Synthesis and electrochemical properties of slipped-cofacial porphyrin dimers of ferrocene-functionalized Zn-imidazolyl-porphyrins as potential terminal electron donors in photosynthetic models†
Dipak
Kalita
,
Mitsuhiko
Morisue
and
Yoshiaki
Kobuke
*
Graduate School of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma 630-0192, Japan. E-mail: kobuke@ms.naist.jp; Fax: (+81)743-72-6119; Tel: (+81)743-72-6110
First published on 10th November 2005
Abstract
A systematic series of ferrocene-functionalized Zn-imidazolyl-porphyrins were synthesized to assemble into the slipped-cofacial porphyrin dimers through imidazolyl-to-zinc complementary coordination as artificial photosynthetic models. Direct substitution at the meso position of the porphyrin ring with ferrocence and octamethylferrocene leads to the characteristic electronic structures, while the ferrocene substituents through phenylene-ethenylene and phenylene-ethylene spacers mitigate the electronic communications. Bathochromic shift of Q band, fluorescence quenching, and redox potentials of porphyrin ring are rationalized by the degree of electron-donating ability of the terminal ferrocenes.
Introduction
The slipped-cofacial arrangement of natural pigments plays an important role in achieving efficient energy and electron transfer reactions with excellent yields in photosynthetic systems. In the natural systems, light energy conversion is carried out by several chromophores that are fixed and arranged closely to each other in the membrane protein.1 The photosynthesis process is initiated by the absorption of sunlight by the light-harvesting antenna complexes where rapid and efficient energy transfer takes place over many pigments until eventually reaching the reaction center. In the reaction center,2 there is a “special pair” of bacteriochlorophyll dimer where the photoinduced charge separation helps convert the harvested solar energy. As shown by the X-ray crystal structures of the bacterial photosynthetic reaction center, the most prominent characteristics of the “special pair” are the slipped-cofacial arrangements of the two bacteriochlorophylls and their electronic coupling.2,3 The beautiful structural arrangement of natural photosynthetic systems have motivated many chemists throughout the world to design artificial photosynthetic models to study its complex energy/electron transfer mechanism and its underlying basic principles.4The introduction of an imidazolyl group at the meso position of a porphyrin led to the spontaneous formation of a “slipped-cofacial porphyrin dimer” by the complementary coordination of the imidazolyls to the central Zn(II) in each porphyrin (Scheme 1).5 The structural model developed by our research group mimics the geometry of natural photosystems by including complementary imidazolyl-to-zinc coordination, and enables two chromophoric π-orbitals to interact strongly in a slipped-cofacial arrangement. It served not only as a special pair model, but also as a building block of circular and linear porphyrin arrays, providing materials for both light-harvesting antenna complexes6 and non-linear optics7 models. However, the slipped-cofacial dimer structure can undergo exchange reactions and dissociate in the presence of competing ligands such as 1-methylimidazole, pyridine and MeOH. These properties enable the structures to be reorganized after their formation and allow the creation of a range of supramolecular systems by self-assembly. These supramolecular approaches have many advantages from the point of view of organic synthesis. Based on a deeper understanding of photoinduced electron-transfer reactions provided by model systems, laboratories hope to design systems for converting solar energy into chemical potential with simpler supramolecular techniques that use ITO (indium-tin oxide) electrode surface. In view of this, Zn-imidazolyl-porphyrin dyads were functionalized with five different types of ferrocenes to give 2-ZnD–6-ZnD as shown in Scheme 1. Methylation increased the electron donating character of the ferrocene core in comparison with that of the unsubstituted ferrocene group and led to potential terminal electron donor groups for model photosynthetic devices. Systematically changing the group linking the ferrocene and the porphyrin may allow the elucidation of the effect of electronic communication between the two chromophores, not only in the present imidazolyl-porphyrins but in porphyrins generally. All electrochemical and spectral analyses of ferrocene-functionalized Zn-imidazolyl-porphyrins (2-ZnD–6-ZnD) were compared to the reference samples 1-ZnD and 1-Zn-Im, which lack ferrocenyl groups (Scheme 1).
 | ||
| Scheme 1 Schematic representation of the dimer–monomer equilibrium of ferrocene-functionalized Zn imidazolyl-porphyrins. | ||
Ferrocene was chosen as the electron donor group in the present study because the electron transfer from ferrocene is known to be very fast8 and its redox active properties and those of its derivatives have been thoroughly investigated by several groups.9 Ferrocene generates a relatively stable cation. The electrochemical potential of which can be tuned by introducing appropriate substituents, thus affording oxidation potentials in the range of −0.2 V to > +0.5 V.10 However, in comparison to the well-developed chemistry of ferrocene and its numerous applications11 in organic synthesis, homogeneous catalysis and materials science, the analogous chemistry of methylated ferrocene derivatives is limited.12
Each of the target porphyrins bears three different meso substituents (AB2C type) including imidazolyl. Two of the porphyrins, 2-ZnD and 3-ZnD, are connected directly to ferrocene or 2,2′,3,3′,4,4′,5,5′-octamethylferrocene by covalent bonds, respectively. The other two porphyrins, 4-ZnD and 5-ZnD, have an intervening phenylene-ethenylene spacer between the porphyrin and the ferrocenyl or 2,2′,3,3′,4,4′,5,5′-octamethyl ferrocenyl substituent, respectively. In compounds 4-ZnD and 5-ZnD, the phenylene bridges are connected directly to the porphyrin ring so that they may serve as spacer groups allowing rotation of the ferrocenyl and octamethylferrocenyl groups while providing a fixed distance between the donor and the acceptor groups. Using Cerius 2 software (Ver. 4.6/Force Field UNIVERSAL 1.02), the center-to-center (Zn–Fc) distance was calculated to be 13.67 Å. In 6-ZnD, direct conjugation between the porphyrin and the ferrocene group was cut off. In each of the five porphyrins, one of the meso-positions is substituted by a 1-methylimidazole group which allows assembly of the molecule into a slipped-cofacial dimer through complementary coordination to the central Zn(II). The remaining two meso-positions (5, 15 positions) are substituted with allyloxy ether groups, this enables the complementary dimer pair to be covalently linked by a ring closing olefin metathesis reaction.6c,17 The ferrocene-functionalized Zn-imidazolyl-porphyrin dyads thus synthesized, 2-ZnD to 6-ZnD, can be tuned by the addition of competing ligands, as shown in Scheme 1.
Although the literature provides the syntheses of several ferrocene substituted porphyrins,13 to our knowledge this is the first time that a sterically hindered octamethylferrocene group has been introduced at the meso-position of a porphyrin by means of a covalent bond or via a spacer unit. Likewise, there have been no previous reports of porphyrins having both imidazolyl and ferrocenyl subunits.
It is intended to use these substituted porphyrins to construct hetero-redox systems on transparent ITO electrodes by the complementary coordination of thiol-substituted Zn-imidazolyl-porphyrins with ferrocene-functionalized Zn-imidazolyl-porphyrins, followed by the formation of a covalent linkages by ring closing metathesis using Grubbs catalyst. This is an extension of our research towards photocurrent generation,14,15 the details of which will be published elsewhere.16 In the present study, we focus on the synthesis of the ferrocenyl-appended Zn-imidazolyl-porphyrins, 2-ZnD to 6-ZnD, and the effect of dimerization on their electrochemical properties. The monomeric reference compounds of these ferrocenyl-appended Zn-imidazolyl-porphyrins, 2-Zn-Im to 6-Zn-Im, were also prepared using 1-methylimidazole, as shown in Scheme 1. The electrochemical behavior of the ferrocenyl-appended Zn-imidazolyl-porphyrins and the monomeric reference compounds were compared to each other.
Results and discussions
Synthesis
The synthesis of the reference sample 1-ZnD was described in our previous report.17 The synthetic routes for the ferrocene-functionalized Zn-imidazolyl-porphyrins, 2-ZnD to 6-ZnD, are shown in Scheme 2. | ||
| Scheme 2 Synthetic route to meso-substituted ferrocene-functionalized Zn imidazolyl-porphyrins (2-ZnD to 6-ZnD). | ||
Following a similar procedure, 2,2′,3,3′,4,4′,5,5′-octamethylferrocenecarboxaldehyde 11 afforded the free base porphyrin 3-H222 in 8% yield, and gave the title compound 3-ZnD in 90% yield on subsequent Zn metalation. The heavy substitution of octamethylferrocene makes it less reactive in comparison to ferrocene itself and this resulted in a significant recovery of the unreacted starting material (37%). When the amount of starting material recovered is taken into account, the yield increases to 12%. Steric hindrance prevented the formation of 5,15-bis(3-allyloxypropyl)-10,20-bis(2,2′,3,3′,4,4′,5,5′-octamethylferrocenyl)porphyrin. This is the first time that a sterically hindered octamethylferrocenyl group has been directly connected to a porphyrin ring by a covalent bond. The neutralization of this acidic reaction medium prior to p-chloranil oxidation should be carried out with care as the octamethylferrocene group is readily oxidized (0.05 V vs Ag/AgCl) when compared to ferrocene itself (oxidation potential 0.48 V vs Ag/AgCl in dichloromethane). TLC and MALDI-TOF MS analysis of the crude reaction mixtures showed no signs of the scrambling of the meso-substituents in any of these reactions. The porphyrins synthesized, 2-ZnD and 3-ZnD, were fully characterized by spectral analysis, including 1H NMR, 13C NMR, UV, IR and MALDI-TOF MS. The analyses of all the compounds were found to be in satisfactory agreement with their suggested structures.
