Synthesis, characterization and catalytic reactivity of ruthenium nanoparticles stabilized by chiral N-donor ligands
Susanna Jansatab, David Picurellia, Katrin Pelzerb, Karine Philippot*b, Montserrat Gómez*a, Guillermo Mullera, Pierre Lecantec and Bruno Chaudret*b
aDepartament de Química Inorgànica, Universitat de Barcelona, Martí i Franquès 1-11, 08028 Barcelona, Spain. E-mail: gomez@chimie.ups-tlse.fr
bLaboratoire de Chimie de Coordination du CNRS, 205, route de Narbonne, 31077 Toulouse Cédex 04, France. E-mail: chaudret@lcc-toulouse.fr; phil@lcc-toulouse.fr
cCentre d’Elaboration des Matériaux et d’Etudes Structurales du CNRS, 29, rue Jeanne Marvig, BP 4347, 31055 Toulouse Cédex 04, France
First published on 7th December 2005
Abstract
The decomposition of the organometallic precursor [Ru(cod)(cot)] (cod = 1,5-cyclooctadiene; cot = 1,3,5-cyclooctatriene) under mild conditions (room temperature, 3 bars H2) and in the presence of optically pure ligands, L*, namely (R)-2-aminobutanol 1, amino(oxazolines) (2, 3), hydroxy(oxazoline) (4) and bis(oxazolines) (5–8), leads to stable ruthenium nanoparticles exhibiting a mean diameter between 1.6–2.5 nm. These nanoparticles can be isolated and re-dispersed. They display different mean sizes, shapes and dispersions depending on the stabilizer nature. These new colloids (Ru1–Ru18) have been characterized by both solid state and molecular chemistry techniques, including TEM/HRTEM, WAXS, elemental analysis, and IR and NMR spectroscopy. To further characterize the surface state of these particles, their catalytic behaviour has been examined in the reduction of organic prochiral unsaturated substrates. Although the asymmetric induction obtained is modest, it reveals the influence of the asymmetric ligand coordinated at the surface of the particles.
Introduction
The synthesis of well-defined metal nanoparticles has experienced tremendous development over the past few years, primarily due to their physical properties and their potential for applications in the field of micro/nanoelectronics.1 Metal nanoparticles have also attracted interest because of their potential in catalysis.2 As such nanoparticles have proven to be efficient and selective catalysts for reactions also catalyzed by molecular complexes, such as olefin hydrogenation or C–C coupling reactions,3 but moreover for reactions which are not, or are poorly catalyzed by molecular species, such as aromatic hydrocarbon hydrogenation.4 Unambiguous distinction between colloidal and true homogeneous catalysis is, however, often very difficult to make.5During the last few years, we have studied the synthesis of metal nanoparticles using an organometallic approach.6 This methodology has led to nearly monodisperse particles of very small size that display remarkable surface coordination chemistry. The use of spectroscopic techniques (IR, NMR in solution or in the solid state) has permitted the study of the coordination chemistry of amines at the surface of ruthenium nanoparticles, as well as their dynamic behaviour in solution.7,8 Furthermore, such studies have evidenced the presence of surface hydrides on these same particles.8 Another approach aimed at characterizing the presence of surface ligands focuses on the reactivity of the resulting nanoparticles, the best example being the selectivity often found in catalytic processes. Despite impressive progress in asymmetric catalysis, however, only a few colloidal systems have been shown to display specific behaviour. The most relevant systems involve Pt or Pd nanoparticles stabilized by cinchonidine, in the hydrogenation of pyruvate derivatives.9
We are therefore interested in metal nanoparticle reactivity, not only with the aim of identifying reactions which are not catalyzed by molecular complexes,10 but also with the goal of characterizing those differences present in well-known reactions catalyzed by either molecular complexes or metal nanoparticles. In this respect, we have recently reported the use of palladium species stabilized by a chiral diphosphite for asymmetric allylic alkylation, and found that the use of nanoparticles leads to slower catalysts but displaying an important discrimination between the two enantiomers of racemic substrates.11
In the present study, we examine another well-known process catalyzed by molecular complexes, namely, asymmetric reduction of unsaturated substrates using dihydrogen or isopropanol in a basic medium as a hydrogen source. Ru nanoparticles were synthesized using nitrogen donor ligands such as β-amino alcohols (1) and chiral oxazolines (2–8), easily prepared from commercially available amino acids and nitrile or carboxylic acid derivatives (Fig. 1). A comparative catalytic study between colloidal and conventional molecular systems is also described.
The coordination chemistry of these chiral ligands, as well as their applications in several enantioselective catalytic processes, has been previously studied: specifically, the reduction of unsaturated substrates (Ru, Rh, Ir),12 allylic substitutions (Pd)13 and olefin epoxidations (Mo).14 β-Amino alcohols, previously shown to be excellent chiral auxiliaries in Ru-catalyzed asymmetric hydrogen transfer processes,15 were also examined. In this paper, we present the synthesis of new Ru nanoparticles, stabilized by both chiral β-amino alcohols and oxazolines, and their catalytic behaviour in the reduction of unsaturated substrates.
