 1994, 116, 7072 CrossRef CAS; (d) R. Kato, S. Nishizawa, T. Hayashita and N. Teramae, Tetrahedron Lett., 2001, 42, 5053 CrossRef CAS; (e) B. Linton and A. D. Hamilton, Tetrahedron, 1999, 55, 6027 CrossRef CAS; (f) T. Gunnlaugsson, P. E. Kruger, T. C. Lee, R. Parkesh, F. M. Pfeffer and G. M. Hussey, Tetrahedron Lett., 2003, 44, 6575 CrossRef.
- (a) B. C. Hamann, N. R. Branda and J. Rebek Jr, Tetrahedron Lett., 1993, 34, 6837 CrossRef CAS; (b) D. H. Lee, H. Y. Lee, K. H. Lee and J. I. Hong, Chem. Commun., 2001, 1188 RSC.
- (a) S. J. Brooks, P. A. Gale and M. E. Light, Chem. Commun., 2005, 4696 RSC; (b) S. J. Brooks, P. A. Gale and M. E. Light, CrystEngComm, 2005, 7, 586 RSC.
- For an example of a 1,2-urea functionalized anthraquinone see: (a) D. A. Jose, D. K. Kumar, B. Ganguly and A. Das, Org. Lett., 2004, 6, 3445 CrossRef CAS; (b) for a macrocyclic system containing a ferrocene and naphthalene see: F. Otón, A. Tárraga, M. D. Velasco, A. Espinosa and P. Molina, Chem. Commun., 2004, 1658 Search PubMed; (c) for a naphthalene based system see: G. Xu and M. A. Tarr, Chem. Commun., 2004, 1050 Search PubMed.
- B. H. M. Snellink-Ruël, M. M. G. Antonisse, J. F. J. Engbersen, P. Timmerman and D. N. Reinhoudt, Eur. J. Org. Chem., 2000, 165 CrossRef CAS.
- A. Shivanyuk, K. Rissanen, S. K. Körner, D. M. Rudkevich and J. R. Rebek Jr, Helv. Chim. Acta, 2000, 83, 1778 CrossRef CAS.
- Z. Yin, Z. Li, A. Yu, J. He and J.-P. Cheng, Tetrahedron Lett., 2004, 45, 6803 CrossRef CAS.
- M. I. Bruce and J. A. Zwar, Proc. R. Soc. London, Ser. B, 1966, 165, 245 CrossRef CAS.
- For a seminal early paper describing the hydrogen bonding ability of ureas with other hydrogen bond acceptors in the solid state see: M. C. Etter, Z. Urbañczyk-Lipkoska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415 Search PubMed.
- For other examples of deprotonation of neutral anion receptors by basic anions see: (a) S. Camiolo, P. A. Gale, M. B. Hursthouse, M. E. Light and A. J. Shi, Chem. Commun., 2002, 758 RSC; (b) P. A. Gale, K. Navakhun, S. Camiolo, M. E. Light and M. B. Hursthouse, J. Am. Chem. Soc., 2002, 124, 11228 CrossRef CAS; (c) S. Camiolo, P. A. Gale, M. B. Hursthouse and M. E. Light, Org. Biomol. Chem., 2003, 1, 741 RSC; (d) T. Gunnlaugsson, P. E. Kruger, P. Jensen, F. M. Pfeffer and G. M. Hussey, Tetrahedron Lett., 2003, 44, 8909 CrossRef CAS; (e) M. Bioicchi, L. Del Boca, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Chem.–Eur. J., 2005, 11, 3097 CrossRef CAS.
- S. Nishizawa, P. Buhlmann, M. Iwao and Y. Umezawa, Tetrahedron Lett., 1995, 36, 6483 CrossRef CAS.
- A. A. Hassan, A.-F. E. Mourad, K. M. El-Shaeb, A. H. Abou-Zied and D. Döpp, Heteroat. Chem., 2003, 14, 535 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR titration curves. See DOI: 10.1039/b511963d |
| ‡ X-Ray crystal data were collected on a Bruker Nonius KappaCCD mounted at the window of an Mo rotating anode following standard procedures. Crystals of the fluoride and chloride complexes of compound 2 yielded poor quality data resulting in poor refinements. The chloride complex was collected several times to try and improve the data quality but some form of twinning that the authors were unable to resolve persisted. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b511963d |
§ Crystal data for the fluoride complex of 2: C32H50N5O2F, Mr = 555.77, T = 120(2) K, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 12.07(3), b = 16.80(3), c = 17.13(3) Å, α = 72.25(16), β = 74.7(2), γ = 71.82(14)°, V = 3087(11) Å3, ρcalc = 1.196 g cm −3, μ = 0.08 mm−1, Z = 4, reflections collected: 61461, independent reflections: 13602 (Rint = 0.137), final R indices [I > 2σ(I)]: R1 = 0.0960, wR2 = 0.1573, R indices (all data): R1 = 0.2021. wR2 = 0.1877. CCDC reference number 283635. , a = 12.07(3), b = 16.80(3), c = 17.13(3) Å, α = 72.25(16), β = 74.7(2), γ = 71.82(14)°, V = 3087(11) Å3, ρcalc = 1.196 g cm −3, μ = 0.08 mm−1, Z = 4, reflections collected: 61461, independent reflections: 13602 (Rint = 0.137), final R indices [I > 2σ(I)]: R1 = 0.0960, wR2 = 0.1573, R indices (all data): R1 = 0.2021. wR2 = 0.1877. CCDC reference number 283635. |
| ¶ Crystal data for the chloride complex of 2: C32H50N5O2Cl, Mr = 572.22, T = 120(2) K, monoclinic, space group P, a = 18.298(5), b = 9.0305(14), c = 20.379(5) Å, β = 101.76(2)°, V = 3296.8(14) Å3, ρcalc = 1.153 g cm −3, μ = 0.150 mm−1, Z = 4, reflections collected: 38786, independent reflections: 7224 (Rint = 0.0903), final R indices [I > 2σ(I)]: R1 = 0.1843, wR2 = 0.4916, R indices (all data): R1 = 0.2671. wR2 = 0.5371. CCDC reference number 283636. |
| || Crystal data for compound 8: C20H18N4S2, Mr = 378.5, T = 120(2) K, monoclinic, space group P21/c, a = 7.9841(3), b = 26.3041(11), c = 8.9855(2) Å, β = 103.535(2)°, V = 1834.68(11) Å3, ρcalc = 1.370 g cm −3, μ = 0.302 mm−1, Z = 4, reflections collected: 23129, independent reflections: 4200 (Rint = 0.1003), final R indices [I > 2σ(I)]: R1 = 0.0522, wR2 = 0.1063, R indices (all data): R1 = 0.1028. wR2 = 0.1242. CCDC reference number 283637. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2006 |