1H NMR of the free base porphyrin is normal in all respects. In compound 2-H222, a sharp singlet at 10.3 ppm was assigned to the β-CH proton of the porphyrin adjacent to the ferrocene moiety. The peak shifted to a lower field due to the increased anisotropic effect and van der Waals deshielding caused by the ferrocenyl group in comparison to the reference sample 1-H222, in which the β-CH protons appeared at 9.54 ppm as a doublet. The inner –NH proton appears in the shielded region at −2.25 ppm, suggesting that 2-H222 retains its aromatic character upon ferrocene substitution at the meso position of the porphyrin. The ferrocenyl group resonates in the region 5.50–4.15 ppm. The ferrocenyl ring protons that are directly linked to porphyrin moiety are inequivalent and appeared as two triplets at 5.50 (J = 1.80 Hz) and 4.84 (J = 1.80 Hz) ppm. In the cyclopentadienyl (Cp) ring which is not linked directly to the porphyrin moiety, all the protons are equivalent and resonate as a sharp singlet at 4.15 ppm. MALDI-TOF mass spectra showed an m/z peak at 771.30 (M + H+).
When Zn is introduced into the free base porphyrin 2-H222 in a non-coordinating solvent, a complementary coordinated cofacial dimer, 2-ZnD, is formed.5 Clear evidence for this can be observed in the UV-visible absorption spectrum shown in Fig. 3, which shows the splitting of the Soret band after Zn introduction. The exceptionally strong interaction between the two stacked porphyrins results in one set of β-CH protons being shifted to higher field due to the strong ring current effect of the facing porphyrin. The affected protons appear as a doublet at 5.43 (J = 4.59 Hz). Due to the shielding effect, the imidazolyl protons are shifted to far higher fields and appear at 5.57 and 2.22 ppm as a pair of doublets. The N–Me group appears as a singlet at 1.70 ppm.
Fig. 1a represents the 1H NMR spectrum of the free base porphyrin 3-H222. The substitution of the 2,2′,3,3′,4,4′,5,5′-octamethylferrocene group at the meso position of porphyrin 3-H222 results in a substantial downfield shift of the β-CH proton adjacent to the octamethylferrocenyl group. This proton appears as a doublet at 11.6 ppm. This shift is quite significant when compared to compound 2-H222 and reference sample 1-H222. The inner –NH proton is also shifted significantly upfield and appears as a broad singlet at −2.39 ppm. All the β-pyrrolic protons are quite distinct, reflecting the strong interaction of the two chromophoric groups. All the methyl protons of the octamethylferrocene appear as eight singlets at higher field from 2.17 ppm to 1.60 ppm. The lone –CH proton of the octamethylferrocenyl group appears as a singlet at 4.19 ppm, this is shifted downfield when compared to its parent compound where it appeared as a singlet at 3.45 ppm. After Zn introduction the protons shifted in the usual manner as described for compound 2-ZnD.
 | ||
| Fig. 1 1H NMR spectra of free base porphyrins 3-H222 (a) and 4-H222 (b) in CDCl3. | ||
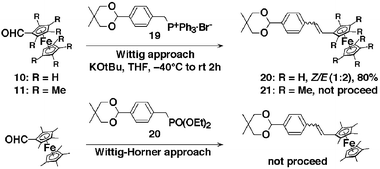 | ||
| Scheme 3 Wittig reactions of 11 and 10 with phosphonium progenitors in THF. | ||
Ferrocenecarboxaldehyde 10 and 2,2′,3,3′,4,4′,5,5′-octamethylferrocenecarboxaldehyde 11 were converted to their corresponding alcohols, 12 and 13, through treatment with LiAlH4 at room temperature in almost quantitative yields. Care should be taken during the addition of the reducing agent, because an excess amount can result in the decomposition of the products. A one-step conversion of these alcohols to Wittig salts 14 and 15 was achieved through treatment with freshly prepared PPh3·HBr22 in toluene with almost quantitative yields. As expected, the Wittig reaction of the ferrocenylmethyl triphenylphosphonium bromide 14 with terephthalaldehyde in the presence of KO-t-Bu resulted in a mixture of cis/trans isomers in 91% yield. The two isomers could be separated by careful chromatography and were obtained in a 1 ∶ 1 cis ∶ trans ratio. The coupling constants for the olefinic protons in the 1H NMR spectra, enabled the unambiguous designation of isomers 16 (cis3JH–H = 12 Hz) and 16 (trans3JH–H = 16.2 Hz). In practice, it is more convenient to treat the cis ∶ trans mixture with 2 eq. of iodine for 3 h at room temperature, which converts cis-16 to trans-16, enabling isolation of trans-16 in 99% yield.
The Wittig reaction of (2,2′,3,3′,4,4′,5,5′-octamethylferrocenyl)methyl triphenylphosphonium bromide 15 with excess terephthalaldehyde (6 eq.) in the presence of KO-t-Bu afforded exclusively the E-“spacered” aldehyde 17 in 67% yield. No cis compound or other by-product could be detected by either TLC or 1H NMR. This may be due to the high steric demand of the octamethylferrocenyl moiety, which favors the E-configuration. The coupling constants for the olefinic protons in the 1H NMR spectra allowed for the unambiguous assignment of isomer 17. The olefinic proton resonance appeared as 16.2 Hz coupled doublets at 7.11 and 6.76 ppm.
Spacer aldehydes 16 and 17 were then employed in porphyrin synthesis through mixed condensation with meso-(3-allyloxypropyl)dipyrromethane 817 and 1-methyl-2-imidazolecarboxaldehyde 918 in the presence of TFA. The resultant intermediate was neutralization with Et3N subsequent p-chloranil oxidation resulted in crude products of the free base porphyrins. Column chromatography was used to separate the three porphyrin products, which resulted in the free base porphyrin 4-H222 and 5-H22 in 26% and 17% yields, respectively. The introduction of zinc generated the title porphyrins 4-ZnD and 5-ZnD in 92% and 90% yields, respectively. Isomerization of the olefinic double bond or scrambling of meso substituents was not observed in these porphyrin syntheses. The exclusive formation of the trans olefin was unequivocally determined by 1H NMR spectroscopy. For compound 4-H222, the olefinic proton resonances appear as 16.2 Hz coupled doublets at 7.26 and 7.11 ppm (Fig. 1b). Similarly, the olefinic proton resonances of compound 5-H22 appeared at 7.24 and 7.09 ppm with a coupling constant of 16.2 Hz. Other porphyrin peaks and the allyloxy ether moiety were also quite distinctive.
UV-visible absorption spectra and fluorescence emission spectra
UV-visible absorption spectral data for all the synthesized porphyrin-ferrocene dyads are listed in Table 1.| Compound | Solvent | Absorption λmax/nm | Emission λmax/nm |
|---|---|---|---|
| a No emission was detected. | |||
| 1-H222 | CH2Cl2 | 648, 591, 549, 515, 416 | 720, 653 |
| 1-ZnD | CH2Cl2 | 619, 565, 435, 413 | 652, 622 |
| 2-H222 | CH2Cl2 | 676, 588, 512, 420 | 724, 656 |
| 2-ZnD | CH2Cl2 | 649, 578, 442, 422 | —a |
| 2-Zn-Py | Pyridine | 639, 575, 432 | |
| 3-H222 | CH2Cl2 | 693, 590, 550, 515, 422 | 723, 654 |
| 3-ZnD | CH2Cl2 | 649, 567, 440, 418 | —a |
| 3-Zn-Py | Pyridine | 649, 564, 432 | |
| 4-H222 | CH2Cl2 | 649,597, 552, 517, 420 | 720, 656 |
| 4-ZnD | CH2Cl2 | 622, 565, 440, 415 | —a |
| 4-Zn-Py | Pyridine | 615, 562, 431 | |
| 5-H22 | CH2Cl2 | 646, 600, 548, 514, 418 | 720, 655 |
| 5-ZnD | CH2Cl2 | 622, 564, 439, 413 | —a |
| 5-Zn-Py | Pyridine | 615, 565, 431 | |
| 6-H22 | CH2Cl2 | 650, 591, 552, 517, 418 | 720, 654 |
| 6-ZnD | CH2Cl2 | 619, 566, 436, 414 | 680, 623 |
| 6-Zn-Py | Pyridine | 612, 562, 430 | |
 | ||
| Fig. 2 Normalized absorption spectra of 1-H222 (solid thin line), 2-H222 (bold dotted line) and 3-H222 (bold solid line) in CH2Cl2. The inset represents the Q band region magnified by 10 times. | ||
 | ||
| Fig. 3 Absorption spectra of 1-ZnD/1-Zn-Im (a), 2-ZnD/2-Zn-Im (b), and 3-ZnD/3-Zn-Im (c) in CH2Cl2. Each spectrum in the Q band region is magnified. The insets show the spectral change in the Soret band region in the course of addition of 1-methylimidazole. | ||
No significant electronic interaction exists between the two chromophores in the ground state when the porphyrin unit is connected to the ferrocenyl unit by an phenylene-ethenylene spacer (4-H222, 5-H22, and 6-H22) (see supplementary data†).