Results and discussion
Syntheses of ligands and stabilized nanoparticles
Four types of N-donor ligands (1–8) were used as stabilizers for the synthesis of Ru nanoparticles (Ru1–Ru8): β-amino alcohol (1), amino(oxazolines) (2, 3), hydroxy(oxazoline) (4) and bis(oxazolines) (5–8) (Fig. 1). The oxazoline derivatives were synthesized following our previously described procedures,12b,13 and obtained in a diastereomerically pure form, with the exception of 8, which was separated as a mixture of two diastereomers due to its axial chirality.13b Phenyl-mono(oxazolines), 2–4, contain alcohols or amines as functional groups in an ortho-position with respect to the heterocycle, capable of interacting with the nanoparticle surfaces.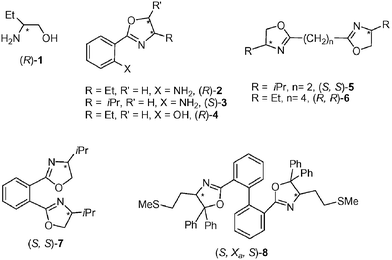 | ||
| Fig. 1 Chiral N-donor ligands: β-amino alcohol 1, mono(oxazolines) 2–4, and bis(oxazolines) 5–8. | ||
Ruthenium colloids (Ru1–Ru8) were prepared following our previously reported procedure, but using the chiral ligands described above as stabilizers (Scheme 1).7 In a typical experiment, the precursor [Ru(cod)(cot)]16 is dissolved at 193 K in a THF or THF–methanol solution containing 0.2 equivalents of the appropriate ligand, L*, in a Fischer–Porter bottle (Scheme 1).
 | ||
| Scheme 1 Synthesis of Ru colloids Ru1–Ru8 stabilized by the corresponding ligands, 1–8. | ||
The resulting yellow solution is then exposed to a dihydrogen atmosphere (3 bar), allowed to warm to room temperature and stirred vigorously for 24 hours. Ligand stability was tested under the same conditions and no imine bond reduction was observed. The colloids obtained (Ru1–Ru8) were purified by precipitation upon the addition of pentane, followed by filtration and drying under reduced pressure, giving rise to a dark brown powder consisting of nanoparticles which could then be re-dispersed in THF or isopropanol. In all cases, the particles were found to be stable over time, and did not show any sign of decomposition under an argon atmosphere. The average yield of these syntheses after work-up was approximately 40%, based on ruthenium.
Characterization of the nanoparticles
The sizes of the nanoparticles were determined by TEM (transmission electron microscopy) and their structures by WAXS (wide angle X-ray scattering), and in selected cases by HRTEM (high resolution transmission electron microscopy). In all instances, well-crystallized hcp nanoparticles of small size and narrow size distribution were obtained.By using (R)-2-aminobutanol, (R)-1, nearly spherical monodisperse nanoparticles of isotropic shape and a mean size near 2.5 nm, were obtained (Ru1). These particles exhibited a strong tendency to self-assemble, as shown in Fig. 2. The coherence length measured on the WAXS spectrum was shorter than the size measured by TEM, a finding consistent with a lack of crystalline order throughout the nanoparticles (Fig. 3).
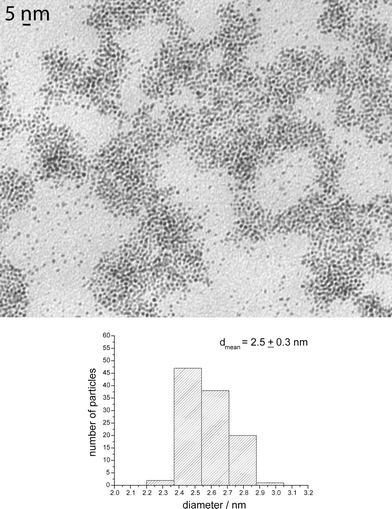 | ||
| Fig. 2 TEM micrograph and size histogram of Ru1. | ||
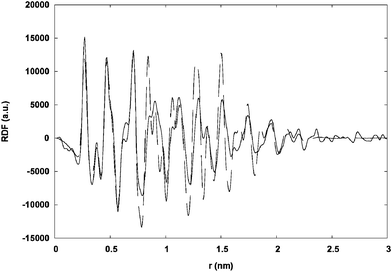 | ||
| Fig. 3 Solid line: experimental RDF (radial distribution function) of Ru1; dashed line: RDF computed from a spherical model according to bulk ruthenium (1.6 nm in diameter; distances in the model were increased by 1.5% for better agreement with the experimental function). | ||
When amino(oxazolines) were used, particle sizes remained near 2.5 nm (Ru2, Ru3). These particles were well separated but showed a tendency to agglomerate on the microscopy grid (Fig. 4). In addition they also displayed a slightly anisotropic shape, which is more visible in Ru3. This, or a lack of crystallinity, may account for the shortening of coherence length, as measured by WAXS, in Ru3 (Fig. 5).