Fig. 4 shows the relative steady state fluorescence spectra in dichloromethane at 25 °C. The effect of introducing ferrocenyl groups is apparent from the sharp drop in fluorescence intensity. The fluorescence of porphyrin 2-H222 and 3-H222 was reduced dramatically to 0.15% that of the free base sample 1-H222 without the ferrocenyl moiety. The phenylene-ethenylene-connected ferrocenyl free base porphyrins 4-H222 and 5-H22 exhibited decreased quenching efficiencies. The decrease of fluorescence intensity in all the ferrocenyl porphyrins is attributed to intramolecular quenching of the porphyrin singlet state by ferrocene. When the conjugation between ferrocene and porphyrin is disrupted, as in compounds 6-H22, intramolecular quenching of the porphyrin singlet state by ferrocene dramatically decreased with a corresponding recovery in the fluorescence (Fig. 5). This result indicates that the ferrocenyl group serves as an effective electron donor. The characteristics of the linking group greatly affect the efficiency of fluorescence quenching. Further investigation of fluorescence quenching of the porphyrin moiety by the ferrocenyl group requires fast dynamic measurements, which are currently under investigation.
 | ||
| Fig. 4 Steady state fluorescence spectra of 1-H222 (solid line) and 1-ZnD (broken line) in CH2Cl2 at 25 °C. Inset shows emission for 2-H222 and 2-ZnD. The intensity was normalized to 0.1 absorbance at the excited wavelength of the Soret band, in each case. | ||
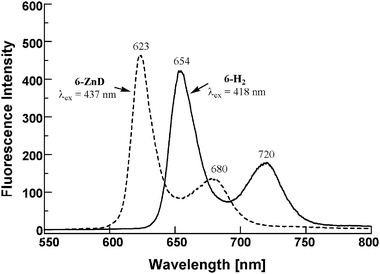 | ||
| Fig. 5 Steady state fluorescence spectra of 6-H22 (solid line), 6-ZnD (broken line) in CH2Cl2 at 25 °C. The intensity was normalized to 0.1 absorbance at the excited wavelength of the Soret band, in each case. | ||
The ferrocene substitution at the meso position of the porphyrin ring leads to a characteristic feature of the electronic structures. Bathochromic shift of the Q bands depends on the redox potential of the ferrocenyl terminal and the degree of π-conjugation though the connecting group. When the linker unit contains an aliphatic chain as in 6-ZnD, the electronic communication is disrupted and the reductive quenching of excited singlet of porphyrin dimer becomes marginal (Fig. 5). Interestingly, the fluorescence was completely quenched in 2-ZnD and 3-ZnD (100%). Quantitative quenching was observed for 3-ZnD and 4-ZnD, too. This is attributed to electron transfer from the directly linked ferrocenyl moiety to the photoexcited singlet state of porphyrin dimer with exceptionally high efficiencies. The efficient fluorescence quenching in the slipped-cofacial dimer may be correlated with the acceleration of the electron transfer by the decreased reorganization energy of environmental solvent molecules.24
Electrochemical measurements
The redox properties of synthesized porphyrin-ferrocene dyads were examined through cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in dichloromethane. The electrochemical investigation employed a conventional three electrode configuration in dichloromethane with 0.1 M tetrabutylammonium hexafluorophosphate (nBu4N·PF6) as a supporting electrolyte at 25 °C.Fig. 6 presents the cyclic voltammograms of the synthesized ferrocene-functionalized Zn-imidazolyl-porphyrin dimers 2-ZnD–5-ZnD, along with the reference dimer 1-ZnD. Fig. 7 shows the differential-pulse voltammograms for the equimolar mixture of the ferrocene-functionalized Zn-imidazolyl-porphyrin dimers 2-ZnD–5-ZnD and their dissociated monomers, which were obtained by the addition of 1-methylimidazole 2-Zn-Im–5-Zn-Im, along with the reference sample 1-ZnD and its monomer 1-Zn-Im.
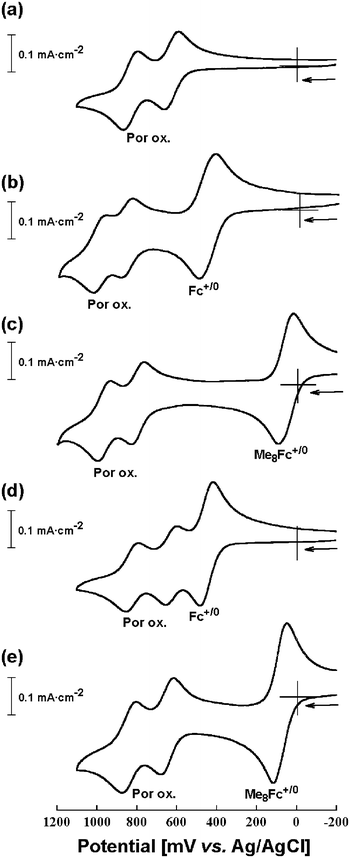 | ||
| Fig. 6 Cyclic voltammograms of 1-ZnD (a), 2-ZnD (b), 3-ZnD (c), 4-ZnD (d), and 5-ZnD (e) in CH2Cl2 containing 0.1 M nBu4N·PF6 supporting electrolyte at 25 °C with a sweep rate of 100 mV·sec−1. | ||
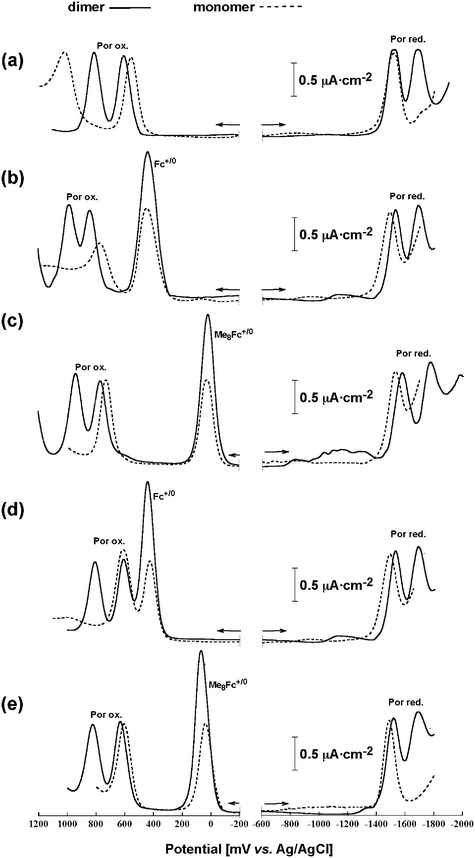 | ||
| Fig. 7 Differential-pulse voltammograms of ferrocene-functionalized Zn-imidazolyl-porphyrins along with reference sample without ferrocenyl unit, solid line; coordination dimer and broken line; dissociated monomer. a) 1-ZnD and 1-Zn-Im, b) 2-ZnD and 2-Zn-Im, c) 3-ZnD and 3-Zn-Im, d) 4-ZnD and 4-Zn-Im, and e) 5-ZnD and 5-Zn-Im in CH2Cl2 containing 0.1 M nBu4N·PF6 supporting electrolyte. | ||
| Compound | Ferrocene oxidation | Porphyrin oxidation | Porphyrin reduction | ||
|---|---|---|---|---|---|
| E 1/2 +/0/V(ΔEp/mV) |
|
|
|
|
|
| E 1/2 = 1/2(Epa + Epc), ΔEp = Epa − Epc (peak potential separation) in CVs Ep = peak potential, Epa = anodic peak potentials, Epc = cathodic peak potential, E1/2 = measured half-wave potential by cyclic voltammetry.a scan rate 10 mV s−1,b Ref. 25. In all cases the scan rate is 100 mV s−1 unless otherwise stated. Solvent: CH2Cl2, supporting electrolyte: 0.1 M nBu4N·PF6 | |||||
| 1-H222 | 1.0 | 1.2 | −1.20(68) | −1.59(64) | |
| 1-ZnD | 0.63(76), 0.83(67) | −1.55(53), −1.70(66) | |||
| 1-Zn-Im | 0.6 | 1.1 | −1.54(81) | ||
| 2-H222 | 0.55(92) | 1.1 | −1.21(62) | −1.62(90) | |
| 2-ZnD | 0.46(86) | 0.87(56), 1.0(50) | −1.5, −1.68 | ||
| 2-Zn-Im | 0.44 | 0.77 | −1.5(106) | ||
| 3-H222 | 0.12(87) | 1.1 | −1.28(61) | −1.64(128) | |
| 3-ZnD | 0.054(70) | 0.80(52), 0.97(60) | −1.57, −1.77 | ||
| 3-Zn-Im | 0.02(69) | 0.73 | −1.53 | ||
| 4-H222 | 0.45 | 0.91 | 1.20 | −1.2(60) | −1.57(75) |
| 4-ZnD | 0.45(63) | 0.63(50), 0.83(57) | −1.50, −1.67 | ||
| 4-Zn-Im | 0.43 | 0.61 | −1.5 | ||
| 5-H22 | 0.076 a(64) | 0.97(57) | 1.18(99) | −1.23(61) | −1.6(73) |
| 5-ZnD | 0.084(59) | 0.65(59), 0.84(65) | −1.52, −1.69 | ||
| 5-Zn-Im | 0.04 | 0.61 | −1.49 | ||
| 6-H22 | 0.39 | 0.9 | 1.21 | −1.21(71) | −1.63(112) |
| 6-ZnD | 0.45(74) | 0.62(56), 0.82(60) | −1.55(77), −1.72(85) | ||
| 6-Zn-Im | 0.40 | 0.60 | −1.54(76) | ||
| H2TPP b | 1.03 | −1.23 | −1.55 | ||
| Zn-TPP b | 0.82 | 1.14 | −1.32 | −1.70 | |
| Fc | 0.48(61) | ||||
| (CH3)8Fc | 0.057(70) | ||||




The significant redox splitting reveals a strong interaction between the redox centers, suggesting that the cation radical generated by the first one-electron oxidation is considerably delocalized over the entire framework of the dimer pair thus raising the potential for the next one-electron oxidation. Further oxidations at more anodic potentials lead to loss of reversibility. Only irreversible processes were observed at 1.2 and 1.5 V. Under controlled anodic potentials, however, dimer 1-ZnD exhibited a completely reversible stepwise one-electron transfer process (Fig. 6a). It is important to note in comparison of the initial oxidation potentials of 1-ZnD and 1-Zn-Im that the dimer formation process does not shift the potential towards easier oxidation. Rather, the second oxidation potential is shifted to a significantly higher value than the first. Similar splittings of the first oxidation wave were observed in the voltammograms of the ferrocene-functionalized Zn-imidazolyl-porphyrin dimers, 2-ZnD–6-ZnD, even though the oxidation of ferrocene moiety occurs by a single oxidation step in the dimers.
In the negative potential region, dimer 1-ZnD exhibited two successive well-defined reversible one-electron reduction waves at 1.55 V and 1.69 V. This also reflects the strong electronic interaction between the two porphyrins in the slipped-cofacial arrangement. Ferrocene-functionalized Zn-imidazolyl-porphyrin dimers, 2-ZnD–6-ZnD, exhibited split successive one-electron reduction waves from −1.5 V to −1.77 V vs Ag/AgCl. The first one-electron reduction of the dimers 2-ZnD–6-ZnD, occurs at the same or at slightly higher potentials relative to reference sample 1-ZnD in a similar manner to the oxidation processes. For all of the dimers, reduction of the porphyrin ring was more difficult than for the parent free base porphyrins 2-H222–6-H22 (Table 2). This is consistent with the fact that free base porphyrins generally serve as electron acceptors from zinc porphyrins. Compound 3-ZnD showed two one-electron reduction waves, at −1.57 V and −1.77 V, representing apparently harder reductions due to the directly connected octamethylferrocene substituents compared to the reference sample 1-ZnD. This effect is mitigated for the spacer-linked ferrocenyl porphyrins 4-ZnD, 5-ZnD, and 6-ZnD.
In conclusion, the slipped-cofacial dimer formation leads to splittings of the first oxidation and the first reduction waves. This is interpreted in terms of the charge-resonance (intervalance) interaction delocalizing the generated cation radical over the entire π-framework of the special-pair porphyrin dimer.23 These properties play crucial roles in photoinduced electron transfer to accelerate the charge-separation but to decelerate the charge-recombination.24 The slipped-cofacial orientation is appropriate as the special-pair model of the natural photosynthesis to provide long-lived charge separation state.
The electron-donating abilities of the octamethylferrocenyl group should be noted. The presence of the electron donating methyl groups caused a shift in the oxidation potential of 3-ZnD by approximately 400 mV to the negative direction compared to 2-ZnD bearing unsubstituted ferrocene. This result indicates that the octamethylferrocenyl group is more susceptible to oxidation than the ferrocene and is oxidized at a very low oxidation potential, consistent with the well-known effects of methyl substitution. Thus, methyl substitution of the ferrocenyl moiety brings increased solubility, lowered oxidation potential, and amplified donor capacity. The accompanying increased stability of the corresponding ferrocenium cations could be exploited for improving materials used in molecular electronics, solar energy conversion systems and others.
In contrast, when separated by a phenylene-ethenylene spacer from the porphyrin, the ferrocenyl moiety had an almost negligible effect on the porphyrin ring oxidation potentials. In the two compounds 4-ZnD and 5-ZnD, the porphyrin oxidation potentials remained almost the same as that of porphyrin without a ferrocenyl group (reference dimer 1-ZnD). The oxidation potentials of the porphyrins are only marginally affected by the presence of the ferrocenium cation when separated by the phenylene-ethenylene spacer. The redox potentials for 6-ZnD were nearly identical to those obtained for compound 4-ZnD, reflecting the minimal perturbation of the porphyrin π-system by the phenylene-ethyl connected ferrocenyl group. Fig. 8 shows the CV and DPV charts for 6-ZnD along with its monomer 6-Zn-Im. The charts were superimposed on the same axis for easy comparison.
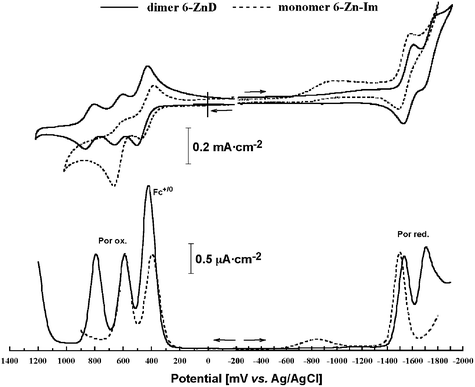 | ||
| Fig. 8 CV and DPV of 6-ZnD and 6-ZnD-Im in CH2Cl2 containing 0.1 M nBu4N·PF6 as a supporting electrolyte. | ||
Judging from the results shown in Table 2, there is no substantial shift of the oxidation potentials of ferrocene or octamethylferrocene moieties by connecting to porphyrin ring except for the directly connected dimers 2-ZnD and 3-ZnD, which showed large anodic shifts for the first ring oxidation of the dimer. This potential shift suggests the existence of the strong electronic communication between ferrocenyl moiety and porphyrin plane with close proximity. The difference between the directly connected case (2-ZnD and 3-ZnD) and the separately connected case (4-ZnD–6-ZnD) is consistent with the spectroscopic properties found in the Q band region.
The degree of metal–metal interaction (electronic communication) between the two ferrocenyl redox centers in the slipped-cofacial zinc-imidazolyl-porphyrin dimers 2-ZnD–6-ZnD can be judged from the oxidation wave patterns of either CV or DPV. In all cases, only a single oxidation wave was observed, therefore there is no mixed-valence interaction between two ferrocene terminals. However the shift of oxidation potentials of porphyrin rings is enhanced by dimer formation in the directly connected cases, 2-ZnD and 3-ZnD, compared with phenylene-ethenylene linked cases, 4-ZnD and 5-ZnD (Fig. 7). This indicates that two terminal ferrocenyl groups in the dimer indirectly and weakly communicate through the porphyrin conjugation chain.
Conclusions
A one-pot conversion of aldehyde precursors to the corresponding free base porphyrins (AB2C type), including imidazole and ferrocene as A,C-meso substituents, was achieved. Zn insertion produced the title ferrocene-functionalized Zn-imidazolyl-porphyrin dimers 2-ZnD–6-ZnD in reasonably good yields. In this series, ferrocene and octamethylferrocene were substituted at the meso position and were connected directly to the porphyrin by phenylene-ethenylene or phenylene-ethyl spacers. The structures were confirmed by spectroscopic analyses and the porphyrins were found capable of serving as a new terminal electron-donor.UV-visible spectra of 2-ZnD–6-ZnD showed splitting of the Soret band, which suggests the formation of slipped-cofacial dimers and strong excitonic coupling between the two porphyrin units. The porphyrin fluorescence was quenched according to the degree of conjugation: almost completely for direct connections (2 and 3), moderately for the phenylene-ethenylenes (4 and 5) and marginally for the phenylene-ethyl linkers (6). Electrochemical studies of the dimer showed splitting of the first one-electron oxidation/reduction potential of the porphyrin units. In compounds 2-ZnD and 3-ZnD, in which the ferrocenyl group is directly connected to the porphyrin, the ferrocenium cation is generated first during oxidation and acts to raise the oxidation potentials of the porphyrin as compared to the reference sample 1-ZnD. On the other hand, when a phenylene-ethenylene spacer is introduced, the oxidation potentials of the porphyrins are only marginally affected by the presence of the ferrocenium cation.
The interaction between the two ferrocenyl units across the coordination dimer is weak and a single oxidation wave was observed in CV and DPV. The ability to supply a total of four electrons from the dimeric arrays indicates possible applications as controlled electron reservoirs or candidates for a terminal electron donor in a potential cascade. Further studies are currently in progress to exploit these properties for application in molecular devices.
Experimental
General
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 5.50 (t, J
= 1.80 Hz, 2H; Cp of Fc), 5.46 (dd, J
= 17.3, 1.89 Hz, 2H; allyl-Htrans), 5.29 (dd, J
= 10.4, 1.7 Hz, 2H; allyl-Hcis), 5.10 (t, J
= 7.02 Hz, 4H; -CH2), 4.84 (t, J
= 1.80 Hz, 2H; Cp of Fc), 4.15 (s, 5H; Fc-H), 4.11–4.06 (m, 4H; -CH2), 3.67 (t, J
= 6.0 Hz, 4H; -CH2), 3.37 (s, 3H; N-Me), 2.82–2.74 (m, 4H; -CH2), –2.26 ppm (s, 2H; inner-NH); 13C NMR (67 MHz, CDCl3): δ 148.5 (Im-C2), 135.0 (CH-allyl), 132.1–126.8 (br, pyrrole-β), 128.2 (Im-C4), 121.3 (Im-C5), 119.2, 118.8 (meso), 116.8 (CH2-allyl), 103.7 (meso), 89.8 (C-Cp), 77.6 (C-Cp), 72.0 (-OCH2), 70.6 (CH-Cp), 69.2 (-CH2-CHCH2), 68.9 (CH-Cp), 37.7 (CH2), 34.4 (N-CH3), 31.5 ppm (CH2). MALDI-TOF MS: found m/z
= 771.30 (M
+ H+), calculated for C46H46FeN6O2 770.30; IR (KBr): ν 2924, 2854, 1474, 1104, 795, 733 cm−1; UV-vis λmax
(CH2Cl2) 420, 512, 588, 676 nm; fluorescence, λEM
(CH2Cl2) 656, 724 nm (λEx 422 nm).