 | ||
| Fig. 4 TEM micrographs of Ru2 (a) and Ru3 (b). | ||
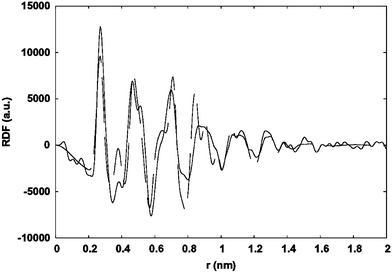 | ||
| Fig. 5 Solid line: experimental RDF of Ru3; dashed line: RDF computed from a spherical model according to bulk ruthenium (1.6 nm in diameter). | ||
Hydroxy(oxazoline) 4 led to well-dispersed and slightly elongated nanoparticles of 2.7 nm mean size (Fig. 6).
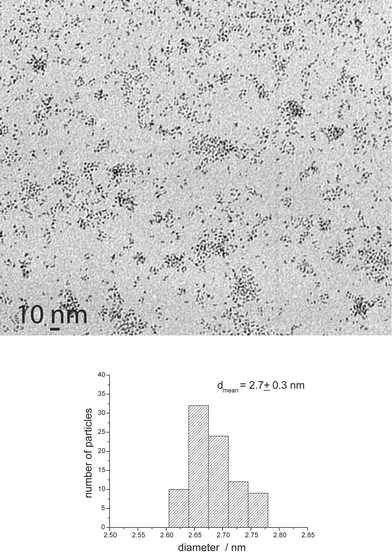 | ||
| Fig. 6 TEM micrograph and size histogram of Ru4. | ||
Concerning the bis(oxazoline) ligands, well-defined nanoparticles of very small size (nearly 1.6 nm) and of isotropic shape (Ru5–Ru8) were obtained in all cases. TEM micrographs of Ru5 and Ru6 are shown as representative examples in Fig. 7. These particles are crystalline, as determined by WAXS, with a coherence length close to 1.6 nm, and exhibiting a strong tendency to aggregate into large and regular mesoscopic super-structures.
 | ||
| Fig. 7 TEM micrographs of Ru5 (a) and Ru6 (b). | ||
We have previously noted that platinum nanoparticles stabilized by oxazoline ligands self-assemble into one-dimensional super-structures (“hairs”), due to the formation of hydrogen bond networks, particularly for ligand 4.17 Although in the present case, formation of a hydrogen bond network may have occurred, no extended super-structures were observed. Both bis(oxazolines) 5 and 6, containing, respectively, two and four methylenic groups within their backbone, gave large spherical super-structures (bowls). This organization may result from the coordination of both ends of the ligands to different nanoparticles, in agreement with the very short interparticle distance observed in these objects (ca. 1 nm). Such super-structures would then result from the statistical connection between nanoparticles, consistent with the apparent lack of organization in these “bowls”.
All of these structural observations denote a good argument for the coordination of the ligands used at the surface of the ruthenium particles.
IR spectra were recorded for all samples. However, due to the low overall content in the ligand, the bands displayed a weak intensity (recording more than 100 scans) and consequently, we were unable to reach any structural conclusions. The same held true for our attempts to characterize colloids by 1H or 13C NMR spectroscopy. In the case of Ru3, broad bands by 1H NMR were observed, which may be attributable to the equilibrium found at the particle surface between free and coordinated ligand molecules, characteristic of the Ru–hexadecylamine system.8 In this instance, some dihydrogen, released in the solution, was detected, which was similarly observed for ruthenium colloids stabilized with heptanol18 or hexadecylamine (HDA).8 The release of dihydrogen suggests that nanoparticles may additionally contain mobile hydrides at their surface.
Catalytic reactivity
To demonstrate the coordination chemistry found at the surface of these particles, we decided to evaluate the catalytic reactivity of these new ruthenium colloids. In this way, the selective metal-catalyzed processes offer us a useful tool. An intrinsic problem in the application of colloids as catalysts is the necessity to distinguish between a true colloidal and a molecular catalyst. We therefore chose reactions well-known to be catalyzed by ruthenium complexes, namely, asymmetric hydrogen transfer and hydrogenation.We will first discuss the results obtained for the asymmetric hydrogen transfer of acetophenone (I), using isopropanol as a hydrogen source under basic conditions, and the ruthenium colloids Ru1–Ru8 as catalysts (Table 1). The reactions were carried out at room temperature and monitored by gas chromatography (Scheme 2). The substrate–metal ratio was 20∶1, whereby the ruthenium amount accounts for all metal atoms included in the nanoparticles (based on the organometallic precursor, [Ru(cod)(cot)]).