), 5.35–5.49 (m, 2H; allyl-Htrans), 5.22–5.31 (m, 2H; allyl-Hcis), 5.12, 4.99 (each t, J
= 7.03 Hz, 4H; -CH2), 4.19 (s, 1H; CH-Cp), 4.10–4.12, 4.03–4.05 (each m, 4H; -OCH2), 3.70, 3.60 (each t, J
= 5.94 Hz, 4H; -CH2), 3.35 (s, 3H; N-Me), 2.81–2.86, 2.67–2.72 (each m, 4H; CH2), 2.16, 2.14, 2.00, 1.92, 1.83, 1.78, 1.72, 1.60 (8s, 24H; (CH3)8Fc), –2.39 ppm (s, 2H; inner-NH); 13C NMR (67 MHz, CDCl3): δ 148.8 (Im-C2), 135.0 (CH-allyl), 132.8, 130.9, 129.5, 129.0, 127.9 (br, pyrrole-β), 128.2 (Im-C4), 121.2 (Im-C5), 119.1, 118.6, 117.9 (meso), 116.7 (CH2-allyl), 103.7 (meso), 91.3 (C-Cp), 85.3 (C-Cp), 84.4 (C-Cp), 80.8 (C-Cp), 80.7 (C-Cp), 80.6 (C-Cp), 80.3 (C-Cp), 80.2 (C-Cp), 80.1 (C-Cp), 72.1 (-OCH2), 71.9 (CH, Cp), 69.3 (CH2, -CH2-CHCH2), 69.2 (C-Cp), 37.8 (CH2), 34.5 (N-CH3), 31.4 (CH2), 12.1, 11.9, 11.4, 11.3, 10.3, 10.3, 9.5, 9.4 ppm; MALDI-TOF MS: found m/z
= 882.42 (M
+ H+), calculated for C54H62FeN6O2 882.43; IR (KBr)
ν 2900, 2850, 1474, 1102, 918, 738 cm−1; UV-Vis λmax
(CH2Cl2) 422, 515, 550, 590, 693 nm; fluorescence, λEM2Cl2) 654, 723 nm (λEx 422 nm).
CH-), 5.76 (t, J
= 1.62 Hz, 2H; Cp of Fc), 5.57 (d, J
= 1.62 Hz, 1H; Im-H5), 5.54 (dd, J
= 10.26, 1.62 Hz, 2H; allyl-Htrans), 5.43 (d, J
= 4.59 Hz, 2H; pyrrole-Hβ), 5.37 (dd, J
= 10.26, 1.35 Hz, 2H; allyl-Hcis), 5.17 (t, J
= 7.03 Hz, 4H; -CH2), 4.93 (t, J
= 1.62 Hz, 2H; Cp of Fc), 4.32 (s, 5H; Fc-H), 4.29–4.22 (m, 4H; -OCH2), 3.94-3.88 (m, 4H; -CH2), 3.10–2.97 (m, 4H; CH2), 2.22 (d, J
= 1.62 Hz, 1H; Im-H4), 1.70 ppm (s, 3H; N-Me); 13C NMR (67 MHz, CDCl3): δ 150.0, 149.5, 148.0 (pyrrole-α), 146.2 (Im-C2), 135.3 (CH-allyl), 132.3, 128.8, 127.1, 127.0 (pyrrole-β), 121.6(Im-C4), 119.1, 118.2 (meso), 117.4 (Im-C5), 116.7 (CH2-allyl), 96.1 (meso), 91.8 (C-Cp), 77.9 (CH-Cp), 72.1 (-OCH2), 70.6 (CH-Cp), 70.10 (-CH2-CHCH2), 68.5 (CH-Cp), 38.5 (CH2), 32.7 (CH3, N-CH3), 31.2 ppm (CH2); MALDI-TOF MS m/z 833.20 (M
+ H+), 1664.7; calculated for C46H44FeN6O2Zn 832.22; IR (KBr): ν 2922, 2853, 1144, 1104, 987, 788 cm−1; UV-Vis λmax
(CH2Cl2) 422, 442, 578, 649 nm.
), 5.47–5.58 (m, 2H; allyl-Htrans), 5.42 (d, J
= 1.35 Hz, 1H; Im-H5), 5.40 (d, J
= 4.6 Hz, 1H; pyrrole-Hβ), 5.30–5.38 (m, 2H; allyl-Hcis), 5.24, 5.15 (each m, 4H; -CH2), 4.62 (s, 1H; CH-Cp), 4.26–4.19 (m, 4H; -OCH2), 3.95-3.87 (m, 4H; -CH2), 3.01–3.04 (m, 4H; CH2), 2.16, 2.11, 2.01, 1.91, 1.78, 1.73.1.65, 1.60 (8s, 24H; (CH3)8Fc), 2.14 (d, J
= 1.08 Hz, 1H; Im-H4); 1.67 ppm (s, 3H; N-Me); 13C NMR (67 MHz, CDCl3): δ 151.1, 149.8, 148.9, 147.4 (pyrrole-α), 146.1 (Im-C2), 135.2 (CH-allyl), 133.8, 131.9, 128.6, 127.4 (pyrrole-β), 121.5 (Im-C4), 118.9, 118.3, 117.1 (meso), 117.5, (Im-C5), 116.5 (CH2, allyl), 96.0 (meso), 81.0 (C-Cp), 80.1 (C-Cp), 77.8 (C-Cp), 72.0 (-OCH2), 71.9 (C-Cp), 70.1 (-CH2-CHCH2), 70.0 (C-Cp), 38.5 (CH2), 34.6 (CH3, N-CH3), 31.6 (CH2) 14.1, 12.8, 11.7, 11.5, 11.3, 10.3, 9.6, 9.5 ppm; MALDI-TOF MS: Found m/z
= 944.34 (M
+ H+), calculated for C54H60FeN6O2Zn 944.34, IR(KBr): ν 2921, 2851, 1459, 1284, 1104, 1074, 986, 787 cm−1; UV-Vis λmax
(CH2Cl2) 418, 440, 567, 649 nm.
C), 6.44 (d, 3Jcis(1H–1H)
= 12.0 Hz, 1H; -CCH), 4.21 (t, J
= 1.62 Hz, 2H; Fc-H), 4.18 (t, J
= 1.62 Hz, 2H; Fc-H), 4.11 ppm (s, 5H; Fc-H); UV-vis λmax
(CHCl3) 318 nm. 1H NMR (270 MHz, CDCl3, trans-isomer): δ 9.97 (s, 1H, CHO), 7.84 (d, J
= 8.1 Hz, 2H, Ar-H), 7.57 (d, J
= 8.37, 2H; Ar-H), 7.10 (d, 3Jtrans(1H–1H)
= 16.2 Hz, 1H; -CHC), 6.75 (d, 3Jtrans(1H–1H)
= 16.2 Hz, 1H; -CCH), 4.51 (d, J
= 1.89 Hz, 2H; Fc-H), 4.36 (d, J
= 1.89 Hz, 2H; Fc-H), 4.16 ppm (s, 5H; Fc-H), 13C NMR (67 MHz, CDCl3): δ 191.37, 143.88, 134.47, 131.38, 130.21, 125.93, 124,43, 82.18, 69.74, 69.36, 67.33 ppm; MALDI-TOF MS: m/z
= 316; IR (KBr): ν 1691, 1593, 1564 cm−1; UV-vis λmax
(CHCl3) 483, 342 nm.
), 5.50 (t, J
= 1.80 Hz, 2H; Cp of Fc), 5.46 (dd, J
= 17.3, 1.89 Hz, 2H; allyl-Htrans), 5.29 (dd, J
= 10.4, 1.7 Hz, 2H; allyl-Hcis), 5.10 (t, J
= 7.02 Hz, 4H; -CH2), 4.84 (t, J
= 1.80 Hz, 2H; Cp of Fc), 4.15 (s, 5H; Fc-H), 4.11–4.06 (m, 4H; -CH2), 3.67 (t, J
= 6.0 Hz, 4H; -CH2), 3.37 (s, 3H; N-Me), 2.82–2.74 (m, 4H; -CH2), –2.26 ppm (s, 2H; inner-NH); 13C NMR (67 MHz, CDCl3): δ 148.5 (Im-C2), 135.0 (CH-allyl), 132.1–126.8 (br, pyrrole-β), 128.2 (Im-C4), 121.3 (Im-C5), 119.2, 118.8 (meso), 116.8 (CH2-allyl), 103.7 (meso), 89.8 (C-Cp), 77.6 (C-Cp), 72.0 (-OCH2), 70.6 (CH-Cp), 69.2 (-CH2-CHCH2), 68.9 (CH-Cp), 37.7 (CH2), 34.4 (N-CH3), 31.5 ppm (CH2). MALDI-TOF MS: found m/z
= 771.30 (M
+ H+), calculated for C46H46FeN6O2 770.30; IR (KBr): ν 2924, 2854, 1474, 1104, 795, 733 cm−1; UV-vis λmax
(CH2Cl2) 420, 512, 588, 676 nm; fluorescence, λEM
(CH2Cl2) 656, 724 nm (λEx 422 nm).
), 5.35–5.49 (m, 2H; allyl-Htrans), 5.22–5.31 (m, 2H; allyl-Hcis), 5.12, 4.99 (each t, J
= 7.03 Hz, 4H; -CH2), 4.19 (s, 1H; CH-Cp), 4.10–4.12, 4.03–4.05 (each m, 4H; -OCH2), 3.70, 3.60 (each t, J
= 5.94 Hz, 4H; -CH2), 3.35 (s, 3H; N-Me), 2.81–2.86, 2.67–2.72 (each m, 4H; CH2), 2.16, 2.14, 2.00, 1.92, 1.83, 1.78, 1.72, 1.60 (8s, 24H; (CH3)8Fc), –2.39 ppm (s, 2H; inner-NH); 13C NMR (67 MHz, CDCl3): δ 148.8 (Im-C2), 135.0 (CH-allyl), 132.8, 130.9, 129.5, 129.0, 127.9 (br, pyrrole-β), 128.2 (Im-C4), 121.2 (Im-C5), 119.1, 118.6, 117.9 (meso), 116.7 (CH2-allyl), 103.7 (meso), 91.3 (C-Cp), 85.3 (C-Cp), 84.4 (C-Cp), 80.8 (C-Cp), 80.7 (C-Cp), 80.6 (C-Cp), 80.3 (C-Cp), 80.2 (C-Cp), 80.1 (C-Cp), 72.1 (-OCH2), 71.9 (CH, Cp), 69.3 (CH2, -CH2-CHCH2), 69.2 (C-Cp), 37.8 (CH2), 34.5 (N-CH3), 31.4 (CH2), 12.1, 11.9, 11.4, 11.3, 10.3, 10.3, 9.5, 9.4 ppm; MALDI-TOF MS: found m/z
= 882.42 (M
+ H+), calculated for C54H62FeN6O2 882.43; IR (KBr)
ν 2900, 2850, 1474, 1102, 918, 738 cm−1; UV-Vis λmax
(CH2Cl2) 422, 515, 550, 590, 693 nm; fluorescence, λEM2Cl2) 654, 723 nm (λEx 422 nm).
CH-), 5.76 (t, J
= 1.62 Hz, 2H; Cp of Fc), 5.57 (d, J
= 1.62 Hz, 1H; Im-H5), 5.54 (dd, J
= 10.26, 1.62 Hz, 2H; allyl-Htrans), 5.43 (d, J
= 4.59 Hz, 2H; pyrrole-Hβ), 5.37 (dd, J
= 10.26, 1.35 Hz, 2H; allyl-Hcis), 5.17 (t, J
= 7.03 Hz, 4H; -CH2), 4.93 (t, J
= 1.62 Hz, 2H; Cp of Fc), 4.32 (s, 5H; Fc-H), 4.29–4.22 (m, 4H; -OCH2), 3.94-3.88 (m, 4H; -CH2), 3.10–2.97 (m, 4H; CH2), 2.22 (d, J
= 1.62 Hz, 1H; Im-H4), 1.70 ppm (s, 3H; N-Me); 13C NMR (67 MHz, CDCl3): δ 150.0, 149.5, 148.0 (pyrrole-α), 146.2 (Im-C2), 135.3 (CH-allyl), 132.3, 128.8, 127.1, 127.0 (pyrrole-β), 121.6(Im-C4), 119.1, 118.2 (meso), 117.4 (Im-C5), 116.7 (CH2-allyl), 96.1 (meso), 91.8 (C-Cp), 77.9 (CH-Cp), 72.1 (-OCH2), 70.6 (CH-Cp), 70.10 (-CH2-CHCH2), 68.5 (CH-Cp), 38.5 (CH2), 32.7 (CH3, N-CH3), 31.2 ppm (CH2); MALDI-TOF MS m/z 833.20 (M
+ H+), 1664.7; calculated for C46H44FeN6O2Zn 832.22; IR (KBr): ν 2922, 2853, 1144, 1104, 987, 788 cm−1; UV-Vis λmax
(CH2Cl2) 422, 442, 578, 649 nm.
), 5.47–5.58 (m, 2H; allyl-Htrans), 5.42 (d, J
= 1.35 Hz, 1H; Im-H5), 5.40 (d, J
= 4.6 Hz, 1H; pyrrole-Hβ), 5.30–5.38 (m, 2H; allyl-Hcis), 5.24, 5.15 (each m, 4H; -CH2), 4.62 (s, 1H; CH-Cp), 4.26–4.19 (m, 4H; -OCH2), 3.95-3.87 (m, 4H; -CH2), 3.01–3.04 (m, 4H; CH2), 2.16, 2.11, 2.01, 1.91, 1.78, 1.73.1.65, 1.60 (8s, 24H; (CH3)8Fc), 2.14 (d, J
= 1.08 Hz, 1H; Im-H4); 1.67 ppm (s, 3H; N-Me); 13C NMR (67 MHz, CDCl3): δ 151.1, 149.8, 148.9, 147.4 (pyrrole-α), 146.1 (Im-C2), 135.2 (CH-allyl), 133.8, 131.9, 128.6, 127.4 (pyrrole-β), 121.5 (Im-C4), 118.9, 118.3, 117.1 (meso), 117.5, (Im-C5), 116.5 (CH2, allyl), 96.0 (meso), 81.0 (C-Cp), 80.1 (C-Cp), 77.8 (C-Cp), 72.0 (-OCH2), 71.9 (C-Cp), 70.1 (-CH2-CHCH2), 70.0 (C-Cp), 38.5 (CH2), 34.6 (CH3, N-CH3), 31.6 (CH2) 14.1, 12.8, 11.7, 11.5, 11.3, 10.3, 9.6, 9.5 ppm; MALDI-TOF MS: Found m/z
= 944.34 (M
+ H+), calculated for C54H60FeN6O2Zn 944.34, IR(KBr): ν 2921, 2851, 1459, 1284, 1104, 1074, 986, 787 cm−1; UV-Vis λmax
(CH2Cl2) 418, 440, 567, 649 nm.
C), 6.44 (d, 3Jcis(1H–1H)
= 12.0 Hz, 1H; -CCH), 4.21 (t, J
= 1.62 Hz, 2H; Fc-H), 4.18 (t, J
= 1.62 Hz, 2H; Fc-H), 4.11 ppm (s, 5H; Fc-H); UV-vis λmax
(CHCl3) 318 nm. 1H NMR (270 MHz, CDCl3, trans-isomer): δ 9.97 (s, 1H, CHO), 7.84 (d, J
= 8.1 Hz, 2H, Ar-H), 7.57 (d, J
= 8.37, 2H; Ar-H), 7.10 (d, 3Jtrans(1H–1H)
= 16.2 Hz, 1H; -CHC), 6.75 (d, 3Jtrans(1H–1H)
= 16.2 Hz, 1H; -CCH), 4.51 (d, J
= 1.89 Hz, 2H; Fc-H), 4.36 (d, J
= 1.89 Hz, 2H; Fc-H), 4.16 ppm (s, 5H; Fc-H), 13C NMR (67 MHz, CDCl3): δ 191.37, 143.88, 134.47, 131.38, 130.21, 125.93, 124,43, 82.18, 69.74, 69.36, 67.33 ppm; MALDI-TOF MS: m/z
= 316; IR (KBr): ν 1691, 1593, 1564 cm−1; UV-vis λmax
(CHCl3) 483, 342 nm.
Alternatively, the cis-product 16 was converted to the trans-product 16 in the following manner. The cis-compound 16 (0.132 g, 0.42 mmol) was dissolved in CHCl3 (20 mL) and then I2 (0.212 g, 0.84 mmol, 2 eq.) was added and stirred at room temperature for 3 h. A saturated solution of Na2SO3 (30 mL) was added and stirred vigorously for 30 min. The organic layer was separated, and the aqueous phase extracted with chloroform (2 × 50 mL). The combined organic layer was washed with water (2 × 100 mL), dried over anhydrous Na2SO4 and solvent removed in vacuo to yield the purple solid. TLC and 1H NMR indicated the complete conversion of cis-isomer to trans isomer. Yield = 131 mg, quantitative. 1H NMR is identical to that of trans-isomer. This material was used without any further purification for the subsequent porphyrin synthesis.
C), 6.76 (d, 3Jtrans(1H–1H)
= 16.2 Hz, 1H; -CCH), 3.33 (s, 1H; CH of Cp), 1.99, 1.83, 1.75, 1.66 ppm (4s, 24H; CH3 of Cp); 13C NMR (67 MHz, CDCl3): δ 191.19 (CHO), 144.85, 134.47, 133.99, 131.99, 130.25, 125.29, 124.45, 82.60, 80.97, 80.81, 79.73, 79.71, 71.40, 11.24, 11.21, 9.93, 9.37 ppm; MALDI-TOF MS: found m/z
= 428.1 (M
+ H+), calculated for C27H32FeO 428.18; IR (KBr): ν 3059, 2966, 2902, 1687, 1590, 1561, 1306, 1218, 1164, 1030, 968, 863, 821, 793, 502, 471 cm−1; UV-vis λmax
(CHCl3) 363, 532 nm.
C), 7.01 (d, 3Jtrans(1H–1H)
= 16.2 Hz, 1H; CCH-), 6.00–6.15 (m, 2H; allyl-CH), 5.44 (dd, J
= 17.01, 1.62 Hz, 2H; allyl-Htrans), 5.27 (m, J
= 10.53, 1.35 Hz, 2H; allyl-Hcis), 5.10 (t, J
= 7.02 Hz, 4H; -CH2), 4.62 (t, J
= 1.89 Hz, 2H; Fc-H), 4.38 (t, J
= 1.62 Hz, 2H; Fc-H), 4.27 (s, 5H; Fc-H), 4.07 (dd, J
= 5.67, 1.35 Hz, 4H; CH2), 3.66 (t, J
= 5.67 Hz, 4H; -CH2), 3.39 (s, 3H; N-Me), 2.83-2.74 (m, 4H; CH2), −2.65 ppm (s, 2H; inner-NH); 13C NMR (67 MHz, CDCl3): δ 148.8 (Im-C2), 140.8 (Ph), 137.2 (Ph), 135.0 (CH-allyl), 134.8 (Ph), 132.0, 130.4, 129.0, 127.6 (br, pyrrole-β), 128.2 (Im-C4), 127.8 (Ph), 125.6 (Ph), 123.9 (Ph), 121.2 (Im-C5), 120.6, 119.2 (meso), 116.7 (CH2-allyl), 104.1 (meso), 83.3 (C-Cp), 77.5 (C-Cp), 72.0 (-OCH2), 69.3 (CH-Cp), 69.2 (-CH2-CHCH2), 68.1 (CH-Cp), 67.1, 37.8 (CH2), 34.5 (CH3, N-CH3), 31.4 ppm (CH2); MALDI-TOF MS: found m/z
= 873.35 (M
+ H+), calculated for C54H52FeN6O2 872.35; IR (KBr): ν 2922, 2852, 1104, 797 cm−1; UV-Vis λmax
(CH2Cl2) 420, 517, 552, 649 nm; fluorescence, λEM
(CH2Cl2) 656, 720 nm (λEx 420 nm).