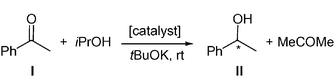 | ||
| Scheme 2 Hydrogen transfer of acetophenone catalyzed by ruthenium systems containing N-donor chiral ligands (L* = 1–8). | ||
| Entry | RuL* | Ru∶L*totalb | Time (h) | Conversion (%)c | Ee II (%)c |
|---|---|---|---|---|---|
| a 0.12 mmol acetophenone and 0.024 mmol t-BuOK in 4 mL of isopropanol, and 6 × 10−3 mmol colloid. Results from duplicated experiments.b Ruthenium–ligand ratio taking into account the total amount of ligand present in the reaction, from the synthesis of colloid with the extra ligand added to the catalytic reaction.c Substrate conversion and ee determined by GC using a chiral column. | |||||
| 1 | Ru1 | 1∶0.2 | 12 | 77 | 0 |
| 2 | Ru2 | 1∶0.2 | 12 | 58 | 0 |
| 3 | Ru3 | 1∶0.2 | 12 | 65 | 10 (S) |
| 4 | Ru4 | 1∶0.2 | 12 | 39 | 0 |
| 5 | Ru5 | 1∶0.2 | 72 | 14 | 0 |
| 6 | Ru6 | 1∶0.2 | 12 | 53 | 0 |
| 7 | Ru7 | 1∶0.2 | 48 | 12 | 0 |
| 8 | Ru8 | 1∶0.2 | 12 | 98 | 0 |
| 9 | Ru1 | 1∶1.2 | 12 | 31 | 0 |
| 10 | Ru1 | 1∶2.2 | 12 | 5 | — |
| 11 | Ru3 | 1∶1.2 | 12 | 15 | 10 (S) |
| 12 | Ru3 | 1∶2.2 | 72 | 20 | 10 (S) |
| 13 | Ru8 | 1∶1.2 | 12 | 79 | — |
| 14 | Ru8 | 1∶2.2 | 12 | 56 | — |
When no further free ligand was added in the catalytic medium (entries 1–8, Table 1), the systems were not selective (except Ru3 which afforded 10% (S) for II), but did exhibit moderate to high activities (up to 98% conversion after 12 hours, entry 8, Table 1). While certain tests were conducted at low temperature (273 K), enantioselectivity did not improve. Optimum activities corresponded with those ruthenium systems stabilized by amino alcohol 1 (entry 1), amino(oxazoline) 3 (entry 3) and bis(oxazoline) 8 (entry 8). In order to observe the influence of ligand concentration, supplemental free ligand was added to the catalytic medium for the more active systems, Ru1, Ru3, and Ru8 (entries 9–14, Table 1). In the three catalytic systems, a remarkable decrease in activity was observed when 1 or 2 equivalents of free ligand (relative to ruthenium) were added (entry 1 vs. 9 and 10; entry 3 vs. 11 and 12; entry 8 vs. 13 and 14), in contrast to the behaviour of the molecular ruthenium catalysts.12,19 This decrease was even more significant when larger amounts of ligand were added (entry 9 vs. 10; entry 13 vs. 14). In only the enantioselective system Ru3, the total ligand amount in the catalytic medium did not induce changes in the enantiomeric excess of II (entries 3, 11 and 12, Table 1).
Analyzing the catalytic behaviour of these Ru colloids, based on the nature of the ligand, we found that those colloids containing amino functional groups (ligands 1–3) were more active than hydroxy(oxazoline) (4) and bis(oxazolines) (5–8). This trend agrees with results published in the literature on molecular Ru catalysts, for which at least one primary or secondary amine moiety must be present in the ligand in order to obtain high yields.20 For bis(oxazoline) systems (entries 5–8, Table 1), an important backbone effect was observed: those bis(oxazolines) containing shorter spacers (5 and 7) were less active than those with longer ones (6 and 8), Ru8 proving to be the most active colloidal catalytic system. Ligands 5, 6, and 8 induced the formation of super-structures, probably by using their two oxazoline rings to link to different nanoparticles, and in the case of 8, to thioether moieties. In contrast, ligands 5 and 7 may have been too firmly fixed on their respective nanoparticles, rendering any approach by the reactants to the metallic surface difficult.
In order to compare the catalytic behaviour between molecular and colloidal ruthenium species, the data corresponding to the molecular systems (RuM1–RuM8)12 and the nanoclusters (Ru1–Ru8), which are stabilized by the same kind of ligands, were collected in Table 2. We observed that for amino alcohol 1 (entries 1 and 2, Table 2) and bis(oxazolines) 5–7 (entries 9–14, Table 2), the colloidal systems were less active than their corresponding molecular ones. This trend changed, however, for amino(oxazolines) 2 and 3, as well as for the thioether bis(oxazoline) 8 (entries 3–6 and 15–16, Table 2).
| Entry | L* | Catalyst | Ru∶L*totalb | Time (h) | Conversion (%)c | Ee II (%)c |
|---|---|---|---|---|---|---|
| a 0.12 mmol acetophenone and 0.024 mmol t-BuOK in 4 mL of isopropanol; 3 × 10−3 mmol [Ru(p-cymene)Cl2]2 with 6 × 10−3 mmol L* for molecular catalysts generated in situ, RuML*; or 6 × 10−3 mmol of colloid catalyst, RuL*. Results from duplicated experiments.b Ruthenium–ligand ratio, taking into account the total amount of ligand present in the reaction.c Substrate conversion and ee determined by GC using a chiral column. | ||||||
| 1 | 1 | Ru1 | 1∶0.2 | 12 | 77 | 0 |
| 2 | 1 | RuM1 | 1∶1 | 1.5 | 93 | 12 (R) |
| 3 | 2 | Ru2 | 1∶0.2 | 12 | 58 | 0 |
| 4 | 2 | RuM2 | 1∶1 | 12 | 15 | 43 (R) |
| 5 | 3 | Ru3 | 1∶0.2 | 12 | 65 | 10 (S) |
| 6 | 3 | RuM3 | 1∶1 | 12 | 55 | 45 (S) |
| 7 | 4 | Ru4 | 1∶0.2 | 12 | 39 | 0 |
| 8 | 4 | RuM4 | 1∶1 | 72 | 0 | 0 |
| 9 | 5 | Ru5 | 1∶0.2 | 72 | 14 | 0 |
| 10 | 5 | RuM5 | 1∶1 | 20 | 88 | 25 (S) |
| 11 | 6 | Ru6 | 1∶0.2 | 12 | 53 | 0 |
| 12 | 6 | RuM6 | 1∶1 | 12 | 90 | 11 (R) |
| 13 | 7 | Ru7 | 1∶0.2 | 48 | 12 | 0 |
| 14 | 7 | RuM7 | 1∶1 | 20 | 42 | 29 (S) |
| 15 | 8 | Ru8 | 1∶0.2 | 12 | 98 | 0 |
| 16 | 8 | RuM8 | 1∶1 | 12 | 62 | 0 |
In addition, the RuM4 molecular system was inactive, while the corresponding colloidal one was active (entry 8 vs. 7, Table 2). This suggests a reaction of the molecular precursor resulting from the presence of the phenoxo group (formation of a σ- or π-bonded phenoxide complex), which would render the molecular system inactive, an outcome not seen in ruthenium nanoclusters.21
It is also noteworthy that Ru8 is the fastest colloidal catalyst, even more so than its molecular counterpart (entry 15 vs. 16, Table 2). This ligand is quite bulky and contains an aromatic biphenyl backbone capable of interacting with the nanoparticle surface and thioether substituents, which are good electron releasing groups.
The activity behaviour of these systems indicates a specific coordination chemistry of ligands at the metallic surface, depending on the nature of the ligand.
With regard to the selectivity, only the colloidal system stabilized by amino(oxazoline) 3 gives asymmetric induction, lower than that of its corresponding molecular system (entry 5 vs. 6, Table 2).
In addition, TEM analysis performed before and after catalysis revealed that the size of ruthenium particles remained unchanged, although some agglomeration was visible.
We also analyzed the behaviour of the sole enantioselective colloidal catalyst, Ru3, for the hydrogen transfer reaction, in the hydrogenation of prochiral olefins such as anisole derivatives (III and V) and dimethyl itaconate (VII).
Concerning the disubstituted arenes, ortho- and para-methylanisole, the molecular system, generated in situ from [Ru(cod)(cot)] and ligand 3 (see Experimental section for details), was not found active, in agreement with the well-known trend for catalytic homogenous systems in the reduction of arenes.3,22 However, under the same conditions (40 bar hydrogen pressure and 50 °C for 6 h), the colloidal system Ru3 led to a more than 50% conversion of the substrate, with the cyclohexane derivatives (IV and VI) being the main products: 57% for substrate III and 87% for V (percentages based on the total amount of detected products). In both cases, the trans-isomer was favoured, up to a trans∶cis ratio of 19∶1 for VI (Scheme 3),23 in contrast to the heterogeneous catalytic systems which favour the cis-derivatives.24 It is important to emphasize that the addition of free ligand (Ru∶3 ratio up to 1∶0.5) rendered the system completely inactive.