), 5.45 (dd, J
= 17.28, 1.62 Hz, 2H; allyl-Htrans), 5.28 (dd, J
= 10.26, 1.35 Hz, 2H; allyl-Hcis), 5.11 (t, J
= 7.02 Hz, 4H; -CH2), 4.08 (dd, J
= 5.4, 1.35 Hz, 4H; -OCH2), 3.66 (t, J
= 6.0 Hz, 4H; -CH2), 3.43 (brs, 1H; CH-Cp), 3.40 (s, 3H; N-Me), 2.75–2.84 (m, 4H; CH2), 2.14, 1.90, 1.85, 1.80 (s, CH3, 24H; (CH3)8Fc), −2.63 ppm (brs, 2H; inner-NH); 13C NMR (67 MHz, CDCl3): δ 148.7 (Im-C2), 140.2 (Ph), 138.2 (Ph), 134.8 (CH-allyl), 134.7 (Ph), 132.0, 130.2, 129.0, 127.4 (br; pyrrole-β), 128.1 (Im-C4), 126.1 (Ph), 123.4 (Ph), 121.2 (Im-C5), 120.8, 120.4, 119.2 (meso), 116.7 (CH2-allyl), 104.0 (meso), 82.0 (C-Cp), 81.0 (C-Cp), 79.5 (C-Cp), 77.1 (C-Cp), 71.9 (-OCH2), 71.4 (CH-Cp), 69.0 (-CH2-CHCH2), 37.7 (CH2), 34.4 (N-CH3), 31.3 (CH2), 11.3, 11.2, 9.9, 9.5 ppm; MALDI-TOF MS: found m/z
= 985.48 (M
+ H+), calculated for C62H68FeN6O2 984.49; IR (KBr): ν 2940, 2898, 2854, 1104, 1002, 964, 796, 740 cm−1; UV-Vis λmax
(CH2Cl2) 418, 514, 548, 646 nm; fluorescence, λEM
(CH2Cl2) 655, 720 nm (λEx 418 nm).
C), 7.11 (d, 3Jtrans(1H-1H)
= 16.0 Hz, 1H; C=CH-), 6.12–6.26 (m, 2H; allyl-CH), 5.52 (dd, J
= 16.47, 1.62 Hz, 2H; allyl-Htrans), 5.49 (d, J
= 1.62 Hz, 1H; Im-H5), 5.41 (d, J
= 4.59 Hz, 2H; pyrrole-Hβ), 5.32 (dd, J
= 10.53, 1.35 Hz, 2H; allyl-Hcis), 5.24 (t, J
= 7.03 Hz, 4H; -CH2), 4.68 (t, J
= 1.62 Hz, 2H; Fc-H), 4.41 (t, J
= 1.89 Hz, 2H; Fc-H), 4.32 (s, 5H; Fc-H), 4.22–4.24 (m, 4H; -OCH2), 3.93–3.94 (m, 4H; -CH2), 2.99–3.11 (m, 4H; CH2), 2.14 (d, 1H, J
= 1.35 Hz; Im-H4), 1.67 ppm (s, 3H; N-Me); 13C NMR (67 MHz, CDCl3): δ 151.0, 149.7, 148.9, 148.0 (pyrrole-α), 146.1 (Im-C2), 142.7 (Ph), 136.7 (Ph), 135.2 (CH-allyl), 132.0, 128.2, 127.2, 127.0 (pyrrole-β), 129.2, 126.1, 121.2 (Im-C4), 119.1 (meso), 117.7 (Im-C5), 116.7 (CH2-allyl), 96.1 (meso), 83.7, 72.1 (-OCH2), 70.1, 69.4 (-CH2-CHCH2), 69.2, 67.1, 38.6 (CH2), 32.7 (N-CH3), 32.2, 31.7 (CH2), 22.74, 14.2 ppm; MALDI-TOF MS: found m/z
= 934.38 (M
+ H+), calculated for C54H50FeN6O2Zn 934.26; IR (KBr): ν 2920, 2852, 1483, 1340, 1104, 1002, 986, 789 cm−1; UV-Vis max
(CH2Cl2) 415, 440, 565, 622 nm.
C), 7.09 (d, 3Jtrans(1H–1H)
= 16.2 Hz, 1H; C=CH-), 6.26–6.12 (m, 2H; allyl-CH), 5.55 (dd, J
= 10.26, 1.62 Hz, 2H; allyl-Htrans), 5.49 (d, J
= 1.62 Hz, 1H; Im-H5), 5.41 (d, J
= 4.86 Hz, 2H; pyrrole-Hβ), 5.35 (dd, J
= 10.26, 1.35 Hz, 2H; allyl-Hcis), 5.24 (m, 4H; -CH2), 4.22–4.24 (m, 4H; -OCH2), 3.91–3.94 (m, 4H; -CH2), 3.52 (brs, 1H; CH-Cp), 3.00–3.12 (m, 4H; CH2), 2.17, 1.92, 1.87, 1.82 (s, 24H; (CH3)8-Fc), 2.14 (d, 1H, J
= 1.35 Hz, Im-H4), 1.67 ppm (s, 3H; N-Me); 13C NMR (67 MHz, CDCl3): δ 150.9 149.7, 148.9, 147.8 (pyrrole-α), 146.0 (Im-C2), 142.1 (Ph), 137.7 (Ph), 135.2 (CH-allyl), 132.0, 129.1, 128.1 126.9 (pyrrole-β), 123.0, 122.9, 121.3, 121.2 (Im-C4), 119.0 (meso), 117.6 (Im-C5), 116.7 (CH2-allyl), 96.0 (meso), 83.7, 72.1 (-OCH2), 70.1, 69.4 (-CH2-CH = CH2), 69.2, 67.1, 38.6 (CH2), 32.7 (N-CH3), 32.2, 31.7 (CH2), 22.6, 15.2, 14.1, 10.7, 10.5, 9.3, 8.7 ppm; MALDI-TOF MS: found m/z
= 1045.48 (M
+ H+), calculated for C62H66FeN6O2Zn 1045.39; IR (KBr: ν 2941, 2900, 2853, 1628, 1428, 1340, 1104, 1002, 986, 788, 713 cm−1; UV-Vis λmax
(CH2Cl2) 413, 439, 564, 622 nm.
), 5.44 (dd, J
= 17.28, 1.62 Hz, 2H; allyl-Htrans), 5.28 (m, J
= 10.26, 1.08 Hz, 2H; allyl-Hcis), 5.13 (t, J
= 7.3 Hz, 4H; -CH2), 4.26 (t, J
= 1.89 Hz, 2H; Fc-H), 4.25 (s, 5H; Fc-H), 4.23 (t, J
= 1.62 Hz, 2H; Fc-H), 4.08 (dd, J
= 5.4, 1.35 Hz, 4H; CH2), 3.68 (t, J
= 5.94 Hz, 4H; -CH2), 3.39 (s, 3H; N-Me), 3.17–3.23 (m, 2H; CH2), 2.95–3.01 (m, 2H; CH2), 2.81–2.76 (m, 4H; CH2), −2.65 ppm (s, 2H; inner-NH); 13C NMR (67 MHz, CDCl3): δ 148.8 (Im-C2), 141.5 (Ph), 139.81 (Ph), 134.9 (CH-allyl), 134.2 (Ph), 132.1, 130.3, 129.0, 127.5 (pyrrole-β), 128.2 (Im-C4), 126.9 (Ph), 126.5 (Ph), 121.2 (Ph), 120.8 (Im-C5), 190.2, 119.4 (meso), 116.7 (CH2-allyl), 104.1 (meso), 88.5 (C-Cp), 76.5 (C-Cp), 71.9 (-OCH2), 69.4 (CH-Cp), 69.1 (-CH2-CH=CH2), 68.6 (CH-Cp), 68.3, 67.3, 54.85, 37.8 (CH2), 34.5 (N-CH3), 32.0 ppm (CH2); MALDI-TOF MS: found m/z
= 874.70 (M
+ H+), calculated for C54H52FeN6O2 874.37; UV-Vis λmax
(CH2Cl2) 418, 519, 550, 649 nm; fluorescence, λEM
(CH2Cl2) 654, 720 nm (λEx 418 nm).
), 5.48 (dd, J
= 16.47, 1.62 Hz, 2H; allyl-Htrans), 5.41 (d, J
= 1.62 Hz, 1H; Im-H5), 5.35 (d, J
= 4.59 Hz, 2H; pyrrole-Hβ), 5.28 (dd, J
= 10.53, 1.35 Hz, 2H; allyl-Hcis), 5.16 (t, J
= 7.03 Hz, 4H; -CH2), 4.26 (t, J
= 1.62 Hz, 2H; Fc-H), 4.22 (s, 5H; Fc-H), 4.17 (t, J
= 1.89 Hz, 2H; Fc-H), 4.15–4.20 (m, 4H; -OCH2), 3.83–3.88 (m, 4H; -CH2), 3.18–3.23 (m, 4H; CH2), 2.90–3.10 (m, 4H; CH2), 2.10 (d, J
= 1.35 Hz, 1H; Im-H4), 1.60 ppm (s, 3H; N-Me); 13C NMR (67 MHz, CDCl3): δ 151.0, 149.7, 149.1 147.9 (pyrrole-α), 146.1 (Im-C2), 141.6 (Ph), 140.8 (Ph), 135.3 (CH-allyl), 134.6 (Ph), 132.0, 129.2, 126.9, 126.4 (pyrrole-β), 128.1 (Im-C4), 126.2, 121.4, 119.0 (meso), 117.7 (Im-C5), 116.7 (CH2-allyl), 96.0 (C, meso), 88.8, 72.1 (-OCH2), 70.2, 68.7 (-CH2-CH=CH2), 68.4, 67.4, 38.7 (CH2), 32.7 (N-CH3), 32.7, 32.2 ppm (CH2); MALDI-TOF MS: found m/z
= 936.57 (M
+ H+), UV-Vis λmax
(CH2Cl2) 414, 437, 565, 619 nm, fluorescence, λEM
(CH2Cl2) 623, 680 (λEx 437 nm).