![Hydrogenation of ortho- and para-methylanisole catalyzed by the Ru3 colloidal system [relative percentage following catalysis (determined by GC) is indicated below each chemical].](/image/article/2006/NJ/b509378c/b509378c-s3.gif) | ||
| Scheme 3 Hydrogenation of ortho- and para-methylanisole catalyzed by the Ru3 colloidal system [relative percentage following catalysis (determined by GC) is indicated below each chemical]. | ||
With regard to dimethyl itaconate, Ru3, as well as the homogenous analogous system RuM3, were found to be active for the hydrogenation of VII (Scheme 4). In this particular case, reactions were carried out at room temperature under 40 bar H2 pressure for 7 hours, using a substrate–metal ratio of 100∶1 (Table 3). When the catalyst was in situ generated from [Ru(cod)(cot)] and the free ligand 3 (Ru∶L* ratio = 1∶2), total conversion was achieved, giving a low ee, 18% (R) for product VIII (entry 1, Table 3). When a colloidal catalyst was used, activity was also high, but without inducing any enantiomeric excess (entries 2 and 3, Table 3). Analogous with the behaviour observed in acetophenone hydrogen transfer, the addition of free ligand in the catalytic medium effected a decrease in activity (entry 2 vs. 3, Table 3).
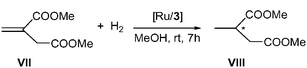 | ||
| Scheme 4 Hydrogenation of dimethyl itaconate catalyzed by ruthenium systems containing ligand 3. | ||
| Entry | Catalyst | Ru∶L*totalb | Conversion (%)c | Ee VIII (%)c |
|---|---|---|---|---|
| a 2 mmol dimethyl itaconate, 2 mL n-butanol (internal standard) and 0.024 mmol t-BuOK in 6 mL of methanol; 2 × 10−2 mmol Ru3 or 2 × 10−2 mmol [Ru(cod)(cot)] and 4 × 10−2 mmol of 3 for molecular catalyst generated in situ, RuM3. Results from duplicated experiments.b Ruthenium–ligand ratio, taking into account the total amount of ligand present in the reaction.c Substrate conversion and ee determined by GC using a chiral column. | ||||
| 1 | RuM3 | 1∶2 | 100 | 18 (R) |
| 2 | Ru3 | 1∶0.2 | 100 | 0 |
| 3 | Ru3 | 1∶0.4 | 80 | 0 |
Conclusions
Ruthenium nanoparticles stabilized by chiral ligands were prepared in an easy and reproducible way. Depending on the stabilizer employed, different shapes and sizes were observed.Surface coordination of ligands not only led to an excellent stabilization of metal particles in solution, but also strongly influenced their catalytic behaviour. Mean diameters ca. 2.5 nm were found using mono(oxazoline) ligands, and ca. 1.6 nm with bis(oxazolines). These trends are consistent with the size decrease observed when ligand concentration increases, as well as with the coordinating behaviour of bis(oxazolines), chelate and bridge effects.
One of the goals of this study was to find evidences for differentiating colloidal from molecular catalysts. In the present work, four clues agree with this objective. First, ligand 5 poisons the molecular system, probably through formation of a Π-bonded phenoxo complex, which was not observed in the colloids. Second, ligand 8, which offers multiple possibilities for interaction with the colloidal surface (nitrogen, sulfur, Π-system), is a better promoter of colloidal than molecular catalysis. Third, in the three studied processes, the addition of excess ligand decreased the activity of the catalytic system without any effect on the selectivity; whereas an increase of ee would be expected in the case of metal leaching to form a molecular catalyst. And fourth, the ruthenium colloidal system stabilized by ligand 3, was active in the reduction of arene derivatives, in manifest contrast to the analogous mononuclear systems. The very low asymmetric induction encountered when colloidal catalysts are used, compared with molecular systems, suggests a fluxional behaviour of the ligands at the surface of ruthenium nanoparticles, as recently described for Ru–amine colloids.
To conclude, this study demonstrates the need to design appropriate ligands, capable of binding to nanoparticles, as well as of performing target reactions. This may well be the case with ligand 8 which displays several sites of coordination. Future work in this field should include this goal in order to better exploit the adaptability of these colloidal systems.
Experimental
General
All operations were carried out using Schlenk tube or Fisher–Porter bottle techniques under argon. RuCl3·H2O (Strem), [Ru(η6-p-cymene)Cl(μ-Cl)]2 (Strem), t-BuOK (Fluka), (R)-(+)-2-aminobutanol (Fluka) and L-valinol (Aldrich) were purchased and used without prior purification. HPLC grade isopropanol was purchased from Fluka. The other reagents were purchased from Aldrich and most of the solvents from SDS. The solvents were distilled under a nitrogen atmosphere. THF was refluxed over sodium benzophenone, pentane over calcium hydride, and methanol over magnesium after activation with iodine. All reagents and solvents were degassed under vacuum at liquid nitrogen temperature by 3 vacuum–argon cycles.[Ru(cod)(cot)] was prepared according to a published procedure.16 It was purified by recrystallization in pentane and the resulting highly sensitive yellow crystals were stored under argon at −30 °C. Their purity was checked by elemental analysis and 1H-NMR spectroscopy. Mono- and bis(oxazoline) ligands were prepared as previously described.12b,13a,13b
Specimens for TEM analysis were prepared by slow evaporation of a drop of colloidal solution deposited under argon onto holey carbon-covered copper grids. The TEM experiments were performed at the “Service Commun de Microscopie Electronique de l’Université Paul Sabatier” using a JEOL 200 CX-T electron microscope operating at 200 kV and a Philips CM12 electron microscope operating at 120 kV, with respective point resolutions of 4.5 and 5 Å. HRTEM observations were carried out with a JEOL JEM 2010 electron microscope working at 200 kV with a resolution point of 2.5 Å. Transmission electron microscopy was used as a standard tool of analysis for determining the mean size of ruthenium particles. Size distribution of the particles was determined by manual analysis of enlarged images by measuring at least 200 particles on a given grid to obtain a statistical size distribution as well as a mean diameter.
Data collection for the wide-angle X-ray scattering was performed on small amounts of powder at the CEMES/CNRS, Toulouse.
NMR spectra of particles were recorded on Bruker DRX 250 or 400 MHz (standard SiMe4) spectrometers, using THF-d8 as solvent. Chemical shifts were reported downfield from standards. IR spectra were recorded on a Perkin Elmer spectrometer GX (FT-IR system). Colloid samples were prepared as KBr pellets, unless otherwise stated.
Elemental analyses were performed in the “Services d’Analyses du CNRS” at the LCC in Toulouse for carbon, nitrogen and hydrogen determinations, and at the “Service Central d'Analyse” in Lyon for ruthenium, sulfur and oxygen determinations.
GC analyses were performed on a Hewlett-Packard 5890 Series II gas chromatograph (50 m Ultra 2 capillary column) with an FID detector. Enantiomeric excesses were determined by GC on FS-cyclodex-β-I/P and FS-cyclodex-α-I/P columns.
General synthesis of Ru particles stabilized by asymmetric organic ligands
In a typical experiment, 150 mg of [Ru(cod)(cot)] (0.476 mmol) was introduced in a Fisher–Porter bottle and left in a vacuum for 30 minutes. 125 mL of solvent (neat THF or THF–methanol mixture), degassed by freeze–pump cycles, was then added. The resulting yellow solution was cooled to 193 K after which a 25 mL solution of THF containing 0.2 equivalents of the ligand was placed in the flask. The bottle was pressurized under 3 bar of dihydrogen and the solution allowed to slowly warm to room temperature under vigorous stirring. After 24 hours a homogenous brown solution was obtained. After elimination of excess dihydrogen, approximately 3 mL of the solution was passed under argon over a small alumina column. The absence of filtrate colour indicated full decomposition of the precursor. The volume of the solution was then reduced to approximately 15 mL. 50 mL of deoxygenated pentane was then added and the resulting mixture cooled to 193 K at which temperature a brown precipitate formed after several hours. Following filtration, the precipitate was washed with pentane (2 × 50 mL of deoxygenated pentane) and dried under reduced pressure. The resulting particles were obtained as dark brown powders. In all cases, ruthenium colloids were found to remain stable over time and did not exhibit any signs of decomposition. They were characterized by IR spectroscopy, TEM, and WAXS analysis.Catalytic reactions
Blank tests were carried out in order to evaluate the stability of the ligands under the catalytic conditions. No hydrogenation reactions were detected in any case.Acknowledgements
The authors are very grateful to V. Collière for his contribution to TEM/HRTEM analysis and the TEMSCAN (Service Commun de Microscopie Electronique de l’Université Paul Sabatier). We would also like to thank CNRS, Egide (PAI Picasso No 04230PL), the Ministerio de Educación y Ciencia (CTQ2004-01546/BQU; Acción integrada HF2001-0024) and the Generalitat de Catalunya for financial support. M. G. would like to thank the Ministerio de Educación, Cultura y Deporte (PR2000-0178 0077107784) for a sabbatical grant.References
- (a) K. J. Klabunde and G. Cardenas-Trivano, in Active Metals Preparation, Characterization, Applications, ed. A. Fürstner, VCH, Weinheim, 1996, p. 237 Search PubMed; (b) Nanoparticles from theory to applications, ed. G. Schmid, VCH, Weinheim, 2004 Search PubMed.
- (a) G. Schmid, Chem. Rev., 1992, 92, 1709 CrossRef CAS; (b) L. N. Lewis, Chem. Rev., 1993, 93, 2693 CrossRef CAS; (c) D. Aiken III and R. G. Finke, J. Mol. Catal. A: Chem., 1999, 145, 1 CrossRef; (d) M. A El-Sayed, Acc. Chem. Res., 2001, 34, 257 CrossRef CAS; (e) H. Bönnemann and R. M. Richards, Eur. J. Inorg. Chem., 2001, 2455 CrossRef CAS.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 191, 187 CrossRef CAS.
- (a) I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 10139 CrossRef CAS; (b) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 191, 317 CrossRef.