Acknowledgements
This work was supported by Grant-in-Aids for Scientific Research (A) (No. 15205020) and for Scientific Research on Priority Areas (No. 16033244, Reaction Control of Dynamic Complexes) from Ministry of Education, Culture, Sports, Science and Technology, Japan (Monbu Kagakusho).References
- (a) J. Deisenhofer and J. R. Norris, The Photosynthetic Reaction Center, Vols. I and II, Academic Press, New York, 1993 Search PubMed; (b) M. E. Michel-Beyerle, The Reaction Center of Photosynthetic Bacteria, Springer, Berlin/Heidelberg, 1996 Search PubMed; (c) H. van Amerongen, L. Valkunas and R. van Grondelle, Photosynthetic Excitations, World Scientific, Singapore, 2000 Search PubMed.
- (a) A. Zouni, H.-T. Witt, W. J. Kern, P. Fromme, N. Krauss, W. Sanger and P. Orth, Nature, 2001, 409, 739 CrossRef CAS; (b) P. Jordan, P. Fromme, H.-T. Witt, O. Kukas, W. Sanger and N. Krauss, Nature, 2001, 411, 909 CrossRef CAS.
- (a) J. Koepke, X. Hu, C. Muenke, K. Schulten and H. Michel, Structure, 1996, 4, 581 CrossRef CAS; (b) K. McLuskey, S. M. Prince, R. J. Cogdell and N. W. Isaacs, Biochemistry, 2001, 40, 8783 CrossRef CAS.
- (a) M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS; (b) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS; (c) J. Vasudevan, R. T. Stibrany, J. Bumby, S. Knapp, J. A. Potenza, T. J. Emge and H. J. Schugar, J. Am. Chem. Soc., 1996, 118, 11676 CrossRef CAS.
- (a) Y. Kobuke and H. Miyaji, J. Am. Chem. Soc., 1994, 116, 4111 CrossRef CAS; (b) Y. Kobuke and K. Ogawa, Bull. Chem. Soc. Jpn., 2003, 76, 689 CrossRef CAS.
- (a) R. Takahashi and Y. Kobuke, J. Am. Chem. Soc., 2003, 125, 2372 CrossRef CAS; (b) Y. Kuramochi, A. Satake and Y. Kobuke, J. Am. Chem. Soc., 2004, 126, 8668 CrossRef CAS; (c) C. Ikeda, A. Satake and Y. Kobuke, Org. Lett., 2003, 5, 4935 CrossRef CAS; (d) R. Takahashi and Y. Kobuke, J. Org. Chem., 2005, 70, 2745 CrossRef CAS.
- (a) K. Ogawa, A. Ohashi, Y. Kobuke, K. Kamada and K. Ohata, J. Am. Chem. Soc., 2003, 125, 13356 CrossRef CAS; (b) K. Ogawa and Y. Kobuke, Angew. Chem., Int. Ed., 2000, 39, 4070 CrossRef CAS.
- C. E. D. Chidsey, Science, 1991, 251, 919 CrossRef CAS.
- (a) D. J. Campbell, B. R. Herr, J. C. Hulteen, R. P. Van Duyne and C. A. Mirkin, J. Am. Chem. Soc., 1996, 118, 10211 CrossRef CAS; (b) S. Creager, C. J. Yu, C. Bamdad, M. Gozin and J. F. Kayyem, J. Am. Chem. Soc., 1999, 121, 1059 CrossRef CAS; (c) T. Kondo, S. Horiuchi, I. Yagi and S. Ye. K. Uosaki, J. Am. Chem. Soc., 1999, 121, 391 CrossRef CAS.
- A. J. Bard and H. Lund, Encyclopedia of Electrochemistry of the Elements, Marcel Dekker, New York, 1979, Vol. 13, p. 3 Search PubMed.
- A. Togni and T. Hyashi, Ferrocenes, VCH Verlagsgesellschaft mbH, Weinheim, Germany, 1995, references cited therein Search PubMed.
- (a) M. Hobi, O. Ruppert, V. Gramlich and A. Togni, Organometallics, 1997, 16, 1384 CrossRef CAS; (b) J. C. Calabrese, L.-T. Cheng, J. C. Green, S. R. Marder and W. Tam, J. Am. Chem. Soc., 1991, 113, 7227 CrossRef CAS; (c) S. Barlow, H. E. Bunting, C. Ringham, J. C. Green, G. U. Bubliz, S. G. Boxer, J. W. Perry and S. R. Marder, J. Am. Chem. Soc., 1999, 121, 3715 CrossRef CAS; (d) S. Barlow and D. O. Hare, Organometallics, 1996, 15, 3885 CrossRef CAS; (e) B. Bildstein, A. Hradsky, H. Kopaca, R. Malleier and K.-H. Ongania, J. Organomet. Chem., 1997, 540, 127 CrossRef CAS.
- Ferrocene porphyrin direct linkage: (a) D. T. Gryko, F. Zhao, A. A. Yasseri, K. M. Roth, D. F. Bocian, W. G. Kuhr and J. S. Lindsey, J. Org. Chem., 2000, 65, 7356 CrossRef CAS; (b) C. Bucher, C. H. Devillers, J.-C. Moutet, G. Royal and E. Saint-Aman, Chem. Commun., 2003, 888 RSC; (c) R. G. Wollmann and D. N. Hendrickson, Inorg. Chem., 1977, 16, 3079 CrossRef; (d) S. W. Rhee, B. B. Park, Y. Do and J. Kim, Polyhedron, 2000, 19, 1961 CrossRef CAS; (e) S. W. Rhee, H. Y. Na, Y. Do and J. Kim, J. Inorg. Chim. Acta, 2000, 309, 49 Search PubMed; (f) J. Kim, S. W. Rhee, H. Y. Na, K. P. Lee, Y. Do and S. C. Jeoung, Bull. Korean Chem. Soc., 2001, 22, 1316 CAS; (g) P. D. W. Boyd, A. K. Burell, W. M. Campbell, P. A. Cocks, K. C. Gordon, G. B. Jameson, D. L. Officer and Z. Zhao, Chem. Commun., 1999, 637 RSC; (h) Ferrocene Linkage with spacer: E. S. Schmidt and T. S. Calderwood, Inorg. Chem., 1986, 25, 3718 Search PubMed; (i) A. K. Burell, W. M. Campbell, D. L. Officer, S. M. Scott, K. C. Gordon and M. R. McDonald, J. Chem. Soc., Dalton Trans., 1999, 3349 RSC; (j) R. Giasson, E. J. Lee, X. Zhao and M. S. Wrighton, J. Phys. Chem., 1993, 97, 2596 CrossRef CAS; (k) N. B. Thornton, H. Wojtowicz, T. Netzel and D. W. Dixon, J. Phys. Chem., 1998, 102, 2101 Search PubMed; (l) A. K. Burrell, W. M. Campbell and D. L. Officer, Tetrahedron Lett., 1997, 38, 1249 CrossRef CAS.
- A. Nomoto, H. Mitsuoka, H. Ozeki and Y. Kobuke, Chem. Commun., 2003, 1074 RSC.
- A. Nomoto and Y. Kobuke, Chem. Commun., 2002, 1104 RSC.
- (a) M. Morisue, S. Yamatsu, N. Haruta and Y. Kobuke, Chem.–Eur. J., 2005, 11, 5563 CrossRef CAS; (b) M. Morisue, D. Kalita, N. Haruta and Y. Kobuke, submitted.
- A. Ohashi, A. Satake and Y. Kobuke, Bull. Chem. Soc. Jpn., 2004, 77, 365 CrossRef CAS.
- L. R. Milgrom, P. J. F. Dempsey and D. Yahioglu, Tetrahedron, 1996, 52, 9877 CrossRef CAS.
- U. T. Mueller-Weaterhoff, Z. Yang and G. J. Ingram, J. Organomet. Chem., 1993, 463, 163 CrossRef CAS.
- (a) C. M. Fendric, L. D. Schertz, V. W. Day and T. J. Marks, Organometallics, 1988, 7, 1828 CrossRef CAS; (b) F. H. Koehler and K. H. Doll, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1982, 37, 144; (c) V. G. Schmitt and S. Ozman, Chem. Ztg., 1976, 100, 143 Search PubMed.
- B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863 CrossRef CAS.
- J. D. Surmatis and A. Ofner, J. Org. Chem., 1963, 28, 2735 CrossRef CAS.
- (a) D. E. Richardson and H. Taube, Inorg. Chem., 1981, 20, 1278 CrossRef CAS; (b) K. Funatsu, T. Imamura, A. Ichimura and Y. Sasaki, Inorg. Chem., 1998, 37, 4986 CrossRef CAS; (c) K. Fukushima, K. Funatsu, A. Ichimura, Y. Sasaki, M. Suzuki, T. Fujihara, K. Tsuge and T. Imamura, Inorg. Chem., 2003, 42, 3187 CrossRef CAS.
- H. Ozeki, A. Nomoto, K. Ogawa, Y. Kobuke, M. Murakami, K. Hosoda, M. Ohtani, S. Nakashima, H. Miyasaka and T. Okada, Chem.–Eur. J., 2004, 10, 6393 CrossRef CAS.
- K. M. Kadish, E. van Caemelebecke and G. Royal, in The Porphyrin Hand Book, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, USA, 2000, Vol. 8, pp. 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: preparation of 11, 1H and 13C NMR spectra, MALDI-TOF mass spectra, UV-Visible absorption and steady state emission spectra of all ferrocenyl porphyrin derivatives. See DOI: 10.1039/b506992k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2006 |