- K. Philippot and B. Chaudret, C. R. Chim., 2003, 6, 1019 Search PubMed.
- C. Pan, K. Pelzer, K. Philippot, B. Chaudret, F. Dassenoy, P. Lecante and M.-J. Casanove, J. Am. Chem. Soc., 2001, 123, 7584 CrossRef CAS.
- T. Pery, K. Pelzer, J. Mathes, G. Buntkowski, K. Philippot, H.-H. Limbach and B. Chaudret, ChemPhysChem, 2005, 6, 605 CrossRef CAS.
- (a) M. Studer, H.-U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45 CrossRef CAS; (b) H. Bönnemann and G. A. Braun, Angew. Chem., Int. Ed. Engl., 1996, 35, 1992 CrossRef; (c) H. Bönnemann and G. A. Braun, Chem.–Eur. J., 1997, 3, 1200 CrossRef CAS; (d) X. Zuo, H. Liu, D. Guo and X. Yang, Tetrahedron, 1999, 55, 7787 CrossRef CAS; (e) J. U. Köhler and J. S. Bradley, Catal. Lett., 1997, 45, 203 CrossRef CAS; (f) J. U. Köhler and J. S. Bradley, Langmuir, 1998, 14, 2730 CrossRef; (g) M. Tamura and H. Fujihara, J. Am. Chem. Soc., 2003, 125, 15742 CrossRef CAS; (h) V. Mévellec, C. Mattioda, J. Schulz, J.-P. Rolland and A. Roucoux, J. Catal., 2004, 1 CrossRef CAS.
- V. Mévellec, A. Roucoux, E. Ramirez, K. Philippot and B. Chaudret, Adv. Synth. Catal., 2004, 346, 72 CrossRef CAS.
- S. Jansat, M. Gómez, K. Philippot, G. Muller, E. Guiu, C. Claver, S. Castillón and B. Chaudret, J. Am. Chem. Soc., 2004, 126, 1592 CrossRef CAS.
- (a) M. Gómez, S. Jansat, G. Muller, M. C. Bonnet, J. A. J. Breuzard and M. Lemaire, J. Organomet. Chem., 2002, 659, 186 CrossRef CAS; (b) M. Gómez, S. Jansat, G. Muller, S. Alvarrez, G. Aullón and M. A. Maestro, Eur. J. Inorg. Chem., 2005, 4341 CrossRef CAS.
- (a) M. Gómez, S. Jansat, G. Muller, M. A. Maestro, J. Mahía, M. Font-Bardía and X. Solans, J. Chem. Soc., Dalton Trans., 2001, 1432 RSC; (b) M. Gómez, S. Jansat, G. Muller, M. A. Maestro and J. Mahía, Organometallics, 2002, 21, 1077 CrossRef CAS; (c) M. A. Pericàs, C. Puigjaner, A. Riera, A. Vidal-Ferran, M. Gómez, F. Jiménez, G. Muller and M. Rocamora, Chem.–Eur. J., 2002, 8, 4164 CrossRef CAS; (d) D. Franco, M. Gómez, F. Jiménez, G. Muller, M. Rocamora, M. A. Maestro and J. Mahía, Organometallics, 2004, 23, 3197 CrossRef CAS.
- (a) J. A. Brito, M. Gómez, G. Muller, H. Teruel, J. C. Clinet, E. Duñach and M. A. Maestro, Eur. J. Inorg. Chem., 2004, 4278 CrossRef CAS; (b) M. Gómez, S. Jansat, G. Muller, G. Noguera, H. Teruel, V. Moliner, E. Cerrada and M. Hursthouse, Eur. J. Inorg. Chem., 2001, 1071 CrossRef CAS.
- M. Henning, K. Püntener and M. Scalone, Tetrahedron: Asymmetry, 2000, 11, 1849 CrossRef.
- P. Pertici and G. Vitulli, Inorg. Synth., 1983, 22, 17.
- M. Gómez, K. Philippot, V. Collière, P. Lecante, G. Muller and B. Chaudret, New J. Chem., 2003, 27, 114 RSC.
- K. Pelzer, K. Philippot and B. Chaudret, Z. Phys. Chem., 2003, 217, 1539 CrossRef CAS.
- D. G. I. Petra, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, A. M. van Loon, J. G. de Vries and H. E. Schoemaker, Eur. J. Inorg. Chem., 1999, 2335 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- D. J. Cole-Hamilton, R. J. Young and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1976,(19), 1995 RSC.
- G. Süss-Fink, M. Faure and T. R. Ward, Angew. Chem., Int. Ed., 2002, 41, 99 CrossRef CAS.
- GC (with α- and β-cyclodextrin columns) and HPLC (with an OD chiral column) analyses were carried out under several conditions, without achieving separation of enantiomers.
- A. Roucoux, J. Schulz and H. Patin, Adv. Synth. Catal., 2003, 345, 222 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2006 |